Introduzione... 3
CAPITOLO 1 - FARMACI BIOLOGICI E BIOSIMILARI...5
1.1 DEFINIZIONE DI FARMACO BIOSIMILARE E DI FARMACO BIOLOGICO DI RIFERIMENTO...6
1.2 SVILUPPO DEL FARMACO BIOSIMILARE...9
1.2.1 PROCESSO PRODUTTIVO...9
1.2.2 IL PRODOTTO È IL PROCESSO...12
1.2.3 SICUREZZA ED IMMUNOGENICITÀ...16
1.3 DIFFERENZE TRA FARMACI BIOSIMILARI E FARMACI BIOEQUIVALENTI. 21 1.3.1 ESERCIZIO DI COMPARABILITÀ...24
1.3.2 INTERCAMBIABILITÀ E SOSTITUZIONE AUTOMATICA: NECESSITÀ DI ELABORARE UN SISTEMA DI NOMENCLATURA UNIVOCO...29
1.4 BIOSIMILI APPROVATI AD OGGI IN EUROPA E PROSSIME SCADENZE BREVETTUALI...33
1.5 BIOSIMILARI UTILIZZATI NELLA PRATICA CLINICA...45
1.5.1 BIOSIMILARI UTILIZZATI A SCOPO TERAPEUTICO...46
Oncologia...46
Nefrologia...52
Dermatologia e reumatologia...52
Pediatria...55
1.5.1.1 IL PRESENTE DEL BIOSIMILARE TRASTUZUMAB...58
1.5.1.2 BIOSIMILARI DI CETUXIMAB, BEVACIZUMAB, ADALIMUMAB, ETANERCEPT E RITUXIMAB IN CORSO DI SVILUPPO...63
1.5.2 BIOSIMILARI UTILIZZATI NELLE TERAPIE DI SUPPORTO...67
1.5.2.1 FATTORE STIMOLANTE LE COLONIE GRANULOCITARIE (G-CSF)...69
1.5.2.2 INTERFERONI...71
CLINICA... 76
2.1 VANTAGGI DEI FARMACI BIOSIMILI...79
2.1.1 VALUTAZIONE FARMACO-ECONOMICA E MERCATO DEI FARMACI BIOSIMILARI...80
2.2 SVANTAGGI DEI FARMACI BIOSIMILI...89
CAPITOLO 3 – ITER REGISTRATIVO E QUADRO
REGOLATORIO...91
3.1 PROCEDURA DI APPROVAZIONE EUROPEA PER UN MEDICINALE BIOSIMILARE...91
3.2 LINEE GUIDA ELABORATE DALL'EMA...94
3.2.1 LINEE GUIDA GENERALI...96
3.2.1.1 PROGRAMMA DI FARMACOVIGILANZA E RISK MANAGEMENT PLAN...105
3.2.2 LINEE GUIDA CLASSE-SPECIFICHE...107
3.3 PROCESSO REGISTRATIVO DEI FARMACI BIOSIMILARI NEGLI STATI UNITI E ASPETTI REGOLATORI NEL RESTO DEL MONDO...122
Conclusioni... 139
Bibliografia... 142
Sitografia... 145
Fin dagli anni Ottanta è stata sfruttata la tecnologia del DNA ricombinante per ottenere molecole proteiche da utilizzare nel trattamento di diverse patologie quali diabete, anemia, neoplasie, sclerosi multipla e patologie infiammatorie. Da allora, grazie alla manipolazione del DNA nei batteri, lieviti e cellule di mammifero, sono state prodotte e adottate nella pratica clinica con grande successo sostanze ancora più complesse come gli anticorpi monoclonali.
Nell'ambito della biotecnologia, grazie all'estinzione della copertura brevettuale, già avvenuta o in procinto di avvenire nei prossimi anni per alcuni dei farmaci biotecnologici in commercio, si è aperto un nuovo scenario: quello dei farmaci biosimilari.
La loro denominazione deriva dal fatto che non possono essere definiti copie esatte del farmaco biologico di riferimento, bensì la loro caratteristica è quella di essere simili, ma non identici rispetto all'originatore. Questa sottile differenza risulta essere fondamentale per la loro caratterizzazione: si tratta infatti di molecole uniche nel loro genere che in definitiva non possono essere considerate come la “versione generica” del corrispettivo farmaco biologico. Essi si distinguono dai farmaci generici equivalenti sotto molteplici aspetti.
Questo concetto è conseguenza della loro complessità strutturale e del processo di produzione sfruttato per la loro messa a punto. Nell'ambito dei farmaci biotecnologici e biosimili, struttura e fabbricazione sono tra loro strettamente interconnesse, tanto che anche piccole variazioni a livello del processo produttivo possono indurre grande variabilità nella struttura della proteina ricombinante ed avere conseguenze sulle caratteristiche biologiche e cliniche del prodotto finale in termini di sicurezza, potenza ed immunogenicità. Per poter individuare anche la più piccola differenza tra farmaco biosimilare e farmaco di riferimento, riuscendo allo stesso tempo a garantire un loro sovrapponibile profilo di qualità, sicurezza ed efficacia, deve essere seguito un preciso e rigoroso iter: perché il prodotto sia approvato e immesso sul mercato, ne deve essere riconosciuta e dimostrata la comparabilità con il farmaco di riferimento, sia dal punto di vista delle proprietà fisico-chimiche, che rispetto a quanto emerge dagli studi preclinici e
l'EMA (European Medicines Agency) ha per prima elaborato nel 2005 delle linee guida generali di riferimento e, a seguire, linee guida classe-specifiche in risposta alle particolari esigenze che col tempo si sono evidenziate per le diverse classi di farmaci. Il suo lavoro è servito da input e da stampo non solo per l'OMS (Organizzazione Mondiale della Sanità), al fine di armonizzare a livello globale la materia, ma anche per altri paesi come l'Australia, il Canada, il Giappone, la Corea e per la FDA (Food And Drug Administration), per disciplinare le varie dinamiche che ruotano attorno all'argomento.
Inizialmente, al contrario di quanto ci si aspettasse, i biosimilari non hanno avuto un grande impatto sul mercato: l'insufficiente informazione e chiarezza in merito hanno fatto nascere dubbi e preoccupazioni nei clinici che spesso e volentieri continuano a preferire affidarsi ai farmaci biologici. L'aumento di prescrizioni e vendite dei medicinali biosimili, grazie al loro minor costo rispetto al farmaco di riferimento biotecnologico, permetterebbe di liberare importanti risorse economiche da reinvestire nella ricerca e produzione di farmaci innovativi e sarebbe vantaggioso sia per ridurre i costi della spesa farmaceutica da parte del sistema sanitario, che oggi si presenta sempre più provato, sia per consentire l'accesso alle cure di un numero ancora maggiore di pazienti. Senza dubbio, la prospettiva è quella di andare incontro ad un futuro in cui questi farmaci finiranno per occupare sempre più ampio spazio nel panorama internazionale, man mano che l'esperienza clinica avanzerà e che il tempo riconoscerà loro effettiva sicurezza ed efficacia.
Con il seguente lavoro si è cercato di definire al meglio quelle che sono le proprietà dei farmaci biosimilari, sottolineandone le differenze rispetto ai medicinali generici equivalenti, e di aggiornare le pratiche regolatorie in continua revisione, sfruttando come fonti i siti web dell'EMA e della FDA.
Negli ultimi anni lo sfruttamento delle biotecnologie, dove per biotecnologia si intende l'insieme dei processi che includono l'uso di sistemi biologici per identificare, sequenziare e manipolare il DNA al fine di produrre molecole terapeutiche e medicinali, ha permesso di ottenere più prodotti biofarmaceutici, conosciuti con il termine di medicinali biologici o biotecnologici. Questo tipo di sostanze è nato con lo scopo di riprodurre proteine endogene quali l'insulina, l'ormone della crescita e l'eritropoietina, per il trattamento di patologie causate dal loro deficit endogeno, ma grazie alle nuove conoscenze raggiunte nel campo sono state messe a punto molecole ancora più complesse come gli anticorpi monoclonali. Il ricorso ai farmaci biotecnologici nella pratica clinica è andato aumentando negli anni; ad oggi, essi costituiscono un'importantissima risorsa per il trattamento di diverse patologie e sono considerati trattamenti di prima linea anche nel caso di malattie autoimmuni, malattie infiammatorie croniche dell'intestino e per patologie rischiose per la vita come i tumori. Derivando da una fonte biologica come le cellule batteriche o eucariote, il loro processo di produzione è particolarmente complesso, laborioso e costoso. Più specificatamente, a questa categoria appartengono gli ormoni (come l'ormone della crescita), l'EPO, l'insulina, che è stata il primo farmaco biologico ad essere messo in commercio nel 1982 come Humulin®, gli immunomodulatori (come gli interferoni), gli emoderivati (come i fattori della coagulazione), gli enzimi, i vaccini, numerosi antibiotici e gli anticorpi monoclonali, che hanno trovato impiego nella terapia oncologica. Così come i medicinali di sintesi chimica, anche questi farmaci sono protetti dalla ventennale copertura brevettuale, che rappresenta lo strumento grazie al quale è riconosciuto al detentore del brevetto il diritto esclusivo di realizzare il farmaco e di commercializzarlo impedendo ad altre aziende di poter fare altrettanto. Poiché il periodo che intercorre tra la presentazione della domanda di brevetto e il momento in cui viene rilasciata l'autorizzazione all'immissione in commercio (AIC) va dai 10 ai 12 anni, sono nati i Certificati Complementari di Protezione (CPC), della durata di massimo 5 anni. Essi hanno lo scopo di allungare i tempi di esclusività della presenza del farmaco in commercio, consentendo di recuperare parzialmente il tempo necessario per l'ottenimento dell’AIC e garantendo in tal modo un maggior ritorno
l'opportunità per le aziende produttrici, o per altre, di intraprendere lo studio e lo sviluppo di farmaci biosimilari, che rappresentino una valida alternativa al trattamento con il farmaco biologico di riferimento, sia per le similari caratteristiche di qualità, sicurezza ed efficacia che per il minor costo.
1.1 DEFINIZIONE DI FARMACO BIOSIMILARE E DI FARMACO
BIOLOGICO DI RIFERIMENTO
Nella stessa denominazione di “biosimilare” è racchiuso il suo significato. Infatti, per biosimilare o biosimile, si intende un prodotto medicinale che è simile, ma non identico, al biofarmaceutico di riferimento. Anche questa versione viene prodotta in laboratorio a partire da linee cellulari e sistemi viventi, tentando di riprodurre una molecola con la stessa identica struttura del farmaco originatore. Mentre in Europa questi medicinali sono conosciuti con l'appellativo di biosimilari, negli USA e in Giappone essi sono denominati Follow-On Biologics, in Australia Similar Biological Medicinal Products (SBMP), in Canada Subsequent-Entry Biologics e dall'Organizzazione Mondiale della Sanità, sono definiti come Similar Biotherapeutic Products (SBPs). L'Agenzia Europea dei Medicinali (EMA) nella sua linea guida “Guideline on Similar Biological Medicinal Products” emanata nel 2005 e successivamente aggiornata, fornisce la seguente definizione di biosimilare: “A biosimilar is a biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product (reference medicinal product). [..] The active substance of a similar biological medicinal product must be similar, in molecular and biological terms, to the active substance of the reference medicinal product. For example, a medicinal product containing interferon alfa-2a manufactured by Company X claiming to be similar to another biological medicinal product should refer to a reference medicinal product containing as its active substance interferon alfa-2a. Therefore, a medicinal product containing interferon alfa-2b could not be considered as the reference medicinal product. The pharmaceutical form, strength and route of administration of the similar biological medicinal product should be the same as
exercise should be provided. Any differences between the similar biological medicinal product and the reference medicinal product will have to be justified by appropriate studies on a case-by-case basis.”1
Negli USA, la Sezione 351(i)(2) del Public Health Service Act (PHS Act) riconosce che: “The term ‘biosimilar’ or ‘biosimilarity’, in reference to a biological product, means that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components; and there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.”2
L'Organizzazione Mondiale della Sanità si limita a stabilire che per SBP si intende: “a biotherapeutic product which is similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product.”3
Allo stesso tempo, le agenzie regolatorie hanno cercato di precisare quali caratteristiche dovesse avere il medicinale biologico di riferimento, conosciuto anche con gli appellativi di medicinale innovatore o originator, per essere considerato tale. Secondo l'EMA “il prodotto medicinale di riferimento deve essere un prodotto medicinale autorizzato nello Spazio Economico Europeo, sulla base di un dossier completo, nel rispetto di quanto stabilito dall'Articolo 8 della Direttiva 2001/83/EC. [..] Comunque, con lo scopo di
1 “Un biosimilare è un prodotto medicinale biologico che contiene una versione della sostanza attiva di un prodotto medicinale biologico originale già autorizzato (prodotto medicinale di riferimento). [...] La sostanza attiva di un prodotto medicinale biologico similare deve essere simile, in termini biologici e molecolari, alla sostanza attiva del prodotto medicinale di riferimento. Per esempio, un prodotto medicinale contenente interferone alfa-2a prodotto dall'Azienda X che viene definito similare ad un altro prodotto medicinale biologico, si dovrebbe riferire a un prodotto medicinale di riferimento contenente come sostanza attiva l'interferone alfa-2a. Di conseguenza, un prodotto medicinale contenente interferone alfa-2b non dovrebbe essere considerato come prodotto medicinale di riferimento. La forma farmaceutica, la potenza e la via di somministrazione del prodotto medicinale biologico similare dovrebbe essere la stessa del prodotto medicinale di riferimento. Quando la forma farmaceutica, la potenza o la via di somministrazione non è la stessa, dovranno essere forniti ulteriori dati nel contesto dell'esercizio di comparabilità. Qualsiasi differenza tra il prodotto medicinale biologico similare e il prodotto medicinale di riferimento dovrà essere giustificata attraverso studi appropriati definiti caso per caso.”
2 “Il termine “biosimilare” o “biosimilarità”, riferito ad un prodotto biologico, significa che il prodotto biologico è altamente similare al prodotto di riferimento malgrado le sottili differenze a livello dei componenti clinicamente inattivi; e che non ci sono differenze clinicamente rilevanti tra il prodotto biologico e il prodotto di riferimento in termini di sicurezza, purezza e potenza del prodotto.”
3 “Un prodotto bioterapeutico che è similare in termini di qualità, sicurezza ed efficacia ad un prodotto bioterapeutico di riferimento già autorizzato.”
clinici in vivo (dove necessari), con un prodotto non autorizzato nello SEE (ovvero una versione non autorizzata del prodotto medicinale di riferimento), che dovrà essere autorizzato da un'agenzia regolatoria con standard scientifici e regolatori uguali a quelli stabiliti dall'EMA. In più, sarà responsabilità del richiedente stabilire che il medicinale con cui è effettuato lo studio comparativo, autorizzato al di fuori dello SEE, sia rappresentativo del prodotto di riferimento autorizzato nello SEE.”4
Con la possibilità di prendere come prodotto di riferimento un medicinale non autorizzato nello SEE, si affaccia la possibilità di poter ridurre i costi di sviluppo di un biosimilare. Su decisione dell'EMA, è dal 2012 che gli studi condotti altrove, su un prodotto biologico di riferimento, possono essere usati comunque nella realtà europea; prima di quell'anno, “gli studi dovevano essere ripetuti su pazienti europei, usando un prodotto di riferimento approvato nell'Unione Europea. L'unico presupposto è appunto che il richiedente dimostri che i lotti utilizzati fuori dallo SEE siano rappresentativi del farmaco di riferimento autorizzato nello SEE, attraverso un'approfondita comparazione analitica. L'EMA richiede dati comparativi farmacocinetici e farmacodinamici per accertare l'adeguatezza dei lotti del prodotto di riferimento.”5
Quindi a livello globale è riconosciuto che il biosimilare è una versione copia di un farmaco biologico innovatore già autorizzato sulla base di un dossier completo, e possiede lo stesso tipo di sostanza attiva biologica intesa come “sostanza che è prodotta o estratta da una fonte biologica e che richiede una combinazione di studi fisico-chimico-biologici insieme al processo di produzione e al suo controllo, per essere caratterizzata e per determinarne la qualità.”6 I due prodotti devono avere lo stesso profilo di sicurezza ed efficacia e, in genere, sono usati con la stessa forma farmaceutica e stessa via di somministrazione, per il trattamento delle stesse condizioni.
4 European Medicines Agency. “Guideline on Similar Biological Medicinal Products”-Draft CHMP/437/04 Rev 1, pag 5/6, 22 Maggio 2013
5 Sito web GaBI online-Generics and Biosimilars Initiative “EMA to accept biosimilar reference medicines from outside
EEA” http://gabionline.net/Guidelines/EMA-to-accept-biosimilar-reference-medicines-from-outside-EEA, 5 Ottobre 2012
6 European Medicines Agency. Q&A: Similar biological products-Question 1-10: Similar-biological-product applications, pag 1/7
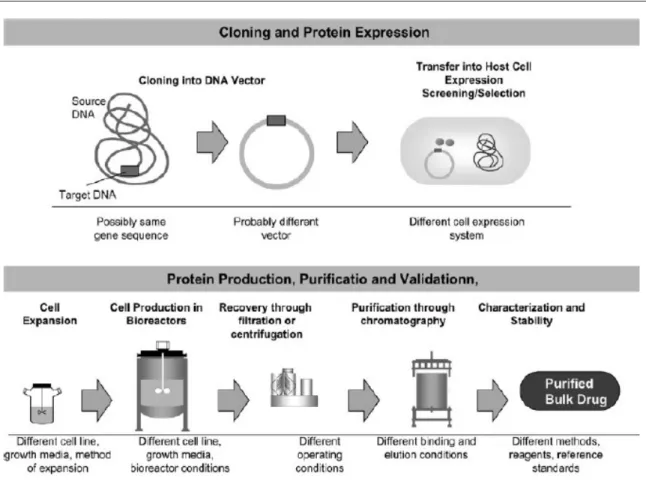
La messa a punto di un farmaco biotecnologico o biosimile che sia, comporta un notevole impegno economico da parte dei produttori, poiché sono sfruttati processi particolarmente lunghi e complessi. Come già illustrato, la sostanza attiva del farmaco biologico, e quindi del biosimile, deriva da organismi viventi che sono preparati ad hoc, attraverso la manipolazione del loro DNA, con lo scopo di generare una grande quantità di principio attivo desiderato. Anche la produzione di questi medicinali deve seguire quanto predisposto dalle GMP, Good Manifacturing Pratice. Queste norme prevedono interventi ispettivi nei laboratori durante i processi di produzione per verificare che il tutto avvenga in osservanza delle linee guida, in ambienti idonei ed in sicurezza.
1.2.1 PROCESSO PRODUTTIVO
Per ottenere il prodotto finale richiesto vengono messe in pratica le conoscenze su cui si basa la tecnologia del DNA ricombinante: introducendo nel DNA dell'ospite (che può essere una cellula batterica o di lievito) la precisa sequenza nucleotidica che codifica per la proteina di interesse, è ottenuta la sostanza attiva. Essa, una volta purificata e analizzata, è inserita nell'opportuna formulazione farmaceutica, confezionata ed immessa in commercio. Le varie fasi del ciclo produttivo comprendono:
l'allestimento del vettore e il suo inserimento nella cellula ospite; l'isolamento e la crescita selettiva delle cellule ospiti;
la produzione della proteina ricombinante; la sua purificazione;
l'analisi della sostanza attiva ottenuta; la formulazione finale del preparato;
confezionamento e conservazione del farmaco.
Nello specifico, il primo passo da compiere è quello di recuperare la sequenza di DNA codificante la proteina di interesse sfruttando enzimi capaci di effettuare precisi tagli a livello del DNA di partenza; la sequenza di DNA viene inserita in uno specifico vettore (ad
dell'ospite permette alla cellula di trascrivere, tradurre e quindi esprimere, oltre ai suoi geni, anche il gene introdotto dall'esterno, codificante per la proteina desiderata. Successivamente, grazie all'uso di specifici marcatori, sono individuate soltanto le cellule che hanno inglobato il DNA esogeno all'interno del loro patrimonio genetico, in maniera tale da poterle fare selettivamente crescere e duplicare in opportuni terreni di coltura, al fine di ottenere linee cellulari pure e geneticamente identiche. La linea cellulare capace di produrre la maggior quantità di proteina desiderata è la fonte primaria della proteina biotecnologica ed è designata con l'appellativo di “banca cellulare master”; viene congelata e conservata. E' da essa che originano le “banche cellulari working”: dalla coltura master sono prelevate delle cellule che una volta espanse in numero, vanno a costituire le cellule sfruttate per la produzione del principio attivo. Per avere grandi quantità di proteina ricombinante, le cellule sono sistemate nei bioreattori, apparecchiature nelle quali, attraverso processi di fermentazione, aumenta notevolmente il volume delle colture cellulari iniziali. Proprio in questi terreni di coltura, in condizioni controllate di pH, temperatura e ossigeno, ciascuna cellula secerne il prodotto ricombinante, che è isolato attraverso successivi passaggi di centrifugazione e filtrazione. Conclusa questa fase occorre sottoporre il filtrato ottenuto al processo di purificazione per eliminare le impurità presenti come elettroliti, zuccheri e prodotti di degradazione. Questa fase risulta essere estremamente delicata: impurità residue alterano la qualità del prodotto finale con potenziali ripercussioni a livello clinico. Per verificare le proprietà della sostanza attiva ricombinante purificata si procede con la sua caratterizzazione, studiandone la struttura e la stabilità; efficacia e sicurezza non possono invece essere definite a questo punto del processo produttivo dato che non sono usati test o metodi di analisi sufficientemente sensibili ad ottenere questo tipo di informazioni. Con l'aggiunta degli opportuni eccipienti si arriva alla messa a punto della forma farmaceutica desiderata, la quale, una volta confezionata, è sottoposta al controllo di qualità finale. Perché nel tempo siano mantenute inalterate le caratteristiche del medicinale è bene indicare le giuste modalità di conservazione, al fine di non alterarne le proprietà farmacologiche (Illustrazione 1).
Dal punto di vista delle tempistiche occorrono, in media, fino a 7-8 anni per arrivare ad avere a disposizione un medicinale già comparabile al biologico innovatore. In particolare, circa tre anni sono necessari per ottenere le banche cellulari; di questi, un anno e mezzo è impiegato per sviluppare il prodotto ricombinante, purificarlo e per produrlo su scala più ampia; servono poi dai 3 ai 5 anni per condurre i test analitici e clinici richiesti per il biosimilare, per dimostrare la sua comparabilità in termini di qualità, sicurezza ed efficacia nei confronti del medicinale biologico di riferimento.
Illustrazione 1 - Processo di produzione di medicinali biotecnologici e biosimilari (Fonte: Dranitsaris G.Drugs 2011)
1-1,5 anni Fase 3: sviluppo del purificazione
Fase 4: produzione su scala più ampia
3,5-4,5 anni Fase 5: test di comparabilità caratterizzazione analitica studi non-clinici e clinici [“Biosimilari
0 1 2 3 4 8 anni La guida”, 2011]
1.2.2 IL PRODOTTO È IL PROCESSO
Esiste una stretta correlazione tra il processo di produzione e le proprietà del medicinale biologico. Esse dipendono così profondamente dalle tecniche produttive che si afferma che “il prodotto è il processo”: la struttura, la funzione e la qualità di un biosimilare sono il diretto risultato del processo produttivo sfruttato per la produzione del farmaco.
Considerando il fatto che le caratteristiche strutturali della proteina si ripercuotono sulla sua funzione, anche minime differenze nella configurazione primaria, secondaria, terziaria e quaternaria della sua struttura amminoacidica, possono avere conseguenze importanti. Ad esempio, l'anomalo ripiegamento della sequenza peptidica può comportare il mancato riconoscimento del target e del sito d'azione, con l'effetto di inefficienza terapeutica del farmaco, oppure una risposta immunitaria da parte dell'organismo che la riconosce come molecola estranea. I fattori legati al processo produttivo che causano la grande variabilità delle sostanze attive di natura biologica sono numerosi; maggiore è la complessità strutturale, maggiore è il numero delle possibili modificazioni. Questa variabilità è conosciuta con il termine di “microeterogeneità”: essa non garantisce la riproducibilità della stessa struttura tra lotti differenti sviluppati all'interno dello stesso sito di produzione (ovvero fabbricati sfruttando le stesse dinamiche produttive), figurarsi allora tra molecole prodotte da laboratori differenti (che pertanto adottano procedure diverse). La microeterogeneità dipende dal tipo di cellula ospite scelta, dalle tecniche usate per il trasferimento del gene di interesse all'interno del suo DNA, dalle condizioni di espansione clonale e dalle tecniche di estrazione e purificazione. Differenti linee cellulari promuovono particolari e specifiche modificazioni post-traduzionali: è fondamentale selezionare la
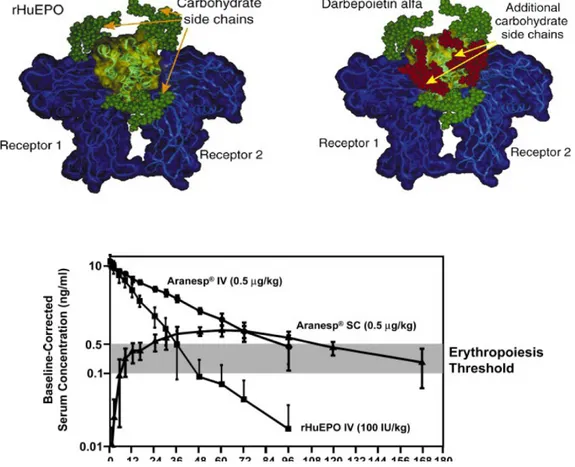
farmacocinetiche del farmaco. Il profilo di glicosilazione, infatti, va ad influenzare numerosi parametri: le proprietà fisiche (come la stabilità, riducendo la suscettibilità alla proteolisi, e la solubilità, proteggendo i residui idrofobici); l'attività biologica (scatenando la citotossicità cellulare anticorpo-dipendente o l'immunogenicità) e le proprietà farmacocinetiche (tra cui l'emivita e la clearance). Un esempio del peso del diverso pattern di glicosilazione sulle caratteristiche farmacocinetiche è rappresentato dal caso della darbaepoetina alfa, una proteina che stimola l'eritropoiesi, analogo dell'eritropoietina umana, da cui si distingue per due siti di N-glicosilazione aggiuntivi, che hanno ripercussione sulla sua attività biologica; come evidenziato nell'Illustrazione 2 questa sottile differenza trova esito clinico nell'emivita dell'epoetina biosimilare, che risulta più lunga di quella dell'eritropoietina endogena.
Illustrazione 2 - Darbapoetina alfa ed eritropoietina umana - Emivita a confronto (Fonte: Elliott
chimerico che ha come bersaglio d'azione la proteina di superficie CD20 dei linfociti B, usato sia in campo tumorale che reumatologico. “Il suo effetto terapeutico dipende dalla funzione effettrice della porzione Fc, a sua volta modulata dal grado di glicosilazione. L'analisi di alcuni lotti con date di scadenza comprese tra settembre 2007 e ottobre 2011 ha evidenziato un cambiamento del profilo di qualità nei lotti in scadenza dal 2010, rilevando una diminuzione del contenuto di lisina C-terminale e di glutamina N-terminale dal 30-50% al 10%. Sebbene questa variabilità non abbia un significato clinico, ha rivelato tuttavia un'alterazione del prodotto associata a un cambiamento nel processo produttivo. Significativo si è invece rivelato l'incremento della quota di glicani non fucosilati e di conseguenza, la citotossicità cellulare anticorpo-dipendente, un fattore essenziale per il meccanismo d'azione di rituximab, e quindi per la sua efficacia clinica.”7 (Illustrazione 3)
7 Redazione Scientifica di Value Relations “Farmaci biotecnologici biosimilari: innovazione e sostenibilità” Edito da AboutPharma pag 19/117
Illustrazione 3 - Citotossicità cellulare anticorpo-dipendente nel caso del rituximab (Fonte: Jefferis R.Trends Pharmacol Sci 2009)
del medicinale biologico. E' opportuno perciò, una volta individuata l'alterazione della struttura, riconsiderare le procedure produttive scelte. Non solo, è ovvio che è ancora più alta la probabilità che si ottengano prodotti diversi se si introducono cambiamenti volontari nelle procedure seguite nell'iter produttivo o nella formulazione.
Il produttore del biosimilare non può fare affidamento sulle conoscenze già utilizzate per lo sviluppo dell'originatore perché al produttore del prodotto biotecnologico di riferimento è riconosciuta la protezione di tali informazioni: ciò porta inevitabilmente allo sfruttamento di diverse linee cellulari, diversi terreni di coltura per la crescita delle cellule, diversi processi di produzione e diverse tecniche di purificazione che avranno come conseguenza l'ottenimento di un prodotto unico nel suo genere, che mai potrà essere esattamente equivalente al biologico di riferimento, ma che verosimilmente sarà il più similare possibile al farmaco originator. Essendo impossibile da parte del produttore del biosimilare accedere ai dati relativi al farmaco innovatore, l'azienda responsabile dello sviluppo del primo deve condurre analisi su più lotti del prodotto di riferimento. Si devono considerare sia lotti prodotti nel recente passato, sia lotti fabbricati durante tutto il periodo post-approvazione, inclusi i lotti che hanno subito cambiamenti nel processo produttivo.
E' importante che i produttori di medicinali biologici e biosimilari abbiano esperienza nel campo, che sappiano quale può essere la conseguenza del cambiamento delle procedure di produzione e che conoscano le opportune tecniche analitiche grazie alle quali è possibile individuare le proprietà delle proteine biosimilari e le loro differenze con il farmaco biotecnologico di riferimento. In uno studio in cui sono state messe a confronto 11 epoetine prodotte in diversi stati al di fuori di Europa e Stati Uniti, è emerso quanto fossero differenti i prodotti similari ottenuti su stampo dell'eritropoietina biologica, e quanto la loro attività in vitro variasse di un intervallo compreso tra il 71% e il 226%. Inoltre, cinque tra queste non rispondevano alle indicazioni specifiche individuali. Indubbiamente le conoscenze messe in campo da questi paesi (dove peraltro non esiste, al contrario dei paesi europei e degli Stati Uniti, una regolamentazione così rigorosa) sono state diverse, così come le metodiche di produzione adottate; questo spiega la non-similarità di ciò che è stato ottenuto rispetto al prodotto endogeno (Illustrazione 4).
1.2.3 SICUREZZA ED IMMUNOGENICITÀ
Sicurezza ed immunogenicità sono fattori di notevole rilevanza nel trattare i medicinali biologici e biosimilari: queste sostanze, data la loro natura proteica, nel caso in cui siano riconosciute come strutture non-self, sono in grado di indurre l'organismo a produrre anticorpi. Questo tipo di risposta varia da farmaco a farmaco, da paziente a paziente e nella maggior parte dei casi non è evidenziabile durante lo svolgimento degli studi clinici pre-approvazione: i test svolti in vitro o in vivo spesso e volentieri non riescono a dare informazioni precise né sul tipo di risposta immunogenica né sulla gravità, e non riescono ad individuare gli eventi avversi rari. Poiché le tecniche analitiche odierne non risultano abbastanza sensibili e sufficienti a definire con assoluta precisione la struttura finale della proteina o ad individuare i piccoli cambiamenti strutturali a cui essa può essere soggetta, l'uso di questa classe di farmaci deve essere seguito e monitorato nei momenti successivi all'entrata sul mercato attraverso un rigoroso studio di farmacovigilanza, volto a ridurre i rischi derivanti dal loro uso, per verificarne la sicurezza e tutelare così la salute dei pazienti. Nel migliore dei casi, la produzione anticorpale non ha alcuna conseguenza sull'attività del medicinale e non porta ad effetti visibili; può invece accadere che si
Illustrazione 4 - Epoetine prodotte in Corea, Argentina, Cina e India: differenze strutturali reciproche e rispetto al campione considerato (Fonte: Sharma B. Biotechnology
caso in cui, specialmente dopo somministrazioni ripetute del farmaco, si hanno conseguenze cliniche caratterizzate dallo sviluppo di allergie, shock anafilattici o gravi patologie (poiché gli NAbs reagiscono in maniera crociata con la proteina endogena). Tra i più noti avvenimenti di questo genere si ricorda il caso Eprex®. Alla fine degli anni Novanta è stato lanciato sul mercato questo farmaco a base di epoetina alfa, una glicoproteina nata per riprodurre l'EPO endogena, utilizzato per il trattamento dell'anemia nei pazienti con patologie croniche renali o in pazienti sottoposti a chemioterapia. Nonostante fosse stata stabilita la comparabilità tra il biosimilare Eprex® e il biologico di riferimento Epogen®, nei pazienti con patologie renali trattati con Eprex® è stato osservato un notevole aumento nell'incidenza della PCRA anticorpo-mediata, ovvero di aplasia delle cellule della serie rossa del sangue, con conseguente necessità di trasfusione. L'insorgenza di tale evento avverso è stata ricollegata a un cambio nella formulazione del prodotto usato per somministrazione sottocutanea: in particolare era stata sostituita l'albumina con uno stabilizzatore, il Polisorbato 80. Il cappuccio della siringa in gomma, non rivestito, aveva interagito con il nuovo elemento, rilasciando residui nella preparazione. Essi, agendo come adiuvanti, avevano indotto la produzione di anticorpi neutralizzanti l'eritropoietina endogena, causando questo tipo di reazione tossica. L'introduzione di un rivestimento in fluororesina ha visto diminuire fin da subito i casi di sviluppo di PCRA. Questa esperienza ha dimostrato come e a quale prezzo il potenziale immunogenico rischi di variare a seguito di cambiamenti nelle procedure di produzione e confezionamento. Altri casi per cui la modifica strutturale della sostanza attiva ha portato ad un aumento del profilo immunogenico del medicinale sono riportati di seguito:
– Interferone beta: per la deglicosilazione dei residui idrofobici non protetti è aumentata l'incidenza di eventi avversi;
– GM-CSF: in seguito all'esposizione di siti antigenici nel materiale derivante dal lievito e glicosilato soltanto ai residui N-legati, è aumentata la risposta immunogenica;
Il potenziale immunogenico di un medicinale biologico o biosimilare è legato a molteplici fattori: è influenzato dalle proprietà strutturali, quindi dalle differenze tra le caratteristiche della proteina biologica e della proteina endogena, e dal profilo di glicosilazione; al contempo, anche altri fattori che si ricollegano ai processi produttivi condizionano l'insorgenza di possibili eventi avversi, come ad esempio il grado di contaminanti e impurità che si ritrovano nella preparazione finale, la formulazione del farmaco, la dose e la via di somministrazione, la lunghezza del trattamento, le caratteristiche personali di ciascun paziente e altri fattori non conosciuti. In merito all'influenza dello stato del paziente sulla risposta al biosimilare si può dire che gli elementi da tenere in considerazione nell'interpretazione dei dati raccolti sono: il suo profilo genetico, il quadro patologico di cui è affetto, le sue condizioni e la possibilità che già in precedenza fosse stato trattato con tale farmaco (Illustrazione 5).
Nemmeno gli studi clinici effettuati in vivo o negli animali possono dare giuste informazioni sull'incidenza di tutti i possibili AEs (eventi avversi) di cui quello specifico biosimilare può essere responsabile. Perciò le agenzie regolatorie richiedono che la sicurezza clinica sia valutata anche nella fase post-marketing: la consegna di report periodici ai corpi regolatori contribuisce a fornire indicazioni sulla tollerabilità di ciascun farmaco, includendo una valutazione sulla frequenza/causalità degli eventi avversi che si manifestano a livello globale. Conoscere queste informazioni consente di realizzare nel più breve tempo possibile se il farmaco costituisce un pericolo per la salute del paziente e, nel caso in cui si dimostri tale, permette alle autorità competenti di prendere i giusti provvedimenti, come il ritiro del farmaco dal mercato. Nell'identificazione delle reazioni avverse, per cui hanno un ruolo centrale i medici e i farmacisti, i report devono comprendere il nome commerciale del biosimilare, il nome INN, il nome dell'azienda produttrice, il numero di lotto e il paese di origine del lotto usato.
Illustrazione 5 - Parametri che influenzano l'immunogenicità: proprietà strutturali, contaminanti ed impurità, formulazione, via e dose di somministrazione, lunghezza del trattamento, caratteristiche del paziente e fattori sconosciuti (Fonte: Schellekens H. Nat Rev
specifiche prendono in considerazione le relative problematiche che la classe può generare indicando le attività di farmacovigilanza più idonee da adottare. (Tabella 1)
Biosimilare Attività di farmacovigilanza addizionali Epoetine
HX 57 (Abseamed®, Binocrit,® Epoetin-alfa Hexal®)
Studio di coorte per monitorare l'incidenza di eventi tromboembolici
Sorveglianza sull'uso sottocutaneo
SB-309 (Retacrit®, Silapo®) Monitoraggio stretto attraverso l'uso di specifici questionari per stabilire eventi di PCRA e tromboembolici
Studio per valutare la sicurezza e la tollerabilità
dell'epoetina zeta somministrata per via endovenosa per il trattamento di mantenimento dell'anemia renale Studio di coorte post-autorizzazione dell'epoetina-zeta per il trattamento dell'anemia renale
Studio prospettico, aperto, non controllato e
multicentrico per valutare la sicurezza e la tollerabilità dell'epoetina zeta somministrata per via sottocutanea per il trattamento dell'anemia in pazienti con il cancro
G-CSFs
XM02 (Biograstim®, Filgrastim Ratiopharm®, Ratiograstim®, Tevagrastim®)
Procedura di rilevazione del segnale per tutti i report di risposta avversa del farmaco da qualsiasi fonte essi provengano; valutazioni anticorpali in caso di sospetta immunogenicità
Cooperazione con SCNIR, analisi dei dati corrispondenti a Biograstim-SCNIR
EP2006 (Zarzio®, Filgrastim Hexal®)
Programma di farmacovigilanza per pazienti con grave neutropenia cronica
Studio di fase 4 di dodici mesi in pazienti con grave neutropenia, seguito da un follow-up di 5 anni sulla
cellule staminali
PLD108 (Nivestim®) Questionari
Follow-up dei pazienti attraverso il registro del SCNIR Programma di cooperazione con i centri trapianto ematologico
Follow-up specializzato per dati a lungo termine Tabella 1 - Attività di farmacovigilanza addizionali8
1.3 DIFFERENZE TRA FARMACI BIOSIMILARI E FARMACI
BIOEQUIVALENTI
Fare chiarezza sulle diversità di farmaci biosimilari e generici, anche conosciuti con il termine di bioequivalenti, diventa importante perché spesso il medicinale biosimile è pensato impropriamente come il generico di un farmaco biotecnologico di riferimento. Come è già stato detto, non può essere definito tale perché il suo grado di complessità nella struttura e nel processo produttivo non lo porterà mai ad essere identico al prodotto di partenza. È ancora più rilevante capire che non può essere considerato generico perché definire un farmaco bioequivalente presuppone la risposta a criteri differenti da quelli validi per stabilire che un prodotto è invece un medicinale biosimilare. Quindi è giusto vedere le due classi come mondi ben separati.
Le prime differenze importanti tra le due categorie risiedono nel tipo di struttura e nel peso molecolare, nonché nel metodo di sintesi: i farmaci generici sono farmaci approvati e commercializzati al termine della protezione brevettuale di medicinali derivati da un processo di sintesi chimica, facilmente controllabile. L'utilizzo degli stessi reagenti, nello stesso ambiente e condizioni di reazione ne consente la riproducibilità, portando sempre ad ottenere lo stesso identico prodotto. Si tratta di piccole e semplici entità chimiche, il cui peso molecolare varia da 50 a 1.000 Da.
8 Edwin Choy, Ira Allen Jacobs “Biosimilar Safety Considerations in Clinical Pratice” Seminars in Oncology, Vol 41, No S1, pag S10, February 2014
di 22.000 Da, l'eritropoietina di 34.000, cetuximab di circa 146.000 Da. Invece, dal punto di vista sintetico i biosimilari sono prodotti da sistemi viventi sfruttando la tecnologia del DNA ricombinante. Sono strutture formate da più livelli di complessità che vanno dalle modificazioni post-traduzionali alla struttura primaria, intesa come sequenza amminoacidica, che si ripiega in maniera precisa formando la struttura secondaria, la quale a sua volta, attraverso le interazioni tra le sue varie parti, dà origine alla struttura terziaria. L'unione tra più strutture terziarie porta alla definizione della struttura quaternaria della proteina.
Altro punto critico è la stabilità: i farmaci generici seguono una cinetica del primo ordine mentre per i biosimili la perdita di stabilità a seguito dell'alterazione della struttura tridimensionale o della denaturazione, a conseguenza di cambiamenti delle condizioni ambientali, può rendere inerte il principio attivo. L'intero processo di produzione deve essere continuamente monitorato, gli eccipienti usati nella formulazione sono da scegliere con attenzione per scongiurare questi avvenimenti e la conservazione del farmaco deve essere idonea a mantenere inalterato il prodotto.
In termini economici, sono i biosimilari a richiedere maggior tempo e maggiori investimenti per essere sviluppati, approvati ed essere immessi sul mercato. Gli investimenti nelle fasi di R&D ( ricerca e sviluppo) per la produzione di un farmaco biosimile sono stimati essere dieci volte maggiori di quelli necessari per un farmaco bioequivalente: la spesa prevista per l'ottenimento di un biosimilare oscilla tra i 75 e 250 milioni di dollari. La complessità degli anticorpi monoclonali rende ancora più alti i costi per il loro sviluppo e la loro produzione.
È raro che le piccole molecole generiche, data la loro natura, facciano insorgere problemi di immunogenicità, pericolo che invece si presenta per i biosimili e per i quali, al momento della presentazione del fascicolo necessario per la loro approvazione, è richiesta la presentazione di un preciso e appropriato piano di farmacovigilanza a garanzia della loro sicurezza d'uso.
caratteristiche di attività e uso a cui è destinato”. In modo simile, l'EMA definisce il generico come “un medicinale che contiene la stessa composizione qualitativa e quantitativa del medicinale di riferimento in termini di sostanza attiva” ed “è usato nella stessa dose per il trattamento delle stesse patologie.”9 Quindi risulta evidente il criterio a cui il farmaco deve rispondere per poter essere definito generico da una parte, e biosimilare dall'altra. Nel primo caso, si parla di “identicità” rispetto all'originatore; nel caso del biosimile, secondo quanto riportato nel paragrafo 1.1, deve essere evidenziata la “similarità” (per le linee guida europee) o “alta similarità” (per le linee guida dell'FDA). I bioequivalenti sono molecole identiche al farmaco originale: identiche nella composizione qualitativa e quantitativa di sostanza attiva, per la forma farmaceutica e la modalità di rilascio. Per la loro approvazione deve essere dimostrata l'equivalenza farmaceutica mediante lo svolgimento di studi fisico-chimici e di biodisponibilità, ovvero il risultato della comparazione dei profili farmacocinetici del medicinale e dell'originatore. Clinicamente, se le concentrazioni plasmatiche di due prodotti risultano uguali o sono comunque molto simili, allora l'effetto terapeutico sarà praticamente lo stesso; gli studi di bioequivalenza mirano a stabilire la non esistenza di differenze clinicamente rilevanti tra il medicinale equivalente e il medicinale di riferimento. Questo si verifica nel momento in cui la differenza di biodisponibilità tra i due rientra nell'intervallo di confidenza compreso tra 80% e 125% dei parametri farmacocinetici di riferimento, quali Cmax, Tmax e rapporto tra le AUC. Nel caso in cui si abbia a che fare con farmaci a basso indice terapeutico i margini dell'intervallo di confidenza risultano inferiori. Per stabilire l'equivalenza di questa classe di medicinali rispetto al prodotto di riferimento non sono richiesti ulteriori studi di efficacia clinica né di tollerabilità.
Al contrario, per i biosimilari la sola dimostrazione di bioequivalenza non può essere sufficiente: essi devono dare prova della loro similarità rispetto al medicinale biologico innovatore in termini di riproducibilità del processo produttivo e, attraverso il cosiddetto 9 Ivo Abraham, Diana Sun, Alaa Bagalagel, Ahmed Altyar, Abdulaziz Mohammed, Soba Tharmarajah e Karen MacDonald. “Biosimilar in 3D-Definition, development and differentiation”. Bioengineered 4:4, pag 203; July/August 2013
non possono sono identici: nonostante e in virtù di questo, l'uno deve dimostrarsi rispetto all'altro, comparabile sia nell'efficacia clinica che nella sicurezza. Le considerazioni sulla similarità devono comprendere analisi strutturali, funzionali, analitiche, di produzione, immunologiche, farmacocinetiche, farmacodinamiche, cliniche e di sicurezza. Quindi come risultato il biosimilare deve mostrare idonea e comparabile efficacia, sicurezza, immunogenicità e qualità rispetto al farmaco biotecnologico.
Nemmeno l'iter di approvazione seguito per i farmaci generici può essere applicato ai biosimilari; perciò, per chiarire e indirizzare il percorso che un produttore di un medicinale biosimilare deve intraprendere, l'EMA e le altre agenzie regolatorie nel mondo hanno sviluppato, con successivi aggiornamenti, linee guida da dover osservare (gli aspetti legislativi saranno affrontati nel capitolo 3.
1.3.1 ESERCIZIO DI COMPARABILITÀ
Il programma di sviluppo di un biosimilare non mira a stabilire quanto il farmaco sia efficace e sicuro di per sé, ma ha l'obiettivo di dimostrare che medicinale di riferimento e biosimile, tra loro, siano sufficientemente similari da poter dire che le proprietà dell'uno e dell'altro sono sovrapponibili. L'obiettivo e i principi secondo i quali viene sviluppato un farmaco biosimilare sono quelli di arrivare ad ottenere un grande livello di similarità con il prodotto biologico di riferimento, in modo che risulti che il biosimilare sia così simile all'originatore come l'originatore lo è nei confronti di se stesso, nel momento in cui si introducono cambiamenti nel processo produttivo. L'esercizio di comparabilità infatti è nato negli anni Novanta per merito della FDA, per gestire le variazioni del processo di produzione dei farmaci biologici adottate all'interno di un'azienda. Successivamente, l'EMA ha esteso la sua applicazione ai farmaci biosimilari, farmaci biologici simili a un prodotto già commercializzato, prodotto da un produttore diverso, con un differente processo di produzione. È impossibile che durante il ciclo di vita di un medicinale biotecnologico il produttore non attui modificazioni nel processo di fabbricazione; questo tipo di cambiamenti possono aver luogo sia durante la fase di sviluppo che nel periodo
sono il frutto dell'innovazione; ogni volta che si operano cambiamenti nel corso del processo di produzione, essi sono finalizzati a migliorare il profilo di qualità del prodotto finale. Secondo quanto stabilito dalle linee guida ICH Q5E “Comparability of Biotechnological/biological products subject to changes in their manufacturing process”, deve essere sempre eseguito l'esercizio di comparabilità tra prodotto pre-cambiamento e il prodotto risultante dall'applicazione del cambiamento. Quindi, con lo scopo di assicurare la stessa qualità, sicurezza ed efficacia del prodotto medicinale post-cambiamento, i dati raccolti attraverso tecniche analitiche sensibili in merito alle proprietà fisico-chimiche, all'attività biologica, alle proprietà immunochimiche, al grado di purezza, impurezze e contaminanti e alla stabilità devono indicare che non esistano ripercussioni dei cambiamenti nel processo produttivo sul prodotto medicinale finito.
Perciò l'approccio scientifico su cui si basa la comparabilità è valido tanto per il prodotto biologico sottoposto a modificazioni nel processo produttivo, quanto per il biosimilare che deve essere approvato, o che vede a sua volta l'introduzione di cambiamenti nel corso della sua produzione.
L'EMA, come requisito fondamentale, richiede che dall'analisi condotta sul biosimilare, confrontato con i dati relativi al prodotto di riferimento, non risultino differenze significative a livello clinico. Infatti, “un biosimilare necessita della dimostrazione di similarità biologica e clinica per assicurare la comparabile qualità, efficacia e sicurezza con il prodotto originale.”10 Secondo quanto riportato dall'EMA, nelle Q&A sugli SBP nel suo sito web, “in accordo con l'Articolo 10(4) della Direttiva 2001/83/EC, quando un prodotto medicinale biologico, similare a un prodotto medicinale biologico di riferimento, non soddisfa le condizioni della definizione di prodotto medicinale generico a causa, in particolare, di differenze attinenti alle materie prime o di differenze nei processi di produzione del medicinale biologico e del medicinale biologico di riferimento, il richiedente è tenuto a fornire i risultati di appropriati test preclinici o delle sperimentazioni cliniche relative a dette condizioni. Il tipo e la quantità dei dati supplementari da fornire 10 Preeta Kaur Chugh, Vandana Roy “Biosimilars: Current Scientific and Regulatory Considerations”, Current Clinical Pharmacology, Vol. 9, No 1, pag 56, 2014
determinare la similarità (o meno) di un dato prodotto medicinale biologico nei confronti di un prodotto medicinale di riferimento. L'esercizio di comparabilità dovrebbe essere una forte e stretta comparazione tra il prodotto medicinale biologico similare e il prodotto medicinale di riferimento eseguita a livello di qualità, sicurezza ed efficacia.”
Sebbene sia analizzato ogni step del processo produttivo e siano tenute sotto costante controllo tutte le condizioni operative, il biosimilare è sottoposto a numerose valutazioni anche come prodotto finito. Inizialmente, in realtà, si parte analizzando più molecole candidate fino ad arrivare a separare quella che dimostra avere il miglior profilo.
In prima analisi si procede con la caratterizzazione della sostanza attiva. Lo studio della caratterizzazione mira a definire:
– le sue proprietà fisico-chimiche: è determinata la struttura primaria, secondaria, terziaria del biosimilare. Incontrare già a questo livello difformità nel peso, nella massa o nella densità del farmaco è indice di possibili disuguaglianze con il medicinale di riferimento;
– l'attività biologica: è individuata la funzione/i della sostanza biologica e quindi la sua attività clinica attraverso la comprensione del meccanismo d'azione; anche a questo livello è verificato che non esistano differenze significative tra il biosimilare e il biologico di riferimento e viene identificata la potenza del farmaco;
– le impurità presenti: con l'uso di tecniche quali l'elettroforesi, la spettroscopia di massa, la cromatografia, emerge qualsiasi impurità correlata al prodotto o al processo di produzione sfruttato; se si notano differenze, è valutato qualsiasi potenziale impatto sull'efficacia o sulla sicurezza d'utilizzo del farmaco;
– la sua stabilità: sono valutate le possibili alterazioni o la degradazione a cui va incontro il biosimilare nel momento in cui viene sottoposto a condizioni di stress (luce, temperatura, agitazione); le differenze rilevanti, qualora emergano nei confronti del medicinale di riferimento, sono identificate e valutate.
comparabilità tra biosimile e biologico conducendo studi sull'attività farmacodinamica, mentre per le analisi in vivo, di confrontare l'attività biologica o farmacodinamica da cui deriva l'indicazione clinica, e di valutare la tossicità non-clinica in specie sensibili al farmaco. Gli studi clinici invece permettono di analizzare i profili farmacocinetici, farmacodinamici e di efficacia del biosimilare, stimando la similarità di questi parametri e del profilo di sicurezza tra il medicinale biosimilare e il biologico innovatore. Gli studi clinici si suddividono in varie fasi, ciascuna delle quali conduce all'acquisizione di dati relativi ad un particolare aspetto della vita e funzione del farmaco: gli studi di fase I mirano a raccogliere dati farmacodinamici e farmacocinetici ricavando la relazione esistente tra esposizione ed effetto. Nel campo dello sviluppo dei biosimilari gli studi di fase II sono di norma saltati per arrivare direttamente a quelli di fase III, nei quali si affrontano efficacia e sicurezza in riferimento alle stesse indicazioni approvate per il medicinale di riferimento.
Esistono due tipi di disegni di studio che possono essere adottati nella valutazione dei biosimilari: studi di equivalenza e studi di noninferiorità. I primi iniziano con la supposizione (ipotesi nulla) che i due prodotti siano nonequivalenti tra loro, cioè o migliori o peggiori. Da qui, l'obiettivo del produttore è quello di mostrare che le differenze tra biosimilare e biologico di riferimento sono piccole, quindi di dimostrare la loro equivalenza. Per fare ciò sono richiesti studi di equivalenza a doppio-cieco che coinvolgano un numero di persone adeguato a stabilire la MCID per l'endpoint primario. Per MCID è intesa “la minima differenza in un significativo endpoint clinico tra due trattamenti, oltre la quale i due farmaci sono considerati essere non-equivalenti.”11 L'ipotesi nulla è che la differenza tra i due trattamenti sia uguale o maggiore alla MCID. Più piccola è la differenza tra i due trattamenti considerata clinicamente importante, maggiore è la grandezza dello studio clinico da dover affrontare.
Gli studi di noninferiorità vogliono dimostrare che il nuovo trattamento non è meno efficace di quello già esistente: l'ipotesi nulla è che il trattamento sia peggiore di quello già esistente (in questo caso con “trattamento già esistente” ci si riferisce al trattamento con il
11 Preeta Kaur Chugh, Vandana Roy “Biosimilars: Current Scientific and Regulatory Considerations”, Current Clinical Pharmacology, Vol. 9, No 1 pag 56, 2014
Come conclusione, può anche risultare che il biosimilare abbia proprietà migliori rispetto al farmaco di riferimento; in quel caso è opportuno studiare il prodotto attraverso un iter diverso rispetto a quello valido per il medicinale biosimile. Gli studi di noninferiorità richiedono una popolazione più piccola rispetto agli studi di equivalenza, ma nonostante questo le agenzie regolatorie preferiscono che siano condotti studi di equivalenza.
“Le linee guida specifiche per le diverse classi di farmaci definiscono quale sia la differenza richiesta per gli endpoint, che può essere considerata clinicamente rilevante per quel prodotto. Per esempio, per un biosimilare interferone alfa-2a, il grado di risposta dopo tre mesi è stato fissato al margine del ±15% nell'epatite C, mentre un biosimilare G-CSF deve dimostrare un margine di ±1 giorno di grave neutropenia successiva alla chemioterapia.”12
Gli studi di fase IV, infine, coincidono con la valutazione della sicurezza e sono necessari per garantire la tutela della salute pubblica: studi osservazionali, con continuo aggiornamento dei database contenenti le possibili reazioni avverse scatenate da un determinato farmaco, consentono di tenere sotto controllo i biosimilari avvertendo le agenzie regolatorie del possibile pericolo che essi possono rappresentare.
Nel corso degli studi clinici, i test effettuati devono essere svolti su una popolazione sensibile ed omogenea così che una qualsiasi differenza tra biosimilare e farmaco originatore possa essere facilmente individuata.
I risultati ottenuti in ciascuna fase dipendono dal tipo di tecnica/analisi usata; “nel corso dell'attuazione di tutte le procedure necessarie a stabilire la biosimilarità è opportuno considerare le limitazioni implicate dalle tecniche analitiche prescelte.”13
Riassumendo, le tappe da intraprendere per arrivare a dimostrare la similarità del farmaco in questione rispetto al farmaco biotecnologico di riferimento sono ben delineate a livello legislativo; lo sviluppo del farmaco biosimile può essere visto come un percorso a piramide. Alla caratterizzazione fisico-chimica e biologica segue la validazione del 12 Preeta Kaur Chugh, Vandana Roy “Biosimilars: Current Scientific and Regulatory Considerations”, Current Clinical Pharmacology, Vol. 9, No 1, pag 56, 2014
intraprendere gli studi clinici (Illustrazione 6).
Nel caso in cui il tutto sia stato affrontato senza mettere in luce differenze rimarcabili, o comunque che possano far presupporre un diverso effetto clinico atteso per il biosimilare rispetto al biologico, è possibile procedere con la presentazione di tutta la documentazione necessaria per l'approvazione e l'immissione in commercio del farmaco in questione.
1.3.2 INTERCAMBIABILITÀ E SOSTITUZIONE AUTOMATICA: NECESSITÀ DI ELABORARE UN SISTEMA DI NOMENCLATURA UNIVOCO
Sulla base della dimostrata bioequivalenza farmaceutica e terapeutica tra medicinale generico e farmaco di riferimento, per i generici valgono i concetti di intercambiabilità e sostituzione automatica, i quali sono ancora oggi argomento di dibattito per i biosimilari,
Illustrazione 6 - Piramide alla base del programma di sviluppo del medicinale
biosimilare (Fonte: L.A.Bui, C.Taylor Developing Clinical Trials for Biosimilars, in “Seminars in
sostituire nella terapia adottata dal paziente. L'intercambiabilità implica che ci sia la possibilità di ricevere o il prodotto biologico o il suo biosimilare, a seconda della valutazione del medico che può, nell'esclusivo interesse della salute del paziente, sottolineare l'insostituibilità del prodotto originale prescritto. Anche la sostituzione automatica si basa sulla qualità di equivalenza terapeutica del generico rispetto al medicinale innovatore. Infatti, al momento della dispensazione, al farmacista è permesso sostituire il prodotto originale con il rispettivo generico, informando e trovando consenso nel paziente che vede in questa scelta, come aspetto vantaggioso, il risparmio economico. Tale pratica è concessa soltanto nel caso in cui quel preciso farmaco sia compreso nelle cosiddette “liste di trasparenza”. Al momento l'AIFA non ha incluso nessun tipo di biosimilare in tali liste, perciò, nonostante non esista alcuna legge in Italia che vieti espressamente la sostituzione di un medicinale biologico con il suo biosimilare, il farmacista non può attuare la sostituzione. L'esclusione dei biosimilari da tali liste deriva dalla preoccupazione che possano insorgere problematiche rilevanti, in particolare a livello del sistema immunitario dei pazienti; pertanto non possono essere sostituiti l'uno con l'altro, almeno non senza l'intervento medico. Infatti esiste la concezione che l'intercambiabilità sia diretta conseguenza dello studio di comparabilità e in fondo, è con questo presupposto che il biosimilare viene introdotto nella pratica clinica rappresentando una valida alternativa terapeutica al medicinale biologico di riferimento. È responsabilità del medico scegliere se prescrivere un farmaco biotecnologico o il suo biosimilare, sulla base delle sue conoscenze dell'uno e dell'altro, anche facendo riferimento a quanto riportato nelle Relazioni di valutazione EPAR pubblicate per ciascun farmaco sul sito dell'EMA. Il farmacista non può che dispensare il farmaco prescritto.
Bisogna considerare che avendo a che fare con prodotti biologici, l'esposizione del paziente a più prodotti contenenti la solita sostanza attiva aumenta il rischio di produzione di anticorpi diretti contro il farmaco, che possono condurre allo sviluppo di reazioni avverse o ridurre l'efficacia del farmaco. Se questo avviene e il paziente è stato trattato con diversi farmaci biologici e biosimilari, risulta impossibile associare l'effetto avverso al farmaco responsabile: deve essere possibile identificare con chiarezza la molecola assunta
Tracciabilità, sostituibilità e farmacovigilanza esigono che sia sviluppato un sistema di nomenclatura più efficiente e preciso, che permetta di individuare un particolare biosimilare senza errori. Al momento, si fa riferimento alla nomenclatura INN (International Nonproprietary Name), conosciuta in Italia come DCI (Denominazione Comune Internazionale). A ogni molecola è assegnato un nome generico che permette di riconoscere il farmaco indipendentemente dall'azienda che lo commercializza. Questo sistema si è rivelato adatto per i farmaci generici bioequivalenti, ma non per i biosimilari, le cui molecole presentano differenze strutturali che nascono durante il processo di produzione. Dal caso delle epoetine si capisce quanto sia problematico questo tipo di nomenclatura nei riguardi dei farmaci biologici e biosimilari: pur trattandosi tutte di epoetine, Abseamed®, Epoetin alfa Hexal® e Binocrit® hanno mantenuto la stessa denominazione di Eprex® come epoetina alfa, mentre Retacrit® e Silapo® sono state identificate come epoetina zeta. E' bene riuscire ad identificare un INN specifico per ciascun prodotto biosimilare, che sia diverso da quello riconducibile al prodotto comparatore, in modo da evitare confusione. In questo caso risulta difficile identificare ciascuna epoetina attraverso il solo INN, perciò molte organizzazioni e associazioni sanitarie a livello internazionale si sono messe in moto per richiedere revisioni del sistema e l'adozione di una nomenclatura che consenta di stabilire un INN distinto per ciascuna molecola.
Nel 2007 l'OMS ha approvato un sistema di nomenclatura che prevede, per ciascuna classe di farmaci, una radice comune definita “stem” (es. per gli anticorpi monoclonali lo stem è “-mab”; per le epoetine è “-poetin”; per gli antagonisti del TNF è “-nercept” e così via). In aggiunta allo stem, usando ancora l'esempio delle epoetine, è prevista l'assegnazione di una lettera greca o un prefisso per differenziarle nel momento in cui la molecola risulta diversa a livello di sequenza amminoacidica o nei siti di glicosilazione (esistono perciò epoetine alfa, beta, gamma, delta, epsilon, zeta, theta, kappa ed omega). L'INN resta lo stesso quando la sequenza amminoacidica rimane uguale, mentre la lettera greca sta ad indicare che il profilo di glicosilazione differisce quantitativamente e/o qualitativamente. “Anche questo approccio ha fatto crescere confusione poiché l'INN è
presentata la domanda per l'autorizzazione al commercio.”14
Perciò continua la necessità di arrivare all'elaborazione di un sistema di nomenclatura esclusivo e, nell'attesa di individuare quello più opportuno da adottare, ci si riferisce al biosimilare con il nome commerciale e l'identificazione del lotto. Nel frattempo, l'Organizzazione Mondiale della Sanità ha fatto un altro passo in avanti in questo senso redigendo una bozza del “Biological Qualifier” (BQ). Su questa base, “è prevista l'aggiunta di un codice di quattro lettere -il BQ- a seguito dell'INN. Il Biological Qualifier è un codice alfabetico assegnato in maniera casuale alla sostanza attiva biologica prodotta a livello di un sito specifico. La scelta delle lettere che vanno a comporre ciascun codice sarà fatta in maniera da facilitarne la trascrizione nelle varie lingue, evitando così che siano usate parole inappropriate e senza senso. L'uso di quattro lettere, vocali escluse, offre più di 160000 codici che dovrebbero essere sufficienti per il prossimo futuro. Il codice sarà applicato a tutte quelle sostanze attive biologiche identificate con un INN. [..] Il codice BQ sarà applicato dal WHO INN Secretariat al momento della presentazione della documentazione per la richiesta dell'approvazione della sostanza attiva all'autorità regolatoria. [..] Le informazioni da includere nella richiesta del codice sono: il nome e l'indirizzo del richiedente, l'INN, il nome commerciale del prodotto, il nome e l'indirizzo del produttore della sostanza attiva per cui è richiesto il codice, il nome e l'indirizzo degli eventuali altri siti di produzione, altre informazioni regolatorie. Lo schema BQ sarà usato e sarà utile per le autorità regolatorie (come database dei siti di produzione delle sostanze attive biologiche, come fonte d'informazione sulle sostanze biologiche approvate, come sistema per identificare in senso univoco sostanze e prodotti, come strumento di farmacovigilanza, per identificare particolari prodotti e la loro autorizzazione, per facilitare la presa di decisioni in merito alla sostituzione e all'intercambiabilità), per le autorità sanitarie (per identificare sostanze e prodotti nell'ambito dei sistemi di rimborso, per la loro prescrizione, per facilitare la decisione in merito a sostituzione e intercambiabilità), per i farmacisti (in ospedale o nelle farmacia del territorio, per identificare un prodotto specifico
14 WHO “Biological Qualifier-An INN Proposal”- Programme on International Nonproprietary Name, INN Working Doc. 14.342 revised draft Luglio 2014, pag. 3/6
identificare problemi nelle risposte dei pazienti a prodotti differenti contenenti sostanze con lo stesso INN), e sarà utile anche per gli stessi pazienti (in particolare, per i pazienti sottoposti a terapia a lungo termine che ricevono prodotti diversi contenenti la stessa sostanza attiva, per rilevare differenze nella loro risposta individuale a questi trattamenti).”15
1.4 BIOSIMILI APPROVATI AD OGGI IN EUROPA E PROSSIME
SCADENZE BREVETTUALI
Le prime approvazioni di farmaci biosimilari da parte della Commissione Europea risalgono al 2006 e hanno visto come protagonisti due medicinali a base di somatropina: Omnitrope® (che ha ricevuto l'autorizzazione il 12 aprile 2006 e a cui è stata rilasciata l'AIC in Italia il 29 ottobre 2007) e Valtropin® (approvato il 24 aprile 2006). Prodotti da Sandoz, il primo, e da Biopartners, il secondo, sono utilizzati nella popolazione pediatrica per il trattamento di disturbi della crescita, e negli adulti nel caso di deficit di tale ormone. Durante la fase di studio dei due prodotti il farmaco comparatore utilizzato non è stato lo stesso: per Omnitrope® è stato scelto Genotropin®, mentre per Valtropin®, Humatrope®.
Con il passare del tempo, la fine della protezione brevettuale di più farmaci biologici (della durata di venti anni, o venticinque nel caso di addizionale copertura brevettuale fornita dai CCP), ha aperto le porte a molti biosimilari, considerando anche il fatto che in commercio, per una stessa sostanza attiva, si trova più di un prodotto biosimilare.
Rispetto al resto del mondo, è in Europa che è stato approvato il maggior numero di farmaci biosimili. Utilizzando come prodotti di riferimento l'ormone della crescita, l'eritropoietina, il G-CSF, l'insulina e gli inibitori del TNF, ad oggi, risultano approvati 18 biosimili in Europa, otto in Australia, quattro in Giappone, uno in Canada e sette in USA, mentre per altri la richiesta di approvazione è stata sospesa o rifiutata.
Le liste dei biosimili approvati sono riportate in Tabella 2 e Tabella 3, rispettivamente
15 WHO “Biological Qualifier-An INN Proposal”- Programme on International Nonproprietary Name. INN Working Doc. 14.342 revised draft Luglio 2014, pag. 5-6/6
Nome del prodotto Sostanza attiva Area terapeutica Data di autorizzazione Nome produttore/azienda Abasria® Insulina glargine
Diabete Opinione positiva del CHMP 26 Giugno 2014, approvata nel Settembre 2014 Eli Lilly/Boehringer Ingelheim
Abseamed® Epoetina alfa Anemia Cancro Insufficienza renale cronica
28 Agosto 2007 Medice Arzneimittel Pütter Alpheon® Interferone alfa-2a Epatite C 2006 Approvazione respinta BioPartners Bemfola® Follitropina alfa Anovulazione (IVF) Opinione positiva del CHMP 23 Gennaio 2014 Finox Biotech Biferonex® Interferone beta-1a Sclerosi multipla remittente 19 Febbraio 2009 Approvazione rifiutata BioPartners GmbH
Binocrit® Epoetina alfa Anemia Cancro Insufficienza renale cronica
28 Agosto 2007 Sandoz
Biograstim® Filgrastim Cancro Trapianto ematopoietico di cellule staminali 15 Settembre 2008 CT Arzneimittel
Neutropenia Epoetin alfa
Hexal®
Epoetina alfa Anemia Cancro Insufficienza renale cronica 28 Agosto 2007 Hexal Filgrastim Hexal® Filgrastim Cancro Trapianto ematopoietico di cellule staminali Neutropenia 6 Febbraio 2009 Hexal Filgrastim ratiopharm® Filgrastim Cancro Trapianto ematopoietico di cellule staminali Neutropenia 15 Settembre 2008 ritirata il 20 Aprile 2011 Ratiopharm
Grastofil® Filgrastim Neutropenia 18 Ottobre 2013 Apotex Insulin Human
Rapid Marvel®
Insulina
umana Diabete mellito 24 Gennaio 2008 Approvazione ritirata
Marvel Life Sciences
Insulin Human
Long Marvel® Insulina umana Diabete mellito 24 Gennaio 2008 Approvazione ritirata
Marvel Life Sciences
Insulin Human 30/70 Mix Marvel®
Insulina
umana Diabete mellito 24 Gennaio 2008 Approvazione ritirata
Marvel Life Sciences
anchilosante Morbo di Chron Artrite psoriasica Psoriasi Artrite reumatoide Colite ulcerosa 2013
Nivestim® Filgrastim Cancro
Trapianto ematopoietico di cellule staminali Neutropenia 8 Giugno 2010 Hospira
Omnitrope® Somatropina Nanismo ipofisario Sindrome di Prader-Willi Sindrome di Turner 12 Aprile 2006 Sandoz Ovaleap® Follitropina
alfa Anovulazione (IVF) 27 Settembre 2013 Teva Pharma Ratiograstim® Filgrastim Cancro
Trapianto ematopoietico di cellule staminali Neutropenia 15 Settembre 2008 Ratiopharm
anchilosante Morbo di Chron Artrite psoriasica Psoriasi Artrite reumatoide Colite ulcerosa 2013 Retacrit® Epoetina zeta Anemia Trasfusione di sangue autologa Cancro Insufficienza renale cronica 18 Dicembre 2007 Hospira Silapo® Epoetina zeta Anemia Trasfusione di sangue autologa Cancro Insufficienza renale cronica 18 Dicembre 2007 STADA R & D Somatropin Biopartners® Somatropina Deficit dell'ormone della crescita 5 Agosto 2013 BioPartners
Tevagrastim® Filgrastim Cancro Trapianto ematopoietico di cellule
15 Settembre
staminali Neutropenia Valtropin® Somatropina Nanismo
ipofisario Sindrome di Turner 24 Aprile 2006 revocata il 10 Maggio 2012 BioPartners
Zarzio® Filgrastim Cancro
Trapianto ematopoietico di cellule staminali Neutropenia 6 Febbraio 2009 Sandoz
*Dati raccolti il 12 Maggio 2011, aggiornati al 4 Luglio 2014 IVF: fertilizzazione in vitro
Fonte: EMA
Tabella 2 - Biosimilari approvati in Europa16
16 GaBi online-Generics and Biosimilars initiative. “Biosimilars approved in Europe”
http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe Posted 08/07/2011, Last update: 4 July 2014







![ricavata da Gipser [6], ottenendo: (B.4) 2 2 0 0 1 i NA N NB C r N C C p C Drum p R R](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)