1
UNIVERSITÀ DEGLI STUDI DI MESSINA
FACOLTÀ DI SCIENZE MM. FF. NN.
Dipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali Dottorato in Biologia Applicata e Medicina Sperimentale
Curriculum: Medicina Sperimentale
- XXX Ciclo -
Coordinatore: Prof. Salvatore Cuzzocrea
STUDIO GENETICO MOLECOLARE DELLE SINDROMI
MALFORMARIVE DEL CERVELLETTO E DEL TRONCOENCEFALO MEDIANTE UTILIZZO DI TECNICHE DI “NEXT GENERATION
SEQUENCING”
Tesi di Dottorato della:
Dott.ssa Alessia MICALIZZI
Tutor:
Ch.mo Prof. Salvatore CUZZOCREA Ch.ma Prof.ssa Enza Maria VALENTE
2
Ad Andrea Poretti (1977-2017)
“Se ho fatto una qualche scoperta di valore, è dovuta più alla paziente attenzione che ad ogni altro talento”
Isaac Newton In te c’erano entrambe!
3
SOMMARIO
INTRODUZIONE GENERALE ... 6
OBIETTIVO DELLA TESI ... 10
MATERIALI E METODI ... 12
RECLUTAMENTO DEI PAZIENTI E VALUTAZIONE CLINICA ... 12
CLASSIFICAZIONE NEURORADIOLOGICA (WP1) ... 12
ESTRAZIONE DEL DNA ... 13
VALUTAZIONE QUALITATIVA E QUANTITATIVA DEL DNA ... 14
POLYMERASE CHAIN REACTION (PCR) ... 14
MLPA-MULTIPLE LIGATION PROBE AMPLIFICATION ... 16
REAL TIME-PCR SU DNA GENOMICO ... 17
PROTOCOLLI NEXT GENERATION SEQUENCING (NGS) PER ANALISI DI PAZIENTI SJ ... 18
Protocollo Targetseq Custom (Life Technologies) su Piattaforma Solid 5500xL ... 18
Protocollo SureSelectQXT Target Enrichment (Agilent Technologies) su Piattaforma MiSeq (Illumina) ... 21
PROTOCOLLI NGS PER ANALISI DI PAZIENTI CON NPCA ... 23
Protocollo TruSeq Custom Amplicon (Illumina) su piattaforma MiSeq (Illumina) ... 23
PROTOCOLLO WES CON SURESELECTXTTARGET ENRICHMENT SYSTEM (CREV1-AGILENT TECHNOLOGIES) SU PIATTAFORMA HISEQ 2500(ILLUMINA) ... 25
PROTOCOLLO CUSTOM HD-CGHMICROARRAY SU PIATTAFORMA SURESCAN MICROARRAY SCANNER (AGILENT TECHNOLOGIES) ... 26
ANALISI BIOINFORMATICA DATI NGS ... 27
CAPITOLO I: SINDROME DI JOUBERT ... 29
INTRODUZIONE ... 29
Quadro clinico ed iter diagnostico della Sindrome di Joubert ... 30
Basi genetiche della sindrome di Joubert ... 33
Overlap clinico e genetico con altre ciliopatie e “mutational load” ... 34
RISULTATIEDISCUSSIONE ... 38
ANALISI MUTAZIONALE DEI PAZIENTI SJ... 38
4
Studio mutazionale del gene CEP120 in pazienti JS (Roosing et al., 2016) ... 44
Identificazione di un nuovo gene-malattia mediante analisi di WES (De Mori et al., 2017)... 46
CAPITOLO II: IPOPLASIA PONTOCEREBELLARE... 51
INTRODUZIONE ... 51
Quadro clinico ed iter diagnostico delle PCH... 52
Basi clinico-genetiche delle PCH ... 53
PCH1 SMA-like ... 54 PCH2-4-5 TSEN-correlate... 56 PCH3 PCLO-correlata ... 57 PCH6 RARS-correlata ... 58 PCH7 TOE1-correlata ... 58 PCH8 CHMP1A-correlata ... 59 PCH10 CLP1-correlata ... 60 PCH11 TBC1D23-correlata ... 61 PCH RELN- VLDLR-correlate ... 62 PCH CASK-correlata ... 63 RISULTATI E DISCUSSIONE ... 65
ANALISI MUTAZIONALE NGS DEI PAZIENTI SJ ... 65
Analisi mutazionale gene EXOSC3 ... 66
Analisi mutazionale gene RARS2 ... 67
Analisi mutazionale gene TOE1 ... 67
Analisi mutazionale gene CASK ... 68
Analisi mutazionale in un paziente con mutazione nei geni PMM2 e ATP2B3. ... 72
Mutazione missenso nel gene VLDLR associato ad una forma lieve di Sindrome da Dysequilibrium (Micalizzi et al., 2016) ... 74
CAPITOLO II: DISPLASIA CEREBELLARE ... 78
INTRODUZIONE ... 78
BASI CLINICHE E GENETICHE DELLE DC... 80
Tubulinopatie ... 86
RISULTATIEDISCUSSIONE ... 88
5
Studio mutazionale in pazienti con diasplasia cerebellare tubulina-correlata (Romaniello et al., 2017) .... 94
CAPITOLO IV: RISULTATI IN PROGRESS* ... 99
MUTAZIONI DEL GENE GSX2 CAUSANO ANOMALIE CONGENITE DELLA GIUNZIONE DIENCEFALO
-MESENCEFALICA ... 100 ATROFIA CEREBELLARE NON PROGRESSIVA NEI DISORDINI BRAT1-CORRELATI ... 102 IDENTIFICAZIONE DI UN POSSIBILE NUOVO GENE-MALATTIA DELLE TUBULINOPATIE... 104 STUDIO MUTAZIONALE DEL GENE ROBO3 IN PAZIENTI CON OFTALMOPLEGIA ESTERNA PROGRESSIVA CON SCOLIOSI PROGRESSIVA (HGPPS) AD ESORDIO PRECOCE ... 106 IDENTIFICAZIONE DELLA VARIANTE ARG480TRP NEL GENE SPTBN2 IN UN PAZIENTE CON UNA FORMA
CONGENITA DI SCA5 ASSOCIATA A DEFICIT COGNITIVO ... 109
6
INTRODUZIONE GENERALE
I difetti congeniti del cervelletto e del troncoencefalo (“Cerebellar and Brainstem
Congenital Defects” - CBCD) rappresentano un gruppo clinicamente e
geneticamente eterogeneo di patologie rare (incidenza stimata 1/5000 nati),
accomunate da un alterato sviluppo embrionale delle strutture della fossa cranica
posteriore. Tale sviluppo è un processo lungo e complesso, che si estende a partire
dalla terza settimana di gravidanza fino a 20 mesi di vita postnatale, ed è
strettamente regolato da molteplici cascate di geni,. Nella maggior parte dei casi
la diagnosi di un CBCD può essere effettuata già in epoca prenatale mediante
ecografia nel secondo trimestre, rappresentando una delle più comuni cause di
interruzione volontaria di gravidanza (Forzano et al. 2007). Il tasso diagnostico di
tali malformazioni è in progressivo aumento grazie ai progressi nelle tecniche di
neuroimaging pre- e post-natale. L'associazione di segni tipici del coinvolgimento
cerebellare, quali atassia congenita non progressiva (“non-progressive congenital
ataxia” - NPCA) e anomalie dei movimenti oculari, con un ampio spettro di segni
neurologici (ritardo dello sviluppo psicomotorio, disabilità intellettiva, alterazioni
comportamentali), e non neurologici (caratterizzati da variabile coinvolgimento
multiorgano) rende queste condizioni altamente disabilitanti (Parisi and Dobyns
2003; Tavano et al. 2007; Millen and Gleeson 2008; Strata et al. 2011). Molti
quesiti riguardo la nosologia e la prognosi dei CBCD restano ancora largamente
irrisolti, così come il tasso di mortalità e l’aspettativa di vita (Barkovich et al.
7
terapie specifiche e la riabilitazione motoria e cognitiva resta l’unica strategia
fondamentale per migliorare la qualità di vita dei pazienti e favorire il loro
inserimento nel contesto sociale.
La presentazione clinica di tali difetti può essere estremamente variabile. A causa
del ruolo cruciale del cervelletto in diverse funzioni non motorie (per esempio
percettivo, neuro-visivo, linguistico, cognitivo, affettivo) (Tavano et al. 2007;
Steinlin 2008), i segni neurologici dei CBCD non sono limitati esclusivamente
all’atassia, ma includono frequentemente l'ipotonia neonatale, la presenza di movimenti oculari anomali (nistagmo, strabismo, aprassia oculomotoria), il
ritardo dello sviluppo psicomotorio e deficit cognitivo di grado variabile. Mentre
tali caratteristiche cliniche spesso non consentono, soprattutto all’esordio, di
distinguere tra loro diverse malformazioni, la risonanza magnetica nucleare
(RMN) cerebrale è spesso l’esame dirimente per un corretto inquadramento
diagnostico dei CBCD (Bosemani et al. 2015).
Negli ultimi decenni, i progressi nel campo della genetica e delle neuroimmagini
hanno portato ad un miglioramento significativo nella definizione dei CBCD. Le
diverse classificazioni ad oggi proposte (che comprendono sia le forme ereditarie
che quelle acquisite) sono basate su criteri di genetica molecolare e di biologia
dello sviluppo o sul fenotipo neuroradiologico (Barkovich et al. 2009;
Jissendi-Tchofo et al. 2009; Doherty et al. 2013).
Una prima classificazione di tali difetti è stata presentata da Barkovich (Barkovich
8
base dei meccanismi di sviluppo genetici e cellulari che regolano l’embriologia
del sistema nervoso centrale:
a. malformazioni secondarie a difetti precoci di differenziazione lungo l’asse
antero-posteriore o dorso-ventrale e malformazioni secondarie a difetti di
specificazione nelle zone germinali del mesencefalo e del romboencefalo;
b. malformazioni dovute a difetti generalizzati dello sviluppo cerebrale, con
particolare coinvolgimento del cervelletto e del tronco encefalico;
c. malformazioni dovute a difetti regionali dello sviluppo cerebrale, con
particolare coinvolgimento del cervelletto e del tronco encefalico (patogenesi
parzialmente conosciuta);
d. difetti secondari ad ipoplasia ed atrofia combinate, in disturbi degenerativi con
possibile esordio prenatale.
Una più recente classificazione è stata proposta da Poretti nel 2016. Il lavoro pone
l’accento sul ruolo chiave che i segni neuroradiologici hanno nel work-up diagnostico dei pazienti con tali difetti. Viene considerato esclusivamente il
pattern neuroadiologico presente nelle condizioni malformative della fossa
cranica posteriore, suddividendole in tre categorie: i) prevalentemente cerebellari;
ii) cerebellari e cerebrali; iii) prevalentemente cerebrali.
Nell’ultimo decennio sono stati identificati numerosi geni correlati ad alcune forme mendeliane di CBCD, come la Sindrome di Joubert (“Joubert Syndrome”, JS) e l’Ipoplasia Ponto-Cerebellare (“Ponto-Cerebellar Hypoplasia”, PCH) (Namavar et al. 2011a). Molti altri CBCD occorrono invece in casi sporadici,
9
suggerendo che i meccanismi patogenetici più comuni siano rappresentati da
mutazioni o riarrangiamenti genomici de novo. Ad oggi, i determinanti genetici
di molti CBCD risultano ancora sconosciuti ed attualmente i test genetici
disponibili riguardano soltanto poche condizioni ad eredità mendeliana, rendendo
10
OBIETTIVO DELLA TESI
Il gruppo di Neurogenetica diretto dalla Prof.ssa Enza Maria Valente si occupa da
anni di CBCD, ed ha apportato importanti contributi scientifici alla comprensione
delle loro basi genetiche. Uno dei progetti, finanziato dall’European Research
Council (ERC), si propone di migliorare le conoscenze sui CBCD attraverso la
creazione di un network italiano multidisciplinare e lo svolgimento di 4
workpackages (WP), che prevedono rispettivamente: 1) raccolta di dati e
campioni di pazienti con CBCD, caratterizzazione neuroradiologica, definizione
delle correlazioni genotipo-fenotipo e follow-up a medio e lungo termine; 2)
studio delle basi genetiche dei CBCD a trasmissione mendeliana (in particolare
JS e PCH) mediante analisi molecolare di geni noti e identificazione di nuovi geni
attraverso tecnologie di “next generation sequencing”(NGS); 3) studio delle basi genetiche dei CBCD sporadici mediante la ricerca di riarrangiamenti genomici
atti ad identificare perdite o guadagni di dose (Copy Number Variations, CNVs)
di uno o più geni che possano essere messi in correlazione col quadro
malformativo (tecnologia SNP-array ad alta risoluzione); 4) studi funzionali
basati sullo sviluppo di un nuovo modello in vitro da cellule staminali embrionali
murine per testare il ruolo dei geni noti o candidati per lo sviluppo del cervelletto
e del troncoencefalo, e valutare l’effetto patogenetico delle varianti identificate.
Scopo del progetto è quello di sviluppare una classificazione neuroradiologica più
dettagliata, identificare nuovi geni causativi e definire la prevalenza e lo spettro
11
disponibili e lo sviluppo di strategie di riabilitazione mirate per le diverse
condizioni.
In particolare, oggetto di questa tesi di dottorato sono stati i risultati del WP2, che
si pone come obiettivo lo studio delle basi genetiche dei CBCD a trasmissione
12
MATERIALI E METODI
Reclutamento dei pazienti e valutazione clinica
Sono state reclutate ad oggi circa 1200 famiglie con differenti forme di CBCD. A
ciascun probando ed a ciascun componente del nucleo familiare è stato attribuito
un numero progressivo riconducibile al soggetto corrispondente inserito
all’interno di uno specifico database, protetto da password e consultabile esclusivamente dal personale impegnato nel progetto di ricerca. I pazienti sono
stati inviati da numerosi centri clinici nazionali ed internazionali, appartenenti a
17 nazioni (Austria, Belgio, Egitto, Emirati Arabi Uniti, Francia, Germania,
Grecia, India, Inghilterra, Irlanda, Islanda, Israele, Italia, Olanda, Pakistan,
Spagna, Portogallo e Turchia). I dati clinici di ogni paziente sono stati raccolti
grazie ad un questionario dedicato che contempla le diverse tappe dell’approccio
diagnostico al paziente e che prevede, pertanto, il lavoro di un team
multispecialistico. Il questionario è articolato in sezioni, ciascuna corrispondente
ad un’area di valutazione clinica specifica suddivisa per organo o apparato, che consente di ottenere informazioni dettagliate sulla funzionalità degli
organi/apparati maggiormente colpiti dalla sindrome quali il SNC, l’occhio, il
rene ed il fegato.
Classificazione neuroradiologica
Per validare ed eventualmente migliorare le classificazioni precedentemente
13
neuroimmagini di ciascun paziente sono state valutate dal gruppo di
neuroradiologi afferenti al progetto. Il fenotipo neuroradiologico è stato definito
mediante l’assegnazione di punteggi specifici, secondo uno schema sviluppato a
partire dalle classificazioni preesistenti (figura 1). Tale approccio ha consentito di
inquadrare i pazienti sulla base dei segni neuroradiologici presenti e ne ha favorito
un rapido indirizzamento verso le indagini cliniche e genetiche più appropriate.
Figura 1: Schema di valutazione neuroradiologica sviluppato dai neuroradiologi afferenti al progetto per la
classificazione dei pazienti CCM
Estrazione del DNA
Il DNA genomico è stato estratto a partire da 2-10 ml di sangue periferico
addizionato ad anticoagulante EDTA e conservato a -20°C. Quando possibile, per
ogni famiglia è stato eseguito il prelievo nel probando, nei genitori ed in eventuali
fratelli affetti o sani. Per ogni soggetto prelevato è stato ottenuto un consenso
informato scritto. Il DNA genomico è stato ottenuto usando un kit di estrazione
commerciale, NucleoSpin® Blood L-XL-QuickPure (MACHEREY-NAGEL).
14
una singola fase di lavaggio. Il DNA genomico è successivamente eluito, in
condizioni di bassa forza ionica, in un tampone di eluizione leggermente alcalino.
Valutazione qualitativa e quantitativa del DNA
Prima dell’utilizzo per l’analisi molecolare, il DNA è stato misurato allo spettrofotometro Nanodrop 1000 (Thermo Scientific) per valutarne la
concentrazione e la purezza nei valori di assorbanza a 260, 280 e 230. Per la
valutazione qualitativa è stata eseguita, inoltre, la corsa elettroforetica su gel
d’agarosio (0,8%) con etidio bromuro, confrontando il DNA estratto con un marcatore di riferimento. La corsa su gel è stata visualizzata sul GelDoc2000
(Bio-Rad). Per esperimenti di NGS, un’ulteriore valutazione quantitativa del
DNA genomico è stata effettuata mediante analisi al Qubit® 2.0 Fluorometer
(INVITROGEN) che utilizza dye fluorescenti per misurare la concentrazione
delle molecole d’interesse specifico, al contrario del NanoDrop® ed altri spettrofotometri UV che usano assorbanza UV e non sono in grado di discriminare
la presenza di acidi nucleici degradati, nucleotidi liberi e di altri contaminanti. La
quantizzazione mediante Qubit® consente anche di leggere concentrazioni molto
basse di DNA o RNA con una sensibilità ed accuratezza maggiore.
Polymerase Chain Reaction (PCR)
L’amplificazione delle regioni codificanti e delle giunzioni esone-introne dei geni analizzati è stata eseguita mediante PCR (Polymerase Chain Reaction - reazione
polimerasica a catena) utilizzando primers specifici per ciascun frammento.
15
Tutti gli esoni sono stati amplificati in un volume finale di 25µl contenenti 5 µl di
buffer reaction 10X, 4 µl dNTPs (200 μM), 5 µl primers senso ed antisenso (10µM), 15-30 ng di DNA genomico e 1U di TaqGo DNA Polymerase
(PROMEGA). Le reazioni di PCR sono state condotte utilizzando i termociclatori
C100 Thermal Cycler e C1000 Touch™ Thermal Cycler (Bio-Rad, Hercules, CA,
USA). Gli amplificati di PCR sono stati sottoposti ad analisi elettroforetica su gel
d’agarosio al 1.8% o Qiexcel (QIAGEN) per verificare la specificità e la resa dei frammenti. I prodotti destinati al sequenziamento diretto sono stati purificati
utilizzando ExoSAP-IT (Affymetrix). Tale metodo di purifica utilizza l’attività di
due enzimi idrolitici, Exonuclease I and Shrimp Alkaline Phosphatase (SAP), che
sono in grado di degradare dNTP in eccesso ed i primers e prodotti di PCR a
singolo filamento presenti nella PCR. L’ExoSAP-IT aggiunto al prodotto di PCR viene incubato a 37°C per 15 minuti e successivamente a 80°C per 15 minuti per
inattivare gli enzimi.
Sequenziamento diretto con metodo Sanger
La reazione di sequenza è stata eseguita in entrambe le direzioni, senso ed
antisenso, utilizzando protocolli standard della chimica del Big Dye Terminator
(Applied Biosystems) e come templato l’amplificato di PCR e sono state eseguite
con un termociclatore C1000 Touch™ Thermal Cycler (Bio-Rad, Hercules, CA,
USA). I prodotti di sequenza sono stati purificati utilizzando le colonnine (DyeEx
2.0 Spin Kit, Qiagen) o le piastre (Montage Seq96, Millipore), come da protocollo
16
miscelato con 20 µl di formammide, denaturato per 2 minuti a 95°C e risolto
mediante elettroforesi capillare sul sequenziatore automatico ABI Prism 3130
Genetic Analyser (Applied Biosystems). La lettura degli elettroferogrammi è stata
effettuata utilizzando i programmi dedicati SeqAnalysis (Applied Biosystems) e
Mutation Surveyor (SoftGenetics) confrontando la sequenza ottenuta con la
sequenza di riferimento presente nel database dell’NCBI
(http://www.ncbi.nlm.nih.gov). Per confermare la patogenicità dei cambi
aminoacidici individuati, è stata studiata la segregazione all’interno del nucleo familiare ed è stata valutata la frequenza nella popolazione (MAF).
MLPA-Multiple Ligation Probe Amplification
Lo screening delle delezioni esoniche del gene CASK è stato effettuato
utilizzando il saggio SALSA MLPA P398-A1 CASK (MRC-Holland,
Amsterdam, The Netherlands) che consente di identificare eventuali
sbilanciamenti di dose del gene CASK. Il saggio è stato eseguito secondo il
protocollo specifico “MLPA® DNA Protocol version MDP-005”. Due μL di
amplificato sono stati miscelati a 5μL di Liz 500 size standard e a 20 μL di formammide e sono stati sottoposti ad analisi elettroforetica mediante un
sequenziatore automatico ABI PRISM 3100 Genetic Analyzer (Applied
Biosystem, Foster City, CA). L’area di ogni frammento è stata quantificata con il programma GeneScan Analysis Software versione 3.7 (Applied Biosystem,
Foster City, CA). I risultati ottenuti sono stati analizzati mediante il software
17
evidenziava dal rapporto tra l’area dei picchi rappresentanti il numero di copie alleliche del paziente rispetto al soggetto di controllo, rispettivamente di <0.7 o
>1.3, mentre un valore di circa 1 indicava l’assenza di queste.
Real Time-PCR su DNA genomico
Tutti i pazienti analizzati tramite CASK-MLPA che sono risultati portatori di
delezioni o duplicazioni esoniche o multi-esoniche sono stati analizzati, per
confermare la mutazione identificata, mediante Real Time-PCR quantitativa
(qRT-PCR) su DNA genomico, utilizzando primers intragenici specifici per la
regione d’interesse. La reazione di qRT-PCR, basata sul saggio SYBR Green (Applied Biosystem), è stata effettuata sull’ABI 7000 (Applied Biosystems).
Sono state eseguite contemporaneamente due reazioni di quantificazione: una per
i geni da testare e l’altra per il gene della telomerasi (TERT), usato come calibratore interno. Le reazioni di PCR sono state effettuate utilizzando 12.5 µl di
SYBR Green 2X (Invitrogen), 0.25 µl di ciascun primer foward e reverse 10μM
e 60 ng di DNA (15 ng/µl), per un volume finale di 25 µl. I cicli di PCR erano: 2’ a 50°C, 2’ di denaturazione a 95°C e 40 cicli ciascuno di 15’’ a 95°C e di 1’ a 58°C. Ogni campione del trios familiare è stato processato con un campione di
controllo disomico che è stato utilizzato per la normalizzazione. Il numero di
copie per ogni gene è stato determinato utilizzando il metodo del calcolo dei valori
di ΔΔCt, come descritto da Carbone e collaboratori (Carbone et al., 2008). Nel confronto delle fluorescenze tra il DNA da testare e il DNA di controllo, utilizzato
18
normalità, un valore inferiore a 0.7 indice della presenza di una delezione e un
risultato superiore o pari a 1.3 indice della presenza di una duplicazione. Ogni
coppia di primers è stata disegnata mediante il software “Primer Express 2.0” (Applied Biosystems).
Protocolli Next Generation Sequencing (NGS) per analisi di pazienti con JS Protocollo Targetseq Custom (Life Technologies) su Piattaforma Solid 5500xL Il protocollo di preparazione “TargetSeq Custom” permette di arricchire specifiche regioni di interesse dal DNA genomico. Per questo progetto sono stati
disegnati due pannelli con un numero totale di 120 geni ciliari, causativi o
candidati per ciliopatie. I due chip coprono tutte le regioni esoniche e 25 basi
introniche fiancheggianti gli esoni e le regioni UTR (“Untranslated region”), per un totale di circa 900 Kb. I campioni sono stati modificati per essere poi
sequenziati su sequenziatore Solid5500_xL (Life Technologies). In questa
preparazione, specifici adattatori “forward” e “reverse” sono stati aggiunti alle
estremità dei frammenti di DNA genomico. Questa prima fase è stata
automatizzata grazie all’utilizzo di uno strumento, AB Libery Builder (Life Technologies), che consente la preparazione contemporanea di 13 campioni. Nel
dettaglio, tre microgrammi di DNA genomico di buona qualità sono stati
frammentati utilizzando un sistema Covaris S2. Questo step permette di ottenere
frammenti di DNA genomico con una grandezza di circa 150-200 paia di basi. Il
DNA frammentato viene poi inserito in specifiche cartucce contenenti tutti i
19
automatico le fasi di “riparazione delle estremità del DNA”, di “A-tailing”, di ligazione e relative purifiche. In questo momento vengono anche inseriti alle
librerie in costruzione i “barcode”, brevi sequenze di 10 nucleotidi che saranno poi necessarie per distinguere ciascuna librerie dopo la creazione di un unico pool.
Una volta terminate queste fasi, i campioni così modificati sono stati amplificati
utilizzando l’enzima “Platinum® PCR Amplification” attraverso una reazione di PCR di 6 cicli. Il prodotto della reazione è stato successivamente purificato
tramite “Agencourt AMPure® XP Reagent”. In questa fase della preparazione le librerie devono essere controllate e quantificate. Per quantificare le librerie e
contemporaneamente controllarne la loro qualità è stato utilizzato lo strumento
2100 Bioanalyzer™ (Agilent Technologies). Dopo aver eseguito questo controllo le librerie sono state unite in un unico pool. La fase successiva è rappresentata
l’ibridazione delle librerie durante la quale vengono “catturate” le regioni genomiche di interesse attraverso sonde a RNA specifiche per queste regioni.
Questo processo avviene attraverso due successive reazioni di ibridazione,
identiche fra loro, nelle quali le sonde a RNA si appaiano con il DNA
complementare. Le sonde appaiate con il DNA d’interesse sono state recuperate, e dopo la loro eliminazione le librerie ibridate sono state purificate e
successivamente amplificate. Attraverso lo strumento 2100 Bioanalyzer™ (Agilent Technologies) è stata verificata l’integrità delle librerie, ne è stata determinata la lunghezza media (circa 260 paia di basi) e la quantità. Si è
20
pool, in cui i precedenti pools sono stati mescolati per raggiungere il totale di 24
campioni. Le librerie così costruite sono state successivamente preparate per
essere sequenziate mediante PCR in emulsione. Riassumendo, le librerie sono
state emulsionate meccanicamente in una miscela di acqua ed olio con delle biglie,
alle quali si attaccano grazie a specifici adattatori. Da questa emulsione si formano
dei “micro-reattori” in fase acquosa ognuno contenente il mix di amplificazione e non più di una biglia ciascuno. A questo punto le biglie emulsionate sono state
sottoposte a PCR per clonare ed amplificare ogni molecola di DNA stampo. La
successiva fase di arricchimento permette il recupero delle biglie che legano il
DNA amplificato (libreria), il loro lavaggio ed il loro arricchimento. In questa fase
è stata anche arricchita la popolazione totale di biglie che portano DNA
amplificato, eliminando le “biglie vuote”. Al termine di questi step le biglie sono
state modificate alla loro estremità 3’ tramite l’enzima Terminal Trasferasi (20 U/μL); questa modifica rende le biglie “appiccicose” e consente la deposizione delle stesse sul flowchip per il sequenziamento. L’emulsione è stata quantificata
tramite lo spettrofotometro NanoDrop® ND-1000, in funzione “Cell Cultures” ad
assorbanza A600, in modo tale da poter calcolare la concentrazione
dell’emulsione e caricare per ogni lane circa 230-250 milioni di beads. Dopo aver misurato la concentrazione dell’emulsione, sono stati prelevati i microlitri corrispondenti ad una concentrazione 230-250 milioni di beads, queste sono state
21
biglie sono pronte per essere deposte sul flowchip, che verrà posizionato nel
sequenziatore Solid5500_xL per essere analizzato.
Protocollo SureSelectQXT Target Enrichment (Agilent Technologies) su Piattaforma MiSeq (Illumina)
Il protocollo di preparazione “SureSelectQXT” permette l’arricchimento di regioni genomiche d’interesse. Nella seconda fase del progetto, si è proceduto con il disegno di un nuovo pannello di dimensioni ridotte rispetto ai precedenti,
includendo un numero totale di 56 geni causativi di JS e sindrome di Meckel. Il
pannello contiene tutte le regioni esoniche e 25 basi introniche fiancheggianti gli
esoni per un totale di circa 304 Kb. Le sonde sono state disegnante utilizzando
SureDesign Software (Agilent Technologies), un sistema automatizzato che
utilizza algoritmi specifici per il disegno delle sonde d’interesse. Il Protocollo SureSelectQXT permette di sequenziare quantità molto basse di DNA. Il DNA
genomico (gDNA) utilizzato per la preparazione delle librerie è pari a 50 ng con
rapporti di qualità 260/280 superiori a 1.8-2.0. Inizialmente, il gDNA è stato
frammentato enzimaticamente, ed alle estremità dei frammenti prodotti sono stati
attaccati degli adattatori, che successivamente sono stati riparati ed amplificati
mediante PCR. Il prodotto è stato purificato tramite “Agencourt AMPure® XP
Reagent”. L’integrità, la qualità e la quantità delle librerie prodotte per ciascun
campione è stata controllata mediante la TapeStation 2200 (Agilent
Technologies). Sulla base della quantizzazione sono stati utilizzati, in accordo con
la dimensione totale delle sonde disegnate del pannello d’interesse (<3Mb), concentrazioni variabili da 500 a 750 ng di ciascun gDNA in un volume totale di
22
12 uL. Le singole librerie sono state ibridate con la Capture Library Hybridization
Mix, costituita dalle sonde target-specifiche del pannello, in una reazione di 60
cicli (65°C 1’- 37°C 3’’).
Dopo l'ibridazione, le molecole arricchite nelle regioni targhettate sono state
catturate mediante l’utilizzo di biglie di streptavidina. Tale passaggio, fondamentale per isolare le regioni d’interesse, è stato effettuato nel mixer a 1800 rpm per 30 minuti. A ciascun DNA post-cattura, legato alle biglie di streptavidina,
sono stati aggiunti index primers specifici, in una reazione di amplificazione di
12 cicli (98°C 30’’- 58°C 30’’ – 72°C 1’). Le librerie di DNA targhettato sono state purificate tramite “Agencourt AMPure® XP Reagent”. La qualità e la
quantità dei campioni arricchiti è stata controllata mediante la TapeStation 2200
(Agilent Technologies). Dopo aver misurato la concentrazione delle librerie (nM),
è stato calcolato il volume da prelevare da ciascuna di esse per preparare il pool
finale, che varia in base alla concentrazione desiderata (tra 2-10nM) ed è
strettamente correlato alla concentrazione iniziale di ciascun campione arricchito.
Infine, è stata caricata sul sequenziatore MiSeq (Illumina) una concentrazione del
pool finale variabile tra 8pM a 12pM. Per il sequenziamento sono stati caricati
pool da 24 campioni utilizzando una cartuccia MiSeq kit v2 (150 cicli) con una
23
Protocolli NGS per analisi di pazienti con NPCA
Protocollo TruSeq Custom Amplicon (Illumina) su piattaforma MiSeq (Illumina)
Per l’analisi di pazienti con altre NPCA non-JS, è stato utilizzato il protocollo di sequenziamento TruSeq Custom Amplicon (TSCA) che consente di analizzare
dalle 2kb alle 650Kb di DNA genomico fino ad un numero massimo di 1536
ampliconi per ciascun campione, permettendo di multiplexare fino a 96 campioni
contemporaneamente. E’ stato disegnato un pannello di 44 geni costituito da geni
noti associati a differenti forme di NPCA con l’esclusione dei geni causativi di
JS. Le sonde dei geni d’interesse sono state disegnate utilizzando DesignStudio
Software (Illumina), un sistema automatico che disegna coppie di probes
specifiche per le regioni esoniche utilizzando un algoritmo che tiene conto di una
serie di fattori, tra cui le regioni ricche in GC, la specificità, l’interazione tra le
sonde ed il coverage. Gli ampliconi candidati (lunghezza 425bp) sono stati
visualizzati e valutati sulla base di uno score assegnato. Per valori bassi di score
si è resa necessaria la modifica di alcuni parametri al fine di migliorare il coverage
delle regioni d’interesse. Nel disegno dello studio, il coverage finale ottenuto è stato del 96%. È stato utilizzato il protocollo TSCA v1.5 (Part # 15027983 Rev.
C-Agosto 2013), partendo da una concentrazione di gDNA di 250ng con rapporti
260/280 superiori a 1.8-2.0. Ciascuna sonda è costituita da sequenze
target-specifiche associati ad adattatori universali che sono stati utilizzati
successivamente nella reazione di amplificazione. Il pool di sonde (Custom
24
utilizzando il buffer di ibridazione ed incubando nell’Heat Block (Hybex Microsample Incubator- Scigene) a 95°C per 1 minuto, poi la temperatura è stata
fatta scendere gradualmente a 40°C in circa 80 minuti, un passaggio fondamentale
per l’ibridazione. Il passaggio successivo è stata l’estensione-ibridazione, reazione che porta alla formazione di prodotti costituiti dalle regioni target; la
DNA polimerasi estende dall’oligonucleotide a monte lungo la regione target e la DNA ligasi esegue la ligazione all'estremità 5' dell’oligonucleotide a valle. Nello step seguente i prodotti dell’estensione-ligazione sono stati amplificati utilizzando
degli index primers (i5 ed i7) e degli adattatori necessari per la generazione dei
cluster (P5 e P7). Gli index aggiunti sono fondamentali per indicizzare i campioni
e consentirne l’unicità quando verranno multiplexati. Le librerie prodotte sono state controllate su gel di agarosio al 2%, successivamente purificate utilizzando
biglie AMPure XP ed etanolo all’80%, ed infine normalizzate con un protocollo basato su biglie che consente di ottenere una distribuzione omogenea della
concentrazione di ciascun campione. Tutte le librerie normalizzate sono state
unite in un'unica libreria prelevandone 5uL da ciascuna; da questo pool finale, il
cui volume dipende dal numero di campioni, 20uL sono stati aggiunti a 580uL di
buffer HT1 (Hybridization buffer) e denaturati a 95°C per 2 minuti. Per la corsa
al MiSeq è stata utilizzata la cartuccia MiSeq kit v3 che aumenta la densità dei
cluster e la lunghezza delle reads (fino a 25M di reads con 600 cicli) e fornisce
25
Protocollo WES con SureSelectXT Target Enrichment System (CRE V1- Agilent Technologies) su Piattaforma HiSeq 2500 (Illumina)
Il “SureSelectXT CRE V1” è un protocollo che consente di analizzare l’intero
esoma, garantendo una profondità di lettura di almeno 20X per i 3654 geni
clinicamente rilevanti (dati OMIM, HGMD e ClinVar). Tale protocollo è stato
utilizzato per analisi Whole Exome Sequencing (WES) su famiglie selezionate.
Sono stati analizzati trios (costituiti da probando, madre e padre) nei casi sporadici
oppure 2 o più soggetti affetti appartenenti ad uno stesso nucleo familiare. Una
concentrazione totale di 250 ng di gDNA di ciascun campione è stato frammentato
utilizzando un sistema Covaris S2. Questo step permette di ottenere frammenti di
DNA genomico con una grandezza di circa 120-150 paia di basi. Il DNA
frammentato viene sottoposto inizialmente ad una reazione di “riparazione delle
estremità”, poi ad una adenilazione al 3’ per rendere le molecole più stabili, ligato con adattori paired-end, ed infine amplificato. Ciascuna delle tre reazioni è
seguita da una purifica con “Agencourt AMPure® XP Reagent”. La libreria
prodotta viene controllata mediante l’uso del Bioanalyzer 2100, per verificarne la qualità e la quantità. Il passaggio successivo si basa sull’ibridizzazione delle
librerie di gDNA preparate con Capture Library target-specifica. Dopo
l'ibridazione, le molecole prodotte coi target specifici vengono catturate con le
biglie di streptavidina e sottoposto ad amplificazione post-cattura. Alle librerie
prodotte vengono aggiunti specifici index per ogni campione e sottoposti ad
amplificazione, seguita da una purifica con “Agencourt AMPure® XP Reagent”.
26
aver misurato la concentrazione delle librerie (nM), è stato calcolato il volume da
prelevare da ciascuna di esse per preparare il pool finale, che varia in base alla
concentrazione desiderata (tra 2-10nM) ed è strettamente correlato alla
concentrazione iniziale di ciascun campione arricchito. Infine, è stata caricata sul
sequenziatore HiSeq 2500 (Illumina) una concentrazione del pool finale variabile
tra 8pM a 12pM. Per il sequenziamento sono stati caricati pool da 8 campioni
utilizzando una cartuccia HiSeq Rapid Run kit v2 (200 cicli) con una lunghezza
delle reads 2x100bp fornendo un output e una qualità dati ottimali.
Protocollo Custom HD-CGH Microarray su piattaforma SureScan Microarray Scanner (Agilent Technologies)
Dall’analisi NGS di alcuni pazienti con JS sono state identificate varianti patogenetiche eterozigoti in singoli alleli. Considerando il limite diagnostico della
tecnica NGS utilizzata, che non permette di identificare CNVs, è stato disegnato
un Array Custom HD 8x15k (Agilent Technologies) ad alta densità sui geni
associati alla sindrome, con obiettivo di individuare eventuali
delezioni/duplicazioni intrageniche patogenetiche anche nel secondo allele del
gene con una sola mutazione eterozigote emersa dalla prima analisi. Per il disegno
è stato utilizzato il software SureDesign (Agilent) con uno spacing delle sonde di
circa 100bp in corrispondenza degli esoni e circa 500bp in corrispondenza delle
sequenze non codificanti. Ogni campione è stato processato utilizzando il
protocollo standard “Agilent Oligonucleotide Array-Based CGH for Genomic
DNA Analysis” (Part Number G4410-90010) utilizzando una concentrazione
27
introdotto nella piattaforma SureScan Microarray Scanner (Agilent
Technologies). Le immagini relative ai fluorocromi di ogni microarray sono state
acquisite ed analizzate utilizzando il software Agilent Scan Control (Agilent
Technologies). I dati sono stati elaborati mediante il software Feature Extraction
(v9.5) ed analizzati tramite il software CGH Agilent CytoGenomics Edition
3.0.6.6 (algoritmo ADM-2; release hg19) (Agilent Technologies). Per ciascun
cromosoma, il programma di elaborazione dati calcola automaticamente le
intensità di segnale di ogni spot, sottrae il rumore di fondo e definisce i rapporti
di intensità dei due fluorocromi relativi ad ogni oligomero, che vengono riportati
su un grafico. Una deviazione da un rapporto modale pari a 0, che indica la linea
di base, costituisce la presenza di un’aberrazione nel numero di copie del DNA genomico del paziente, che si traduce rispettivamente in una delezione per ≤ a
-0.57 e in una duplicazione per valori ≥ a +-0.57.
Analisi bioinformatica dati NGS
Le short-reads prodotte dal sequenziamento dei campioni sono state allineate al
genoma di riferimento hg19 tramite tre software distinti: Lifescope, Shrimp e
Bowtie. Considerando punteggi di qualità sito-specifici e relativi al mapping
globale delle reads, è stato scelto il miglior allineamento e, quindi, sono stati
selezionati i migliori BAM files. Di questi, sono stati esaminati preliminarmente
i valori di coverage per-base e per-esone ed è stata prodotta una stima delle regioni
potenzialmente “deboli”, cioè maggiormente soggette ad ospitare varianti “false positive”. I BAM files sono stati successivamente scansionati da tre diversi
28
variant callers: DiBayes & SOLiD Small Indel Tool, GATK e SAMtools. Alle
varianti ottenute dall’analisi sono stati assegnati dei punteggi in base alla loro frequenza di ricorrenza nella popolazione (dbSNP, 1000 Genomes, Exome
Variant Server (EVS), Exome Aggregation Consortium(ExAC), genome
Aggregation Database (GnomAD)), all’impatto sulla proteina codificata
(ANNOVAR, snpEff) ed infine al loro grado di conservazione (phyloP, GERP++,
SiPhy). La patogenicità di ogni singola variante è stata inoltre verificata in silico
tramite sei software di predizione: PolyPhen-2, SIFT,
MutationAssessor,MutationTaster, Provean, CADD. Le varianti così annotate
sono state sottoposte a filtraggio progressivo in modo tale da escludere
preliminarmente tutte le varianti non-geniche, introniche e sinonime non
occorrenti in siti canonici di splicing. Le varianti risultanti dal filtraggio sono state
ridotte ulteriormente di numero, esaminando solo quelle non presenti nei controlli
in-house, e con valori di MAF ≥0.01 nei database on line dbSNP 138, EVS, ExAC
e GnomAD ed escludendo quelle predette come benigne da tutti i predittori di
29
CAPITOLO I: SINDROME DI JOUBERT
INTRODUZIONE
La sindrome di Joubert (JS) rappresenta la più comune forma di atassia cerebellare
congenita ad eredità autosomica recessiva o X-linked. La diagnosi viene effettuata
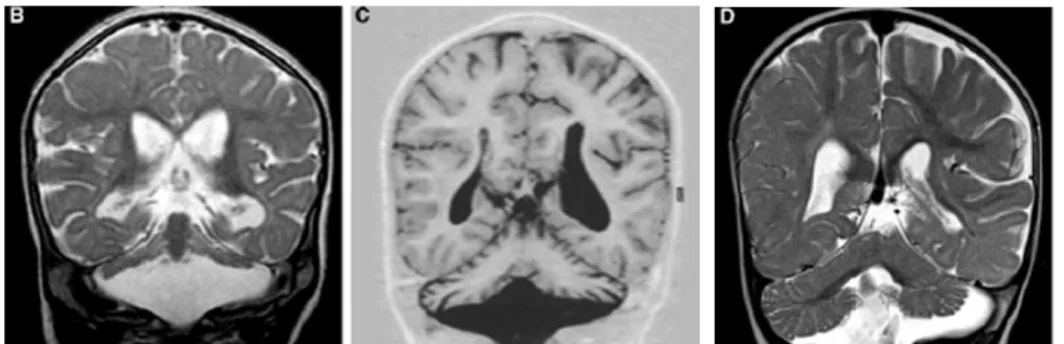
mediante l’identificazione in RMN di una malformazione complessa e peculiare, denominata “segno del dente molare” (molar tooth sign, MTS) (Maria et al. 1997).
Tale segno caratteristico deriva dalla coesistenza di: 1) ipodisplasia del verme
cerebellare; 2) ispessimento ed orizzontalizzazione dei peduncoli cerebellari
superiori; 3) assottigliamento dell’istmo ed approfondimento della fossa interpeduncolare (figura 2). Nelle sezioni assiali di RMN encefalo, al livello della
giunzione ponto-mesencefalica, tali anomalie conferiscono al ponte l’aspetto di
un dente, da cui il termine “MTS”. La JS è una condizione clinicamente e
geneticamente eterogenea, ma che riconosce un meccanismo patogenetico
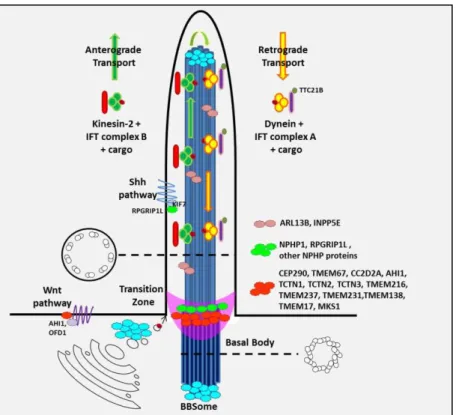
comune nella disfunzione di un organello subcellulare quasi ubiquitario, il cilio
primario (figura 3). In molti tessuti adulti, il cilio primario rappresenta un sensore
di segnali extracellulari e li trasduce all'interno delle cellule al fine di regolarne la
stabilità, la polarità o la proliferazione. L’alterazione di queste funzioni sensoriali a livello di cellule specializzate, quali l'epitelio renale, l’epitelio del dotto biliare o dei fotorecettori retinici, spiega molti dei difetti a carico di vari organi osservati
30
Figura 2: Neuroimmagini di un bambino di due anni con sindrome di Joubert pura (pannelli superiori) confrontati
con un bambino sano (pannelli inferiori). (A) L’immagine mediosagittale T1-pesata mostra una moderata ipoplasia e displasia del verme cerebellare (frecce) con una distorsione secondaria ed ampiamento del quarto ventricolo con spostamento rostrale del fastigium (punta della freccia). Si può osservare anche un’approfondimento della fossa interpeducolare. (asterisco). (B) Immagine Parasagittale T1-pesata mostra i peduncoli cerebellari superiori inspessiti, allungati e orizzontalizzati (freccia). (C) L’immagine assiale T1-pesata alla giuzione pontomesencefalica mostra il segno del dente molare con approfondimento della fossa interpeduncolare (punta della freccia) e peduncoli cerebellari superiori inspessiti, allungati e orizzontalizzati (frecce). Inoltre il verme cerebellare sembra essere ipoplasico. (D) L’immagine coronale T1-pesata mostra i peduncoli cerebellari superiori inspessiti (frecce) (Romani et al, 2013).
Figura 3: Rappresentazione schematica della struttura del cilio primario e dei suoi complessi proteici (Romani
et al, 2013)
Quadro clinico ed iter diagnostico della Sindrome di Joubert
Il sospetto di JS deve essere posto in tutti i neonati che nei primi giorni di vita
31
oculomotoria, nistagmo orizzontale o rotatorio o movimenti oculari erratici)
associati a disturbi del pattern respiratorio, in particolare episodi alternati di apnea
e tachipnea che si presentano in epoca neonatale e nella maggior parte dei casi
tendono a scomparire nei primi anni di vita. Può essere presente anche strabismo,
ptosi ed ambliopia. L’ipotonia muscolare, che si osserva per lo più nella prima infanzia, evolve successivamente in atassia. È inoltre presente quasi sempre un
ritardo nell’acquisizione delle tappe psicomotorie e in molti pazienti anche una disabilità intellettiva (Braddock et al. 2006). Tuttavia, le capacità cognitive si
collocano all’interno di uno spettro variabile che va dal riscontro di un quoziente intellettivo normale ad una marcata compromissione delle competenze motorie,
del linguaggio e delle funzioni adattive. Il deficit cognitivo spesso si associa ad
alterazioni comportamentali e dello spettro autistico e a disturbi del sonno
(Brancati et al. 2010). Il quadro clinico della JS può essere ulteriormente
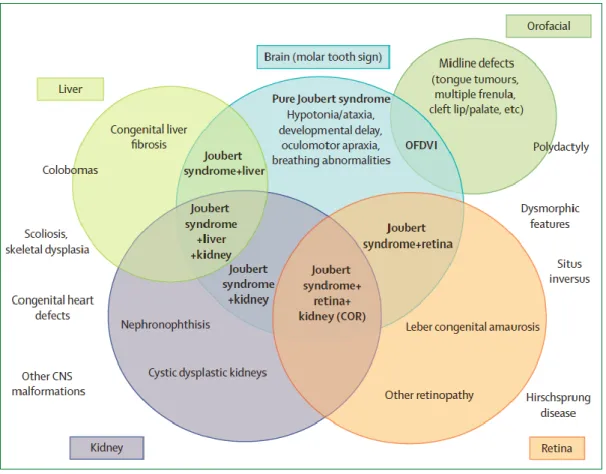
complicato dal possibile coinvolgimento di alcuni organi, in particolar modo la
retina, i reni, il fegato e lo scheletro. Il coinvolgimento renale e del fegato può
causare elevata morbilità e mortalità e necessita di un regolare follow-up.
Caratteristiche cliniche più rare includono colobomi corioretinici o del nervo
ottico, malformazioni cardiache congenite, situs inversus, scoliosi, displasia
scheletrica, malattia di Hirschsprung, difetti oro-faciali della linea mediana come
labio e/o palatoschisi, lingua lobulata con frenuli multipli e tumori molli della
lingua. In particolare, l'associazione della JS con polidattilia mesassiale e/o alcuni
32
superiore) definisce la Sindrome oro-facio-digitale di tipo VI (OFDVI) (Poretti et
al. 2012). La variabilità delle manifestazioni cliniche associate all’MTS non
rappresenta sindromi cliniche distinte, ma rientra in un vasto spettro di fenotipi
che sono caratteristici della sindrome stessa (figura 4).
Nei feti affetti l’esame ecografico mirato allo studio morfologico, eseguito tra la 20a e la 22a settimana di gestazione, è in grado di rilevare un’eventuale ipoplasia del verme cerebellare, che può essere anche associata ad altre anomalie come la
polidattilia, l’encefalocele occipitale o entrambi. Posto dunque il sospetto
ecografico di una malformazione cerebellare, si rende necessario come
approfondimento diagnostico l’utilizzo della risonanza magnetica fetale da cui sarà possibile rilevare la presenza dell’MTS (Fluss et al. 2006; Saleem and Zaki
2010; Pugash et al. 2011). Ad oggi, la diagnosi genetica prenatale di JS può essere
offerta a tutte le famiglie con un figlio affetto, in cui sia stata precedentemente
identificata la mutazione responsabile della malattia. Tali mutazioni possono
essere ricercate nel campione di DNA fetale ottenuto mediante prelievo di villi
coriali (tra l’11a e la 13a settimana di gestazione) o di liquido amniotico (tra la 16a e 18a settimana di gestazione). In epoca perinatale, la prognosi è relativa alla
gravità delle alterazioni respiratorie, in particolare, episodi ricorrenti e prolungati
di apnea possono portare alla necessità di assistenza con ventilazione meccanica
33
si risolvono spontaneamente nei primi mesi di vita, sebbene disturbi respiratori
sonno-correlati possano persistere fino all'infanzia (Kamdar et al. 2011).
Figura 4: Spettro di organi coinvolti nella sindrome di Joubert e classificazione dei sottogruppi clinici (in
grassetto) (Romani et al, 2013)
L’approccio, sia per quanto riguarda la diagnosi che la gestione del paziente, deve essere di tipo multidisciplinare, deve pertanto coinvolgere più specialisti non solo
per la valutazione neurologica e neuropsichiatrica ma anche per la valutazione
della funzionalità degli organi principalmente interessati.
Basi genetiche della sindrome di Joubert
Le basi genetiche della JS sono estremamente complesse e ad oggi comprese
soltanto in parte, nonostante il notevole contributo alla scoperta di nuovi
geni-malattia apportato negli ultimi anni dalle tecniche di NGS. Ad oggi sono stati
34
sono ad oggi identificabili soltanto in circa il 60-70% dei pazienti, suggerendo
ulteriore eterogeneità genetica.
Overlap clinico e genetico con altre ciliopatie e “mutational load”
L'elevata variabilità clinica della JS acquisisce ulteriore complessità se si
considera il notevole grado di sovrapposizione clinico-genetica con altre
ciliopatie come la nefronoftisi isolata (NPHP), la sindrome di Senior-Loken
(SLS), la sindrome di Bardet-Biedl (BBS), la sindrome di Meckel (MKS) e
ciliopatie scheletriche come le “short rib polydactylies” (Tabella 1). Il principale
esempio di questa sovrapposizione è rappresentata dalla MKS, una grave
condizione malformativa, spesso letale in utero, che si presenta con encefalocele
ed altre anomalie della fossa cranica posteriore, malformazione dei dotti epatici,
rene policistico e polidattilia. Per alcuni geni causativi sia di JS che di MKS (es.
TMEM67), sono emerse interessanti correlazioni genotipo-fenotipo, con una
maggiore occorrenza di mutazioni troncanti nei pazienti MKS ed una prevalenza
di mutazioni missenso con conservata produzione della proteina mutata nei
pazienti JS. Ad oggi sono noti ben 19 geni mutati in entrambe le ciliopatie,
(Brancati et al. 2007; Delous et al. 2007; Mougou-Zerelli et al. 2009; Tallila et al.
2009; Iannicelli et al. 2010) (Valente et al. 2010; Dowdle et al. 2011; Thomas et
al. 2012; Romani et al. 2014; Lambacher et al. 2016). L’elevata variabilità clinica
e genetica nelle ciliopatie può essere in parte spiegata da un modello di eredità
oligogenico, in cui mutazioni, varianti rare e polimorfismi in loci differenti
35
(Zaghloul and Katsanis 2010). Ad esempio, la variante p.R830W nel gene AHI1
è stata associata ad un aumentato rischio di sviluppare danni retinici nei pazienti
con nefronoftisi portatori della delezione omozigote del gene NPHP1 (Tory et al.
2007). Analogamente, la variante p.A229T nel gene RPGRIP1L sembra
aumentare il rischio di sviluppare degenerazione retinica nei pazienti affetti da
ciliopatie con mutazioni in altri geni (Khanna et al. 2009). Le varianti in
eterozigosi nei geni ciliari possono essere, quindi, responsabili dell’elevata
variabilità fenotipica, supportando il concetto del “mutational load”, che è la somma di tutte le mutazioni ed alterazione genomiche che contribuiscono a
36
JBTS locus Gene Protein Prevalent JS phenotypes Genetic overlap with other
ciliopathies
JBTS1 9q34 INPP5E Inositol polyphosphate-5-phosphatase JS±retina; (JS+fegato) MORM
JBTS2 11q12 TMEM216 Transmembrane protein 216 JS± rene; (JS+fegato, OFDVI) MKS2
JBTS3 6q23 AHI1 Jouberin JS±retina (JS+rene) -
JBTS4 2q13 NPHP1 Nephrocystin 1 JS+ rene NPHP1, SLSN1
JBTS5 12q21 CEP290 Centrosomal protein 290kDa JS+retina+ rene MKS4, NPHP6*, SLSN6, BBS14,
LCA10
JBTS6 8q22 TMEM67 Meckelin JS+ fegato (JS+rene) MKS3, NPHP11, BBS*
JBTS7 16q12 RPGRIP1L Protein fantom JS+ rene; (JS+ fegato) MKS5, NPHP8
JBTS8 3q11 ARL13B ADP-ribosylation factor-like 13B JS -
JBTS9 4p15 CC2D2A coiled-coil and C2 domain containing protein 2A JS±retina MKS6
JBTS10 Xp22 OFD1 oral-facial-digital syndrome 1 Fenotipo variabile OFD1, SGB2
JBTS11* 2q24 TTC21B tetratricopeptide repeat protein 21B -- MKS§, NPHP12, ATD4
JBTS12 15q26 KIF7 Kinesin-like protein 7 JS, (OFDVI) ACS, HLS2, MEDF
JBTS13 12q24 TCTN1 Tectonic-1 JS±retina -
JBTS14 2q33 TMEM237 Transmembrane protein 237 JS+ rene MKS§
JBTS15 7q32 CEP41 Centrosomal protein 41kDa JS MKS§
JBTS16 11q12 TMEM138 Transmembrane protein 138 Fenotipo variabile MKS§
JBTS17 5p13 C5Orf42 Uncharacterized protein C5orf42 JS±retina±caratteristiche OFD MKS§ + OFDIV
JBTS18 10q24 TCTN3 Tectonic-3 JS± caratteristiche OFD OFD4+ caratteristiche MKS
JBTS19 16q12 ZNF423 Zinc finger protein 423 JS+ rene NPHP14
JBTS20 16q23 TMEM231 Transmembrane protein 231 JS+retina+ rene MKS11
JBTS24 12q24 TCTN2 Tectonic-2 JS MKS8
JBTS21 8q13 CSPP1 Centrosome and spindle pole-associated protein 1 JS MKS-JATD
JBTS22 2q37 PDE6D Retinal rhodopsin-sentitive cGMP 3’, 5’-cyclic phosfodiesterase
subunit δ JS+ rene +retina -
JBTS§ 1q42 EXOC8 Exocyst complex component 8 JS -
JBTS28 17q22 MKS1 Meckel syndrome, type 1 JS±retina MKS1, BBS13
JBTS27 17p11 B9D1 B9 Domain-Containing Protein 1 JS MKS9
JBTS§ 19q13 B9D2 B9 Domain-Containing Protein 2 JS MKS10
JBTS25 1p36 CEP104 Centrosomal protein 104kDa JS -
JBTS 17p13 TMEM107 Transmembrane protein 107 JS+retina MKS13, OFDXVI
JBTS26 16p12 KIAA0556 Katanin-Interacting Protein 0056 JS -
37
Tabella 1: *mutazioni riportate in eterozigosi; § locus non assegnato: JS: solo caratteristiche neuroradiologiche. Abbreviazioni: ACS: Acrocallosal syndrome; ATD:
Asphyxiating thoracic dystrophy (Jeune syndrome); BBS: Bardet-Biedl syndrome; HLS: Hydrolethalus syndrome; LCA: Leber congenital amaurosis; MEDF: macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance; MORM: mental retardation, truncal obesity, retinal dystrophy, and micropenis; MKS: Meckel syndrome; NPHP: Nephronophthisis; OFD: Oro-facio-digital syndrome; SGB: Simpson-Golabi-Behmel syndrome; SLSN: Senior-Löken syndrome; JATD: Jeune asphyxiating thoracic dystrophy.; SRTD: Short-rib thoracic dysplasia;TCDOE: Cerebellar dysraphia with occipital encephalocele; SHH:Sonic Hedgehog
JBTS§ 11q13 C2CD3 C2 Domain-Containing Protein 3 JS+OFD OFDXVI
JBTS§ 1p36 NPHP4 Nephrocystin 4 JS+ rene NPHP4, SLSN4
JBTS§ 5q23 CEP120 Centrosomal protein 120kDa JS TCDOE, MKS, JATD, OFD
JBTS23 14q23 KIAA0586 Katanin-Interacting Protein 0086 JS SRTD14
JBTS§ 10q24 SUFU Suppressor of Fused Homolog JS+SHH-related disorders -
JBTS§ 2p15 TMEM17 Transmembrane protein 17 JS+OFD OFD
JBTS§ 12q21 POC1B POC1 centriolar protein homolog B JS -
38
RISULTATI E DISCUSSIONE
Analisi mutazionale dei pazienti JSCome parte del presente progetto di dottorato, è stata impiegata la tecnologia NGS
di Target Resequencing per effettuare l’analisi simultanea e rapida di un ampio
numero di geni coinvolti in ciliopatie in pazienti affetti da JS. La casistica oggetto
di studio include tutti i pazienti inseriti nel database tra il 2004 ad oggi con
mutazioni non precedentemente diagnosticate.
Sono stati disegnati due pannelli (“chip”): il primo costituito da 120 geni ciliari
(geni JS noti, geni JS candidati identificati con WES in progetti paralleli o
collaborazioni internazionali e geni codificanti per proteine che fanno parte dei
pathways di Shh, Wnt e PCP) è stato utilizzato nelle prime fasi di screening dei
pazienti, mentre il secondo (in uso negli ultimi 12 mesi) è costituito soltanto da
56 geni noti associati a JS e MKS.
Il totale dei probandi con JS ad oggi analizzati è di 416. L’analisi di filtraggio si
è basata sulla ricerca di varianti nonsenso, missenso non sinonime e di frameshift
con MAF (Minor Allele Frequency) < 0.01 in regioni esoniche o di splicing.
Mutazioni patogenetiche, sia in omozigosi che in eterozigosi composta sono state
identificate in 239 pazienti. Per confermare le mutazioni riscontrate, sono stati
effettuati studi di segregazione nel nucleo familiare di ciascun paziente, ed i
genitori sono risultati tutti portatori sani come atteso. La percentuale dei mutati
per gli esperimenti eseguiti è pari al 57% (239/416), in accordo con i dati della
39
Grafico 1: Distribuzione della percentuale di geni mutati riscontrati nella casistica totale raccolta e la loro
correlazioni con i fenotipi clinici.
L’impiego della tecnologia NGS ha consentito di diagnosticare in tempi rapidi un numero elevato di mutazioni patogenetiche, rispetto alle tecniche tradizionali
utilizzate in precedenza che richiedevano tempi di analisi molto più lunghi.
In 66 pazienti sono state identificate varianti patogenetiche in singoli alleli.
Recentemente Lindstrand e collaboratori hanno descritto 72 pazienti affetti da
BBS studiati per la ricerca di CNV patogenetiche mediante l’utilizzo di un Array
Custom 4x180k costituito da 94 geni di cui 17 associati a BBS e la restante parte
correlati ad altre ciliopatie. Lo studio ha permesso l’identificazione di
duplicazioni/delezioni muti-esoniche in 17 pazienti affetti (18,5%) (Lindstrand et
al. 2016). Alla luce di tale risultato e considerando il limite diagnostico della
tecnica NGS utilizzata, che non permette di identificare CNV, è stato disegnato
un Array Custom HD 8x15k (Agilent Technologies) ad alta densità sui geni
associati alla JS con l’obiettivo di individuare eventuali delezioni/duplicazioni
40
eterozigote emersa dalla prima analisi. L’analisi dell’Array Custom è tuttora in
corso.
Tra le famiglie negative all’analisi di Target Resequencing sono state sottoposte
a WES 5 famiglie. L’analisi dei dati preliminari ha permesso, ad oggi,
l’identificazione del difetto molecolare responsabile del fenotipo patologico in un nucleo familiare in cui è stata evidenziata la presenza di due varianti in eterozigosi
composta nel gene SUFU.
Studio mutazionale del gene C5orf42 in pazienti JS (Romani et al. 2015)
Il gene C5orf42 è stato identificato dal gruppo di Srour e collaboratori in 10
pazienti con un fenotipo neuroradiologico e clinico di JS pura, in un solo caso
associato a polidattilia pre- e post-assiale (Srour et al. 2012). Successivamente
sono stati descritti altri pazienti con fenotipo JS puro associato in un caso a
retinopatia ed in un altro ad encefalocele occipitale (Alazami et al. 2012; Ohba et
al. 2013). Quest’ultimo difetto cerebrale è stato riportato in un feto mutato in
C5orf42 in associazione con labioschisi e palatoschisi, dotto arterioso pervio e
idrocefalo in assenza di polidattilia o altri segni oro-facciali.
Nel 2014, Lopez e collaboratori hanno eseguito WES in 11 famiglie con sindrome
OFDVI individuando mutazioni nel gene C5Orf42 in 9 pazienti (82%),
suggerendo che questo gene potrebbe rappresentare la principale causa genetica
di tale condizione (Lopez et al. 2013). Precedentemente mutazioni rare nei geni
41
OFDVI (Coene et al. 2009; Valente et al. 2010; Darmency-Stamboul et al. 2013),
ma la definizione genetica di questa sindrome è rimasta a lungo non determinata.
Dei 12 pazienti mutati riportati da Lopez e collaboratori, soltanto 4 erano viventi
e rientravano nei criteri diagnostici di OFDVI: tutti avevano MTS associato a
multipli frenuli linguali con o senza amartomi (criterio diagnostico) e polidattilia
pre-assiale, variabilmente associata ad altri difetti di tipo oro-facio-digitali. Due
di essi mostravano anche amartomi ipotalamici e altri difetti a carico del sistema
nervoso centrale (porencefalia, eterotopia nodulare, polimicrogiria, agenesia del
corpo calloso). Gli altri otto pazienti (da 6 famiglie) erano feti con anomalie
cerebellari assimilabili all’MTS e varie combinazioni di polidattilia pre-, meso- o
post-assiale; in tali feti le caratteristiche orali erano assenti o difficilmente
valutabili (solo un paziente presentava frenuli multipli). Inoltre, alcuni pazienti
presentavano anomalie scheletriche associate, difetti cardiaci, utero setto e
microftalmia (tabella 2) (Lopez et al. 2013).
Ad oggi, dei 416 probandi analizzati sono state identificate mutazioni
patogenetiche nel gene C5orf42 in 36 pazienti, che rappresentano il 9% della
casistica. Tale risultato indica un coinvolgimento rilevante del gene C5orf42 nella
patogenesi della JS.
Nell’ambito del progetto di dottorato, nel 2015 è stato oggetto di una pubblicazione sulla rivista Human Genetics (Paper Allegato) un studio condotto
su 17 pazienti della casistica (fino al 2014) con diagnosi di OFDVI (Romani et al.
42
Dei diciassette probandi analizzati soltanto tre (17,6%) sono risultati portatori di
varianti patogenetiche nel gene C5orf42, una percentuale molto più bassa di
quella descritta da Lopez (9/11 - 82%). I tre pazienti mutati presentavano MTS
associato ad amartomi linguali e polidattilia meso e/o post-assiale; in un paziente,
inoltre, erano presenti frenuli multipli e la RMN mostrava la presenza di
amartoma ipotalamico, corpo calloso sottile e polimicrogiria bilaterale (paziente
11 in (Poretti et al. 2012)). Lo screening ha inoltre consentito di identificare una
mutazione emizigote nel gene OFD1 (c.2315dupT; p. P772Tfs*4) in un quarto
paziente (COR211) con sindrome OFDVI (JS con frenuli linguali multipli e
polidattilia post-assiale delle mani). Nessun’altra mutazione patogenetica è stata
riscontrata nei rimanenti 14 pazienti con fenotipo OFDVI.
I risultati ottenuti confermano che le mutazioni di C5Orf42 possono causare
OFDVI di gravità variabile, ma indicano anche che il fenotipo OFDVI ha una più
ampia eterogeneità genetica rispetto a quanto descritto da Lopez.
Oltre ai tre casi con fenotipo OFDVI, sono stati individuati altri 19 pazienti
portatori di mutazioni in C5Orf42, di cui 18 con fenotipo JS puro ed uno soltanto
con retinopatia. Le caratteristiche cliniche e neuroradiologiche sono descritte in
tabella 2.
L'identificazione di mutazioni C5Orf42 in pazienti con un fenotipo di JS pura è
pienamente in linea con i risultati precedentemente riportati in letteratura. Infatti,
considerando i pazienti finora riportati con mutazioni patogenetiche di C5Orf42
43
soltanto in due casi), mentre meno di un terzo presenta i criteri diagnostici
d’inclusione della sindrome OFDVI.
Il coinvolgimento renale o epatico non è stato mai riportato in associazione con
le mutazioni in C5Orf42, mentre la polidattilia, di qualsiasi tipo, sembra essere un
evento più frequente, presente nel 42% dei pazienti.
Fino ad oggi non sono stati pubblicati screening del gene C5Orf42 in feti con
MKS. Un ruolo di questo gene nella patogenesi di questa sindrome letale sembra
improbabile sia per l’assenza di associazione con la malattia cistica renale che la fibrosi epatica congenita, che sono due principali caratteristiche diagnostiche
della MKS, sia per la rara presenza di encefalocele occipitale, che è stato descritto
in percentuali molto basse. Non è possibile, ad oggi, definire chiare correlazioni
genotipo-fenotipo, poiché mutazioni troncanti e missenso del gene C5Orf42 sono
distribuite lungo la sequenza dell’intero gene e sono state riscontrate sia in pazienti con un JS pura che con OFDVI indipendentemente dalla severità del
quadro clinico. È possibile ipotizzare, come già suggerito per altre ciliopatie, che
ulteriori varianti non ancora identificate in geni differenti possono agire come
modificatori genetici del fenotipo ed influenzare, di conseguenza, la penetranza e
l'espressione delle caratteristiche ora-facio-digitali dei pazienti portatori di
mutazioni nel gene C5Orf42. Allo stato attuale, l'identificazione di tali
modificatori, quindi, rappresenta una delle più grandi sfide che ha come fine una
44 Srour AJHG Srour JMG Alazami HuMu Ohba Neurogen Lopez HumGen Shaheen EJHG Present study TOTAL (%) Nr. di pazienti 10 1 3 2 12 1 22 51 Fenotipi clinici#: - JS pura 10 1 2 2 - - 19 34 (66%) - JS con retina - - 1 - - - 1 2 (4%) - JS con rene - - - - - COR - - - - - JS with fegato - - - - - OFDVI (inclusi feti) - - - - 12(8 feti) - 3 15 (31%) - feti MKS-like - - - 1 - 1 (2%) Caratter. cliniche§: - segni neurologici 10 1 3 2 4 - 19 39 (100%) - retinopatia - - 1 - - - 1 2 (5%) -coinvolgimento rene/fegato - - - 0 - caratteristiche oro-facciali - - - - 6 1 3 10 (21%) - amartoma linguale / frenuli multipli* - - - - 5 - 3 8 (17%) - altre oro-facciali** - - - - 4 1 - 5 (10%) - polidattilia 1 - - - 12 - 7 20 (42%) -polid.mesoassiale* - - - - 6 - 2 8 (17%) - polid.preassiale 1 - - - 12 - 2 15 (31%) - polid.postassiale 1 - - - 9 - 4 14 (29%) - altri difetti SNC - - 1 - 9 1 4 15 (31%) - amartoma ipotalam* - - - - 5 - 1 6 (12%) - encefalocele occip. - - 1 - - 1 1 3 (6%) - altri difetti SNC*** - - - - 4 - 2 6 (12%) - altri difetti cong.**** - - - 1 7 1 3 12 (25%)
Tabella 2: Prevalenza delle caratteristiche cliniche dei pazienti mutate in C5Orf42 descritti fino ad oggi: #come
descritto in Romani et al, 2013;§ non sono conteggiati nelle percentuali i 3 pazienti senza questionario clinico*sufficiente per la diagnosi di OFDVI in associazione con MTS (Poretti et al, OJRD 2013); **labio e/o palate schisi, alterazioni dentarie, lingua lobulata, frenula corti; ***porencefalia, eterotropia nodulare, polimicrogiria, alterazioni del corpo calloso, idrocefalo, arinencefalia; ****alterazioni delle ossa corte e lunghe, cubito valgo,difetti cardiac ed aortici, utero setto, microftalmia, morbo di Hirschsprung, scoliosi.
Studio mutazionale del gene CEP120 in pazienti JS (Roosing et al. 2016)
Recentemente, mutazioni in CEP120 sono state riportate in quattro individui con
45
ciliopatia scheletrica (Shaheen et al. 2015). Nell’ambito di questo dottorato, il
lavoro svolto in collaborazione con il gruppo del prof. Gleeson ha permesso
l'identificazione di mutazioni nel gene CEP120 in uno spettro di fenotipi che
vanno dalla JS pura a condizioni più gravi con caratteristiche fenotipiche che si
sovrappongono con altre ciliopatie distinte come MKS, JATD e OFD. Inoltre,
abbiamo descritto un paziente con disrafia tetto-cerebellare ed encefalocele
occipitale (tectal and cerebellar dysraphia with occipital encephalocele –
TCDOE) con varianti patogenetiche in questo gene, identificando la possibile
causa genetica di tale condizione. Lo scopo dello studio è stato di descrivere la
frequenza e lo spettro fenotipico delle mutazioni nel gene CEP120, oggetto della
pubblicazione su Journal of Medical Genetics (Roosing et al. 2016) (Paper
Allegato).
L’analisi è stata condotta su un numero totale di 491 pazienti di cui: 346 provenienti dal nostro screening di target resequencing di 120 geni ciliari, e 145,
di cui 21 feti affetti da MKS e uno da TCDOE, analizzati nel laboratorio Gleeson
mediante WES. Mutazioni bialleliche sono state individuate in 4 probandi affetti
da una forma di JS pura (0,8%) e due feti con diagnosi di MKS e TCDOE. Sono
stati, inoltre, analizzati 15 pazienti con fenotipo ODFVI, tutti risultati negativi
all’analisi mutazionale del gene suggerendo che mutazioni di CEP120 non contribuiscano a questo specifico fenotipo. In conclusione, tale studio aggiunge
CEP120 alla lista di geni causativi di ciliopatie clinicamente distinte ma