1 MIELOMA MULTIPLO
1. 1 Descrizione
Il Mieloma Multiplo deriva dalla trasformazione neoplastica delle plasmacellule di tipo B, della linea linfoide nel midollo osseo. E' caratterizzato dall'accumulo di plasmacellule nel midollo osseo, dalla produzione e secrezione di gamma globuline (IgG o IgA) nel sangue e lesioni osteolitiche.
Nonostante sia stato proposto un modello di MM de novo, è diffusamente accettato che la maggior parte, anche se non tutti i MM evolvono attraverso uno stadio di proliferazione clonale prima della manifestazione sintomatica della patologia .
Un aspetto unico di queste plasmacellule maligne è che la loro sopravvivenza e proliferazione dipende dall'interazione con le cellule dello stroma e le citochine mielomiche sia autocrine che paracrine, dentro l'ambiente del midollo spinale, fino allo stadio finale della malattia in cui avviene il rilascio di plasmacellule nel sangue (Plasma cell Leukemia, PCL) o si verifica in siti extramidollari (EMM).
La Gammopatia monoclonale di significato indeterminato (MGUS), è lo stadio clinicamente precoce di espansione clonale delle plasmacellule. MGUS è caratterizzato da bassi livelli di plasmacellule nelle infiltrazioni midollari (<10%) e bassa concentrazione di proteina gammaglobuline monoclonale nel sangue (<3g/dl). Il Mieloma Multliplo asintomatico (SMM) è uno stadio intermedio tra MGUS e MM sintomatico, le differenze biologiche tra MGUS e SMM non sono ben definite, si distinguono per un livello più elevato di plasmacellule nelle infiltrazioni (>10%) ed una concentrazione più elevata di gamma globuline nel sangue (>3g/dl) per ciò rappresenta lo stato asintomatico del MM
1. 2 Sviluppo delle plasmacellule normali
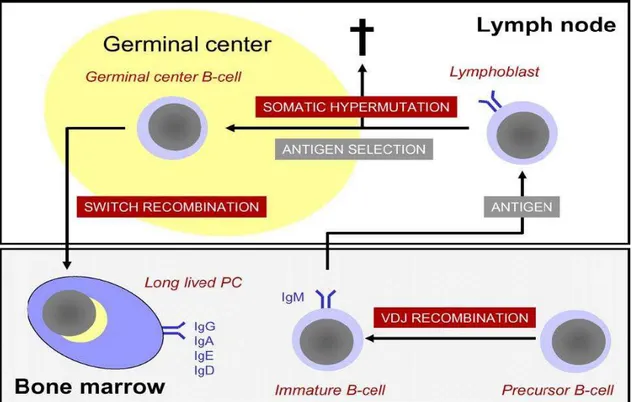
In condizioni fisiologiche normali, un linfocita B immaturo si differenzia dentro il midollo osseo fino allo stadio i cui avviene il riarrangiamento dei geni delle catene leggere (IgL) e di quelle pesanti (IgH) nel locus delle immunoglobuline. Dopo l'espressione di una IgM funzionale sulla superficie, un linfocita B migra come cellula navetta nei tessuti linfoidi secondari, dove la stimolazione con l'antigene porta alla proliferazione e al differenziamento di un plasmoblasto.
I plasmoblasti differenziano in plasmacellule di vita breve e generalmente esprimono solo IgM (anticorpi a bassa affinità); le plasmacellule stimolate entrano anche nei centri follicolari dei linfonodi dove vanno incontro al cambiamento di classe delle sequenze genomiche delle IgH e alla ipermutazione delle IgL. Questo processo porta alla formazione di un clone che esprime una Ig ad alta affinità che può entrare nel circolo sanguigno come cellula della memoria o può differenziare ulteriormente in un plasmoblasto postgerminale che produce generalmente IgG o IgA (occasionalmente IgE o IgD) oppure migrare nel midollo osseo dove dà origine a una plasma cellula di lunga vita, che sopravvive da più di 30 giorni fino ad anni e secerne elevate quantità di anticorpi.
1. 3 Le cellule B del centro follicolare sono le cellule di origine.
Figura 1. Sviluppo delle plasmacellule e meccanismi di modificazione del DNA ( Peter Liebisch, Hartmut Dohner)
Nei pazienti con MM, le plasmacellule mostrano caratteristiche tipiche delle cellule di lunga vita del midollo osseo. Esprimono generalmente l'antigene di superficie CD138 (syndecan 1), CD 38, ed altri marcatori immunofenotipici. Le plasmacellule B del MM presentano estese ipermutazioni somatiche dei geni delle IgH riarrangiati, e nella vasta maggioranza dei casi, esprimono un isotipo di Ig diversa dalla IgM, fatto che sta ad indicare un origine postfollicolare.
Queste cellule sono state esposte a tre meccanismi specifici di modificazione del DNA: ricombinazione VDJ, scambio (switch recombination) delle catene Igh ed ipermutazione somatica.
2 RILEVANZA PATOGENETICA DELLE ANOMALIE CROMOSOMICHE
Il MM è caratterizzato da anomalie cariotipiche; l'analisi citogenetica mostra che la malattia può essere suddivisa in due categorie citogenetiche:
1) a cariotipo non iperdoploide (4445 cromosomi)/pseudodiploide(da 44 -45 fino a 46 - 47 cromosomi); in questa categoria sono inclusi il cariotipo quasi tetraploide (>75 cromosomi, dovuto alla frequente perdita cromosomica nelle cellule di mieloma tetraploidi) e quello iperdiploide (>46-47 cromosomi);
2) a cariotipo iperploide, definito dalla presenza di trisomie cromosomiche multiple (più frequentemente dei cromosomi 3, 5, 7, 9, 11, 15, 19 e 21). L'analisi comparativa di cellule in interfase mediante ibridazione in situ fluorescente (FISH) mostra delle differenze notevoli nelle anomalie strutturali specifiche tra le due categorie, eg. l'incidenza di aneuploidie del cromosoma 13 (monosomia del 13 e delezione della banda 13q14) è significativamente più elevata nei tumori non iperdiploidi. In più le traslocazioni primarie che coinvolgono il cromosoma 14 nel locus delle immunoglobuline (14q32) sono ragionevolmente meno frequenti nel MM iperdiploide (<40%) mentre sono presenti nell’ 84% delle cellule non iperdiploidi.
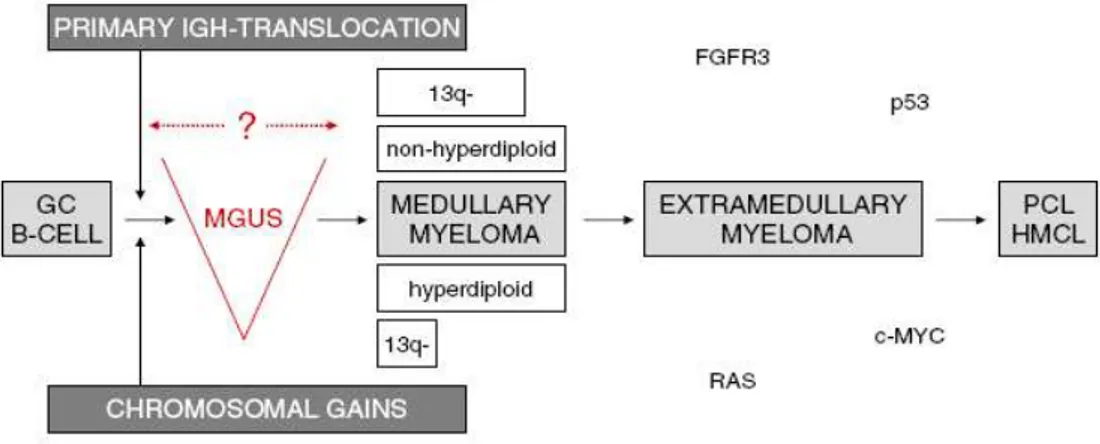
2. 1 Modello genetico di mielomagenesi
Nonostante le caratteristiche patologiche similari dei tumori del MM con o senza traslocazioni nel locus dei geni delle IgH, c'è una stretta associazione tra le tre traslocazioni principali [t(11;14)(q13;q32); t(4;14)(p16,3;q32); t(14;16)(q32;q23)] e il MM non-iperdiploide. Al contrario, altri tipi di traslocazioni nei loci delle immunoglobuline, diverse dalle tre più ricorrenti, avvengono con incidenza simile, e anche maggiore, nel MM iperdiploide. Il modello genetico di mielomagenesi implica due meccanismi diversi nella patogenesi iniziale della malattia, per cui, basandosi sulle osservazioni fatte è stato sviluppato un modello dicotomico (Liebisch and Dohner 2006).
La elevata incidenza delle traslocazioni primarie nel locus delle IgH nei MM non-iperdiploide suggerisce che i riarrangiamenti della regione cromosomica 14q32 con la conseguente attivazione di oncogeni, siano obligatori affinchè le plasmacellule acquisiscano l'immortalizzazione. Nel MM iperdiploide, è l'acquisizione di funzione di geni critici, che rappresenta l'evento chiave di trasformazione genetica.
La progressione della malattia da MGUS a MM ed infine a PCL è accompagnata da un aumento nel tasso di modificazioni genomiche secondarie, come mutazioni che portano all'attivazione di N-Ras, K-Ras e FGFR3, inattivazione di p53 mediante delezione o mutazione, e traslocazione di c-myc.
Il ruolo delle anomalie cariotipiche del cromosoma 13 in questo contesto rimangono un argomento di dibattito.
Figura 2. Modello genetico di mielomagenesi (Peter Libisch, Hartmut Dohner). Nel MM non-iperdiploide le tre traslocazioni principali nel locus delle Ig sono responsabili dell'attivazione di oncogeni mentre nel MM iperdiploide è l'acquisizione di funzione di geni critici l'evento chiave.
2. 2 Traslocazioni nel locus delle IgH
Una delle anomalie cariotipiche più frequenti nel MM, coinvolge il locus delle IgH nella regione 14q32, che normalmente prende parte in traslocazioni. Diversamente dal processo fisiologico in cui le sequenze geniche delle immunoglobuline vengono assemblate durante il cambio di classe, le traslocazioni in 14q32 nel MM sono caratterizzate dalla giustapposizione di sequenze geniche delle IgH con sequenze di DNA non-immunoglobuliniche (riarrangiamento illegittimo). Analisi mediante FISH in interfase, mostrano che queste traslocazioni sono presenti in circa il 50% dei pazienti con MGUS, nel 60-75% dei pazienti con MM, ed in più del 80% dei pazienti con PCL (plasma-cell leukaemia)
Si osserva una eterogeneità di cromosomi partners nelle traslocazioni; le regioni 11q13, 4p16.3, 16q23 e 6p21 sono i siti cromosomici ricorrentemente coinvolti nelle traslocazioni con 14q32. I quattro tipi di traslocazioni sono mutualmente esclusive e comprendono circa il 60% delle traslocazioni totali nel locus delle IgH. Questo
processo risulta nell'attivazione di oncogeni che vengono così sottoposti all'influenza delle regioni enhancer del locus dei geni delle IgH.
11q13
La traslocazione t(11;14)(q13;q32), con spostamento del gene della ciclina D1 contiguo al locus delle immunoglobuline, si trova in circa il 15% dei pazienti e provoca una sovraespressione della proteina ciclina D1; dato che i punti di rottura nella banda 11q13 non sono raggruppati in un cluster di traslocazione principale ma sparsi lungo una regione di 330 kb in posizione centromerica rispetto al locus della ciclina D1, la traslocazione t(11;14) porta anche alla deregolazione di un secondo gene (myeloma overexpressed gene, MYEOV), centromerico rispetto a ciclina D1. La traslocazione t(11;14) è particolarmente frequente nei casi di MM di tipo IgM, IgE e senza secrezione.
4p16.3
La traslocazione t(4;14)(p16;q32) è presente in circa il 15% dei pazienti ed è la causa dell'espressione alterata di due geni traslocati, il recettore del fattore di crescita dei fibroblasti 3 (FGFR3) sul cromosoma der14, e il locus SET del mieloma multliplo (MMSET) sul cromosoma der4.
16q23
La traslocazione t(14;16)(q32;q23), presente in circa il 5% dei pazienti, provoca una espressione estensiva di c-MAF, un fattore di trascrizione delle cellule linfoidi, coinvolto nella regolazione dell'espressione dell'interleuchina 4. Il suo ruolo nella patologia molecolare deve ancora essere determinato.
6p21
E' una traslocazione rara, presente in circa il 4% dei pazienti che causa la sovraespressione della ciclicna D3.
3 MONOSOMIA 13/ DELEZIONE 13q (-13/ 13q-)
3. 1 Incidenza biologica
Le anomalie cariotipiche del cromosoma 13 sono presenti nel 40-50% dei campioni; e coinvolgono la regione cromosomica 13q14-q21 ricorrentemente deleta sia nel MM che in altri tipi di patologie delle plasmacellule. Mediante analisi di citogenetica convenzionale, la delezione nelle regioni del cromosoma 13, in pazienti con un cariotipo informativo, ha una incidenza che varia dal 10% al 20%. La delezione è associata ad altre alterazioni frequenti; nei casi in cui sia presente la traslocazione t (4;14)(p16.3;q32) oppure la traslocazione t(14;16)(8q32;q23) l'incidenza di del(13) è approssimativamente del 90% (Liebisch and Dohner 2006) . Questa anomalia è più frequente nei casi con una copia extra del cromosoma 1q (65% vs 30%) (Liebisch, et al. 2005) e nei tumori non.iperdiploide rispetto a quelli iperdiploidi (65% vs 25-35%) (Fonseca, et al. 2004; Fonseca, et al. 2003). Nonostante che la delezione di 13q sia presente in tutti gli stadi, la sua incidenza varia nelle diverse condizioni, per cui ci sono dati diversi rispetto alla presenza di del(13) nel MGUS. Alcuni autori rilevano un'incidenza molto minore nel MGUS rispetto al MM (circa 25%) (Avet-Loiseau, et al. 1999), mentre altri mostrano una prevalenza simile a quella del MM (circa 50%) (Fonseca, et al. 2002a). Il primo dato implicherebbe che la delezione del(13) sia coinvolta nella progressione da MGUS a MM, mentre il secondo suggerirebbe fortemente che la delezione del(13) sia un evento iniziale e non progressivo. Questa anomalia è associata ad una minore risposta al trattamento chemioterapico(Desikan, et al. 2000; Fonseca, et al. 2002b; Shaughnessy, et al. 2003), all'aggressività della malattia e quindi ad una prognosi peggiore. L'impatto prognostico delle diverse strategie utilizzate per la rivelazione della delezione del(13) è controverso; un’ analisi comparativa tra il metodo di citogenetica convenzionale e quello di citogenetica
molecolare mediante ibridazione in situ fluorescente in interfase (i-FISH), evidenzia che la presenza di metafasi anomale è associata ad una prognosi peggiore, che si aggrava ulteriormente se sono presenti le delezioni del(13) o del(p53) o la traslocazione t(4;14) , ma solo la presenza della delezione del(13) rimane costante tra i diversi tipi di analisi: i pazienti portatori della delezione del(13) rilevata citogeneticamente oppure mediante iFISH, hanno una peggior prognosi. Tuttavia, quando i casi in cui la delezione del(13) viene rivelata da entrambi i metodi, vengono separati da quelli ottenuti tramite iFISH soltanto, la gravità della prognosi di questi ultimi scompare, mostrando un decorso paragonabile a quelli non portatori di del(13). L'effetto netto sulla prognosi, quando la delezione del(13) viene presa in considerazione esclusivamente come fattore prognostico, è maggiore se la delezione viene rilevata mediante analisi di citogenetica convenzionale che non attraverso FISH interfasica (Chiecchio, et al. 2006; Fonseca, et al. 2002b) . La situazione cambia quando vengono presi in considerazione solo casi di MM iperploide (H-MM). Il mieloma iperploide è associato al sesso maschile, al sotto-tipo di immunoglobulina kappa, a patologie delle ossa ed a una prognosi migliore in confronto al MM non-iperdiploide. In un vasto campione di casi di MM iperploide studiati mediante FISH per la ricerca di anomalie cromosomiche comuni, la sopravvivenza è minore quando sono presenti traslocazioni del locus delle catene pesanti delle Ig (IgH), specialmente quelle che coinvolgono partners sconosciuti, mentre la rivelazione della delezione del(13) mediante FISH non compromette significativamente la prognosi. In conseguenza di ciò, il Mieloma Multiplo iperdiploide costituisce un sotto gruppo individuale di MM associato ad una prognosi migliore (Chng, et al. 2006) .
3. 2 Regione minima deleta in 13q14
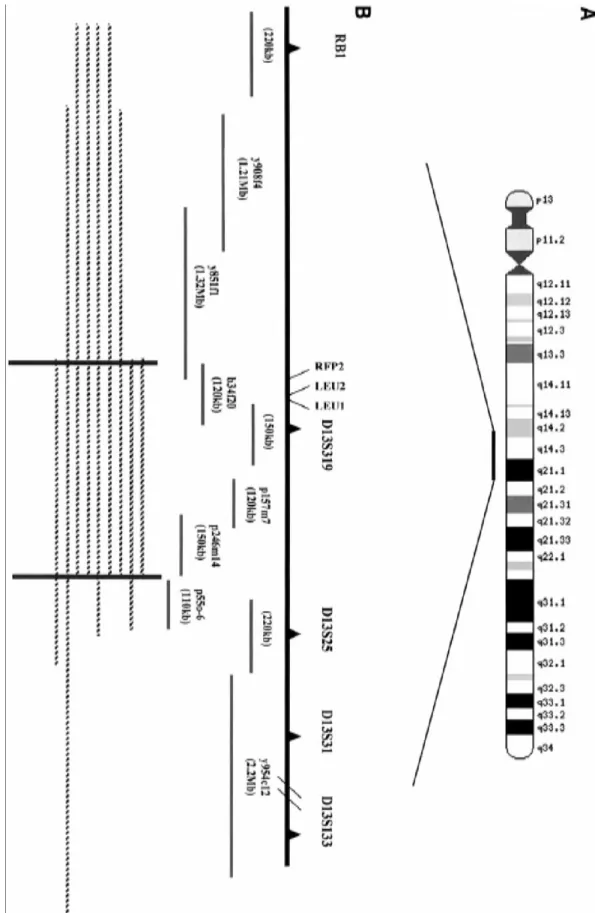
L'area minima comune deleta sul cromosoma 13q14, non è stata ancora completamente mappata. Inizialmente mediante dual-color FISH in interfase e utilizzando tre sonde per coprire la regione (RBI, D13S319 e D13S25), in 29 su 82 (35.4%) casi di MM sono state osservate delezioni con almeno una delle tre sonde, di cui tutte tranne una sono mono alleliche. In seguito, con l'impiego di YACs, PACs, e un BAC contigui lungo la regione 13q14-q21 in aggiunta ad una sonda telomerica del 13q sono state mappate numerose delezioni. Nel 55% dei casi sono state trovate grandi delezioni fino alla regione 13q34, mentre il 13,8% dei casi ha mostrato una perdita delle regioni 13q34 e 13q14 con conservazione della banda 13q21. E’ stata quindi identificata una regione comune deleta in 13q14 (Elnenaei, et al. 2003) di, approssimativamente, 350kb. La regione all' estremità prossimale, di 120kb, in posizione centromerica rispetto D13S319, è costituita da un area ricca in EST, che contiene anche i geni DLEU1, DLEU2 e RFP2, da un isola CpG in D13S319 ed da un EST (SGC32580) prossimale al limite telomerico.
La zona identificata corrisponde a quella del gene RFP2; attraverso l'analisi mutazionale mediante sequenziamento diretto della ORF (sequenza di lettura aperta) di questo gene, non sono state trovate mutazioni nei campioni analizzati. Se RFP2 è coinvolto, lo è per opera di altri meccanismi, possibilmente epigenetici, come la metilazione del DNA e mediante aploinsufficienza.
Due nuovi geni identificati nella regione deleta 13q14.3, RFP2/LEU5 e DLEU2 sono implicati anche in altre patologie (Corcoran, et al. 2004). Non sono state trovate mutazioni disattivanti specifiche, in questi geni, suggerendo insieme ai dati riportati sopra, un meccanismo di soppressione di tumore atipico. Il gene DLEU2 codifica per un RNA antisenso, con un esone sovrapposto direttamente al primo esone del gene
RFP2/LEU5 orientato in senso contrario. In più, i trascritti di RFP2/LEU5 possono
subire uno splicing alternativo, generando sia trascritti monocistronici diversi, che bicistronici codificanti due sequenze separate con modulo di lettura aperta, aumentando la complessità del locus. Il fatto che questa struttura sia conservata nel topo, sottolinea ulteriormente il significato inusuale di questo tipo di organizzazione e suggerisce una funzione biologica per DLEU2 nella regolazione di RFP2/LEU5.
Figura 3. Mappatura delle delezioni in 13q14-13q21 (da Manal O. Elnenaiei et al. 2003)
4. ASINCRONISMO REPLICATIVO
4. 1 Meccanismi epigenetici di modificazone del DNA
Dal momento che la ricerca di mutazioni in questi geni candidati a svolgere la funzione di soppressori di tumore ed associabili ad un fenotipo tumorale ha dato esiti negativi, i meccanismi coinvolti devono essere in grado di modificare l'espressione genica senza alterazioni nella sequenza nucleotidica. In questo contesto rientrano i fenomeni epigenetici come la metilazione del DNA e l'acetilazione degli istoni. Nelle cellule di mammifero, la metilazione del DNA opera generalmente come marker per la repressione trascrizionale. Consiste in una modificazione chimica catalizzata da una DNA metiltransferasi in grado di trasferire un gruppo metile dalla S-Adenosil-Metionina al carbonio 5 dei residui di citosina presenti nei dinucleotidi CpG, questo gruppo meC può essere facilmente riconosciuto da fattori specifici. I diversi profili di metilazione del DNA, primariamente nelle isole CpG, dove questo dinucleotide ha una frequenza elevata, sono mantenuti dalla attività della DNMT1 (DNA metiltransferasi 1), che durante la replicazione (fase S), copia l'assetto di metilazione già esistente sul filamento parentale. Circa il 50% delle isole CpG sono associate ai geni housekeeping e sono ipometilate in tutti i tessuti, quelle associate ai geni tessuto-specifici mostrano un profilo di metilazione più variabile ma, generalmente non sono metilate. In contrasto con DNMT1, le altre due DNA metiltransferasi di mammifero, DNMT3a e DNMT3b, facilitano la metilazione de
novo del DNA, e sono localizzate diversamente durante il ciclo cellulare (Bachman,
2001). In fibroblasti di embrioni di topo, durante il ciclo cellulare, la DNMT3a si trova insieme a HP1 (proteina eterocromatinica 1) e la MeCP2 (proteina che riconosce il DNA metilato) associate all'eterocromatina pericentromerica replicante
contro nelle cellule staminali embrionali, DNMT3b si localizza nei siti pericentromerici insieme a DNMT3a e HP1. Quindi, la modifica della normale localizzazione di queste DNA metiltransferasi de novo durante uno stadio di sviluppo precoce, potrebbe contribuire a creare un profilo di metilazione che verrà poi mantenuto dalla DNMT31 (McNairn and Gilbert 2003) .
L'acetilazione degli istoni è associata tipicamente alla cromatina trascrizionalmente attiva. Nonostante l'assemblaggio della cromatina sia mediato dalle stesse proteine chaperon istoniche, sia durante la fase-S tarda che precoce, avviene un cambiamento nello stato di acetilazione tra gli istoni incorporati in tarda fase-S che non si verifica durante la replicazione precoce. Anche se l'istone H4 quando entra nel nucleo è acetilato sui codoni per lisina 5 e 12, e si accumula nei siti di replicazione tardiva, questi gruppi acetile vengono rimossi approssimativamente venti minuti dopo che la sintesi del DNA è completata. Questa deacetilazione viene impedita mediante il trattamento delle cellule con TSA (tricostatina) e l'inibitore della HDAC (Taddei, et al. 1999; Verreault 2000).
La metilazione aberrante dell'estremità 5' del promotore di un gene è il meccanismo principale mediante il quale avviene il silenziamento di un gene soppressore di tumore in diversi tipi di patologie tumorali (Takahashi, et al. 2004); come evidenzia un lavoro realizzato sullo stato di metilazione delle tre forme principali di patologie ematopoietica e linfoide ( MM, Leucemia e Linfoma) in cui queste mostrano un profilo di metilazione alterato; tutte e tre confermano la ipermetilazione di dieci dei quattordici geni noti o sospetti essere GST, e questa è tumore specifica, con profili individuali.
4. 2 Controllo temporale della replicazione ed espressione genica
Un altro degli aspetti importanti della fedeltà nella replicazione del DNA consiste nel controllo temporale del processo. In particolare, i loci espressi in modo costitutivo (geni "housekeeping"), generalmente vengono replicati precocemente dal momento in cui ha inizio la fase S; i geni tessuto specifici si replicano durante la fase S precoce nei tessuti in cui vengono espressi, invece in quei tessuti dove sono inattivi si replicano nella fase S tardiva; mentre i loci non espressi tendono ad avere una replicazione molto tardiva, tipicamente associata alla completa inattività trascrizionale, caratteristica dell'eterocromatina.
E' stata evidenziata la correlazione tra il tempo di replicazione e le caratteristiche genomiche con rilevanza biomedica in dieci dei quindici geni soppressori di tumori/oncogeni noti su 11q e 21q, i quali risultano localizzati all'interno o nelle vicinanze delle regioni di transizione da replicazione precoce a tardiva. Ulteriormente, ventuno geni associati a patologie bene caratterizzate sono stati ritrovati nelle zone di transizione del tempo di replicazione, indicando che queste regioni sono ricche in geni responsabili di malattie e suggerendo che proprio per questa caratteristica replicativa tali geni sarebbero più instabili (Watanabe, et al. 2002) .
4. 3 Asincronismo replicativo
In un genoma diploide, lo spazio temporale di replicazione degli alleli di un locus con capacità trascrizionale, dipende dal modello di espressione. Le coppie di alleli di un gene espresso in modo biallelico, come descrive l'eredità Mendeliana, si replicano in modo sincrono. In contrasto le coppie di alleli soggette ad un qualunque meccanismo che porti al silenziamento di uno degli alleli del gene espresso (espressione monoallelica, espressione allele-specifica), quali imprinting, innattivazione del X o esclusione allelica, generalmente si replicano in modo asincrono. Ci sono diversi studi nei quali si accerta la modificazione della modalità regolare di espressione allelica da parte di cellule tumorali; in cellule derivanti da CML e Linfoma le larghe differenze temporali nella replicazione tra quattro paia di loci omologhi, indicano alleli con replicazione precoce e altri tardiva; il profilo di replicazione dei loci p53 e 21q22 nelle cellule di carcinoma alla cervice mostra che il tasso di asincronismo è significativamente elevato già nelle lesioni intraepiteliali avanzate e ancor più alto nel tumore allo stadio invasivo (Amiel, et al. 1998a; Amiel, et al. 1998b). In cellule non maligne provenienti da individui affetti, come nei
linfociti periferici di pazienti affetti di cancro alla prostata, le analisi dimostrano la perdita dell'ordine temporale di replicazione allelica accompagnata da aneuploidia, il risultato di una segregazione cromosomica anomala (Dotan, et al. 2000). Persino in individui con elevata suscettibilità al cancro per anomalie cariotipiche il tasso di asincronismo è elevato, come mostrano gli studi eseguiti su individui portatori di inversioni 2 e 9 e di traslocazoni bilanciate ereditarie (Amiel, et al. 2001a; Amiel, et al. 2001b).
In maniera simile, è stata descritta l'associazione tra una replicazione allele specifica con il fenomeno di aneuploidia nelle cellule del sangue di pazienti con disordini ematici (Korenstein-Ilan, et al. 2002) e di pazienti affetti di tumore alla prostata (Dotan, et al. 2004); inoltre nelle cellule di donne affette da tumore ereditario all' ovaio, elevati livelli di aneuploidia sono stati collegati ad una perdita del sincronismo replicativo degli alleli di una sequenza non codificante appartenente a regioni di DNA satellite dei centromeri (Litmanovitch, et al. 1998), il componente fondamentale per la corretta disgiunzione dei cromosomi.
Dunque, si può assumere, che la perdita della fedeltà inerente all'ordine temporale di replicazione di una coppia allelica costituisca una risorsa per la realizzazione di numerosi eventi genetici (aneuploidie) ed epigenetici (inattivazione allelica) richiesti per conferire un fenotipo maligno, suggerendo che l'asincronismo possa riflettere una espressione monoallelica, presente in una parte delle cellule. Alcuni autori ritengono che, se la replicazione allele specifica riflette l'espressione allele specifica, da un punto di vista funzionale l'asincronismo replicativo possa essere equivalente alla perdita di eterozigosi, caratteristica nello sviluppo tumorale; diversamente da questa la perdita di funzione dovuta alla replicazione asincrona è reversibile e potrebbe essere implicata sia nella progressione sia nella regressione del cancro
(Korenstein-In uno studio recente, il silenzimento allelico della banda 13q14.3 sia nelle cellule B-CLL (Chronic lymphocytic leukemia) che in quelle di donatori sani, suggerisce un meccanismo di soppressione di tumore epigenetico (Mertens, et al. 2006). E' stato trovato che le due regioni omologhe della regione critica replicano in modo asincrono, suggerendo un diverso stato di impacchettamento della cromatina. Nonostante sia stato trovato un meccanismo di silenziamento monoallelico dei geni localizzati nella zona critica, l'espressione monoallelica che ne deriva ha origine in entrambe le regioni omologhe sia materna che paterna, escludendo un meccanismo di imprinting. L'analisi sullo stato di metilazione delle sequenze di DNA in qusta regione eseguite in linee cellulari di origine ematopoietica, rivela che una delle isole CpG della regione è metilata. La demetilazione dell'isola CpG e l'acetilazione degli istoni induce un'espressione biallelica; altresi, la modalità di replicazione asincrona non varia. Il tempo di replicazione differenziato potrebbe rappresentare una marcatura epigenetica precoce che differenzia le due regioni omologhe di 13q14.3, e in conseguenza di ciò, produce un impacchettamento diverso della cromatina e l'espressione monoallelica. In accordo, la delezione della regione attiva di 13q14.3, avrebbe come risultato un significativo abbassamento dell'espressione dei geni candidati e la perdita di funzione.
L'aspetto peculiare di questo meccanismo di esclusione allelica è che, guardando i casi singolarmente, si osserva che l'attività genica ha una base cromosomica fortemente sbilanciata, essendo espresso preferenzialmente il cromosoma materno o quello paterno. Ciò indica che un sottogruppo di geni localizzati nella banda 13q14.3 vengono espressi solamente da una delle regioni cromosomiche omologhe e silenziati nell'altra.
L'espressione monoallelica della regione critica ha delle profonde conseguenze nel meccanismo patologico di 13q14.3, dato che la perdita dell'allele attivo del gene
potrebbe essere sufficiente per provocare la perdita di funzione. Per testare questo modello, sono state isolate cellule T da pazienti affetti da CLL eterozigoti per uno SNP localizzato in RFP2OS. In queste cellule, benigne, la copia attiva di RFP2OS è stata identificata, dopo di che sono state analizzate le cellule B, che rappresentano le cellule tumorali. Nei due casi informativi, la copia genica attiva di RFP2OS presente nelle cellule T, risultava deleta nelle cellule B.