1
PhD Program in
“Translational Medicine”
Gene Therapy with miR-199a-3p for
Myocardial Infarction
Candidate: Thesis Advisor:
2 LIST OF CONTENTS
ABSTRACT……….7
Chapter 1. INTRODUCTION ... 9
1.1 Cardiomyocyte proliferation, a capacity lost after birth ... 12
1.2 Gene therapy to stimulate cardiomyocyte proliferation ... 16
1.3 MiRNA biogenesis and mechanism of action ... 17
1.4 MicroRNAs in Cardiovascular Disease………. 20
1.4.1 MiRNAs in cardiovascular disease, historical perspective ... 20
1.4.2 Control of cardiomyocyte proliferation by microRNAs ... 22
1.5 The need for large animal models to test new therapies ... 27
1.5.1 Pig model of myocardial infarction ... 28
1.5.2 MiRNA delivery………..30
1.5.2.1 Adeno associated viral vectors ... 33
1.5.3 AAV delivery techniques ... 35
1.5.4 Direct AAV injection in infarcted hearts ... 41
1.6 Diagnostic imaging of myocardial infarction by cardiac magnetic resonance (CMRI) ... 42
1.6.1 Tagging MRI – quantifying myocardial regional wall motion ... 43
1.6.2 Late gadolinium enhancement method the quantification of myocardial infarction size ... 45
1.6.3 Myocardial Edema, Area at Risk and myocardial salvage... 46
1.6.4 Myocardial Perfusion ... 48
Chapter 2. OBJECTIVE OF THE STUDY ... 51
Chapter 3. METHODS ... 52
3.1 Production and purification of recombinant AAV vectors and miRNA ... 52
3.2 Surgical instrumentation ... 52
3.2.1 Myocardial infarction model ... 52
3.2.2 Testing the different AAV serotypes ... 56
3.3 Experimental protocol ... 57
3.4 Cardiomyocyte proliferation assay by BrdU ... 57
3
3.6 Hemodynamic measurements and tissue sampling ... 59
3.7 Tissue analysis ... 61
3.8 CMRI data analyses and representation ... 64
3.9 Hemodynamic data analyses ... 72
3.10 Statistical analyses ... 72
Chapter 4. RESULTS ... 73
4.1 Mortality rate and completed protocols ... 73
4.2 Selection of the AAV serotype for pig myocardial transduction ... 77
4.3 hsa-miR-199a-3p expression ... 78
4.4 Hemodynamics and cardiac function ... 80
4.5 Cardiac volumes and remodeling ... 81
4.6 Regional LV contractile performance ... 85
4.6.1 LV wall thickening ... 85
4.6.2 LV Radial and Circumferential shortening ... 87
4.7 Infarct size ... 92
4.7.1 Infarct scar size ... 92
4.7.2 Gray zone ... 98
4.8 Myocardial Edema, AAR and myocardial Salvage ... 100
4.9 Myocardial Perfusion ... 101
4.10 Tissue samples analysis ... 104
4.10.1 Cardiomyocyte proliferation ... 104
4.10.2 Cardiomyocyte size ... 106
4.10.3 ANP, BNP, MYH6/7 gene expression ... 107
4.11 Preliminary Results………..……….………...110
4.11.1 Purkinje cell proliferation……….…....110
4.11.2 miR-199a target gene expression……….………111
4.11.3 Expression of YAP-activated genes………...………..112
Chapter 5. DISCUSSION ... 113 5.1 Study limitations ... 117 5.2 Conclusions ... 118 5.3 Future directions ... 119 Appendix of tables ... 120 BIBLIOGRAPHY ... 133 AKNOWLEDGEMENTS ... 144
4 LIST OF ABREVIATIONS AND ACRONYMS
AAPC = Average annual percentage change AAR = Area at risk
AAV6 = Adenoassociated virus serotype 6 AD = Adenovirus
AGO = The Argonaute protein ANP = Atrial natriuretic peptide AOP = Aortic blood pressure ATP = Adenosine triphosphate
BIRC5 = Baculoviral repeat-containing 5 BNP = Brain natriuretic peptide
BRDU = Bromodeoxyuridine
CARP = Cardiac ankyrin repeat protein CLIC = Clathrin Independent Carriers
CLIC5 = Chloride intracellular channel protein 5 CMRI = Cardiac magnetic resonance imaging CO = Cardiac output
CTGF = Connective Tissue Growth Factor CX = Circumflex branch of left coronary artery CYR61= Cysteine-rich angiogenic inducer 61 DALY = Disability adjusted life years
DAPI= 4',6-diamidino-2-phenylindole
DGCR8 = DiGeorge syndrome chromosomal region 8 Ea = arterial elastance
ECC = Circumferential strain
ECG = Electrocardiogram ECM = Extracellular martrix EDP = End-diastolic pressure
EDPVR=End dystolic pressure volume relationship EDU = 5-ethynyl-2′-deoxyuridine
EDV = End-diastolic volume
EDWT = End-diastolic wall thickness EF = Ejection fraction
ELL = Longitudinal strain
5 ERα = Estrogen receptor α
ESPVR=End systolic pressure volume relationship ESV = End-systolic volume
ESWT = End-systolic wall thickness FRMD6 = FERM-domain-containing-6
GEEC = GPI-anchored protein Enriched Endocytic Compartment HARP = Harmonic phase analysis
HNRNP A1 = Heterogeneous nuclear ribonucleoprotein A1 HOMER1 = Homer protein homolog 1 protein
HOPX = Homeodomain-only protein HR = Heart rate
IAP = Inhibitor of apoptosis IR = Inversion recovery IVS = Interventricular septum
KSRP = The KH-type splicing regulatory protein LAD = Left anterior descending coronary artery LATS = Large tumour suppressor
LV = Left ventricle
LVM = Left ventricle mass LVP = left ventricular pressure LVV = and left ventricular volume LVWT = Left ventricle wall thickening MHC = Myosine heavy chain
MI = Myocardial infarction MiRNA = MicroRNA
MOB1 = Mps-one binder kinase activator-1 MRI = Magnetic resonance imaging
MRNA = Messenger RNA
MST1, MST2 = Mammalian sterile-20-like kinases type 1 and type 2 NCRNA = Small non-coding RNA
NF2 = Neurofibromatosis type II protein (Merlin)
NHLBI = National Heart, Lung, and Blood Institute NPRA = Natriuretic peptide receptor A
ORF = Open reading frame
PACT = Protein kinase RNA activator PAS = periodic acid-Schiff's stain
6 PBS = Phosphate-buffered saline
PCI = percutaneous coronary interventioon PET= Positron emission tomography PH3=Phospho-histone-3
Pre-miRNA = Precursor miRNA
PRSW=preload recruitable stroke work Pri-miRNA = Primary microRNA P53 = Tumor protein 53
P1 = Postnatal day 1
Ran-GTP = GTP-binding nuclear protein Ran RCA = Right coronary artery
RISC = RNA-induced silencing complex
ROS = Reactive oxygen species RV = Right ventricle
RYR = Ryanodine receptor SAV1 = Salvador homolog 1 SCD = sudden cardiac death SE = Echo sequence
SEM = Standard error of mean SI = signal intensity
SI-RNA = Short interfering RNA
SPAMM = Spatial modulation of magnetization
SPECT = Single-photon emission computed tomography SV = Stroke volume
TARBP = Transactivation response RNA binding protein
TEAD = TEA domain-containing sequence-specific transcription factor TIMI = Thrombolysis in myocardial infarction
TNF = Tumor necrosis factor VF = Ventricular fibrillation VG = Vector genomes VT = Ventricular tachicardia
WGA = Lectin Wheat Germ Agglutinin YAP = Yes-associated protein
7 ABSTRACT
One of the striking differences between the adult and the embryonic heart in mammals is the incapacity of cardiomyocytes to proliferate in response to damage. It has been previously shown that microRNA hsa-miR-199a-3p promotes cell cycle re-entry of rodent adult cardiomyocytes, ex vivo, and favors almost complete recovery of cardiac functional parameters when injected in infarcted mouse hearts by stimulating cardiac regeneration.1
The aim of this study was to test the therapeutic action of miR-199a in a pre-clinical, large animal model of myocardial infarction (MI). The precursor DNA encoding for hsa-mir-199a was encapsidated in serotype 6 adeno-associated viral vectors (AAV6) for cardiac delivery, in vivo. MI was induced in pigs by occluding the left anterior descending artery (LAD), for 90 minutes, immediately below the 1st diagonal branch, and a total of 2x1013 AAV6-hsa-miR-199a-3p (AAV6-miR-199a group; n=10) or 2x1013 AAV6 containing an empty polylinker (AAV6-control group; n=9) was injected intramyocardially during reperfusion in 20 different sites along the infarct border zone. Sham operated pigs (n=6) were used as non-infarcted, normal controls. Pigs underwent cardiac magnetic resonance imaging (MRI) without and with gadolinium-delayed contrast enhancement at 2 days, 1, 4 and 8 weeks post-MI. The infarct area was not significantly different between the two infarcted groups at 2 days post-MI. However, at 4 weeks, the scar involved 23.0±2.5% of the total left ventricle myocardium in the AAV6-control group, and only 11.49±1.28% in the AAV6-miR-199a group. Consistently, ejection fraction was 65.3±2.0% (n.s. vs normal control) in the AAV6-miR-199a group, markedly preserved compared to 53.7±3.04% in the AAV6-control group (P<0.05). Finally, a 8-segment analysis of left ventricular circumferential and radial shortening, as measured by tagging-MRI, was performed to generate a 8-point curve. The area under the curve (in arbitrary units) for Ecc (circumferential shortening) was -259.3±19.8 in the AAV6-miR-199a group (n.s. vs sham operated group) and -177.6±26.2 in the empty-AAV6 group (P<0.05 vs sham operated group, and AAV6-miR-199a group). The AUC for ERR (radial shortening) was 338.15±67.7 in the AAV6-miR-199a group, compared to 213.4±35.18 (P<0.05) in the AAV6-control group. In both infarcted groups, the AUC of ERR was lower compared to sham operated animals (540±24.6, P<0.05).
8 Histology revealed that AAV6-has-miR-199a-3p treatment did not increase cardiomyocyte cross sectional area at 4 weeks after MI. Bromodeoxyuridine and phospho histone 3 quantification assays, markers of cell proliferation, did not show any difference between the two infarcted groups. The expression of genes typically upregulated in the failing heart, such as atrial natriuretic peptide and brain natriuretic peptide decreased in both infarcted groups over time and the difference between AAV6-miR-199a and AAV6-control groups were found at 8 weeks. The myosin heavy chain6/7 ratio gradually increased in the AAV6-miR-199a group and became significantly higher compared to the AAV6-control group at 8 weeks after MI.
All the above-mentioned parameters indicate extensive recovery of regional contractility in hearts receiving miR-199a-3p. These are the first data to show, in a clinically relevant animal model, the efficacy of a new therapeutic strategy for acute MI based on cardiac delivery of a small regulatory RNA.
9 Chapter 1: INTRODUCTION
Ischemic heart disease ranks among the leading causes of mortality 2 (table 1), although it has substantially declined over the past 30 years 3,4 (figure 1). This improved outcome has been mainly due to the advances in pharmacological therapies and devices, the progress in percutaneous and surgical interventions on coronary vessels and the identification of risk factors, such as obesity, smoking, diabetes and hypertension, with consequent refinement of prevention strategies.
Table 1: Changes in rankings for 15 leading causes of DALYs (Disability Adjusted Life Years), 2002-2030 (baseline scenario). Table taken from reference2
However, the increased survival after myocardial infarction (MI) necessarily implies an increased number of patients with some degree of heart dysfunction eventually evolving towards failure. The search of innovative therapies that, rather than attenuating the symptoms of cardiac failure and/or delaying its progression, can effectively repair the infarcted heart, has been perceived as one of the top priorities in the cardiovascular field over the past two decades. 5,6,7
The acute MI, particularly if large and transmural, can produce profound morphological alterations in both infarcted and non-infarcted regions. Chamber remodeling can notably affect ventricular function and is a complex phenomenon involving inflammation, accelerated extracellular matrix (ECM) turnover, myocyte hypertrophy and vascular and muscular tissue regeneration.8,9These alterations can
10 be well characterized, ex vivo, utilizing classical methods of histological and molecular analysis.
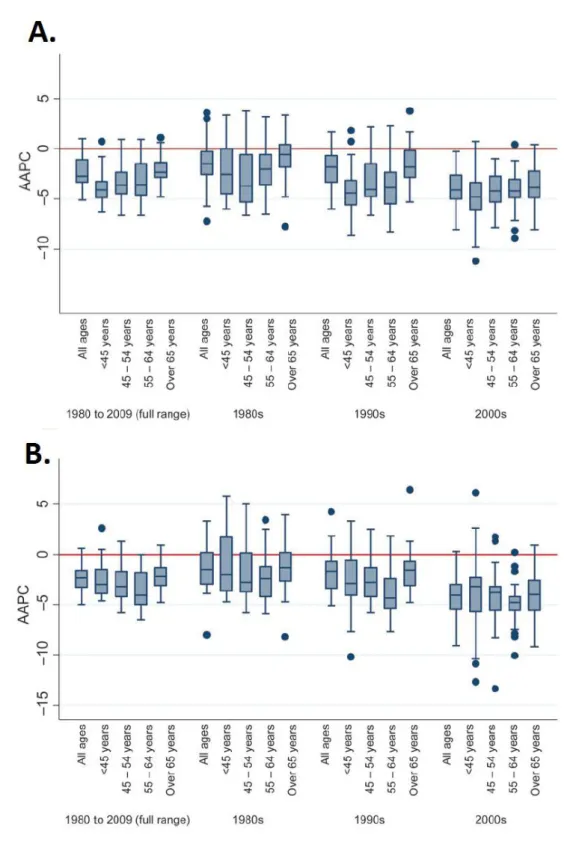
Figure 1. Boxplot of distribution of average annual percent changes in coronary heart disease among men (A) and women (B) overall and for each decade across age groups in Europe. AAPC-Average annual percentage change. Figure taken from reference 3
11 After the onset of ischemia, myocardial cell death starts within 20-45 minutes, and the main losses occur over the first 6 to 24 hours in case of permanent coronary occlusion or prolonged occlusion (>120 min) followed by reperfusion. Cell death involves also the peri-infarct zone, albeit to a lesser extent, and is initially the result of residual ischemia, then continues during cardiac remodeling.10 This latter affects peri-infarct and remote zones and stimulate myocyte apoptosis due to several factors, including neurohormones, increased mechanical strain, inflammatory cytokines and reactive oxygen species.11 Chronic LV remodeling with chamber dilation and impaired systolic pump function is temporally associated with increased myocyte apoptosis in myocardium remote from the area of initial ischemic damage. This process continues for months in the remote myocardium as remodeling progresses.10,11
At the start of ischemia, the mitochondrial electron transport stops functioning and energy metabolism shifts to anaerobic glycolysis. Together with concurrent lysosomal activation, this alteration causes ATP depletion and intracellular accumulation of lactic acid and calcium. Other deleterious changes include free radical production, increase in cell osmolarity and acidosis. The final effect is cell dysfunction (which can be initially reversible) and later cell death.12
Studies in canine models of MI have determined the progression of tissue sufferance at different time points after coronary occlusion: at 15 minutes, ischemia could be totally reversible, while after 40 minutes focal or confluent subendocardial necrosis are observed; after 3 hours MI will start developing with the involvement of mid- or subepicardial layers. Necrosis is usually transmural after coronary occlusion lasting longer than 6 hours.13,14In porcine models of MI, necrosis is already evident after 2 hours, due to the lack of collateral coronary vessels.15
One of the typical features of mammalian myocardial biology is the absence of cardiomyocyte proliferation in response to acute or chronic myocardial injury. Cells rather undergo very limited division cycles, which are inadequate for regaining normal cardiac anatomy and function. This process is much slower in the heart compared with the digestive system, blood or skin. Despite some evidence that the rate of cardiomyocyte renewal may increase slightly after injury in mammals, including humans,16,17 such response is insufficient to replace the ≈1 billion cardiomyocytes that may be lost during a typical MI, with consequent progressive
12 cardiac dysfunction. Therefore, developing novel therapeutic strategies that could enhance the normal regenerative potential of the adult mammalian heart would have an enormous clinical impact.
1.1 Cardiomyocyte proliferation, a capacity lost after birth
The heart is the first organ to be formed during embryonic development, when cardiomyocytes have the ability to undergo cytokinesis. After birth, cardiomyocytes stop dividing and this process is accompanied by the uncoupling of DNA synthesis and from cytokinesis.18 It was found that cell cycling in postnatal life, under physiological conditions and after injury, leads to polyploidy, but also to new diploid and mononucleate cardiomyocytes. For example, multinucleated cardiac cells are formed in mice between the postnatal period P4 and P14.18 Eighty percent of cardiomyocytes in adult mice, rats, rabbits, and guinea pigs and 45% of cardiomyocytes in dogs and cows are binucleate.19 In pigs, cardiomyocytes may contain up to 32 nuclei.20 In humans, 75% of adult cardiomyocytes remain mononucleate.21 This observation might be relevant, considering that mononucleated cardiomyocytes hold greater potential for cell division upon stimulation.22,23
During development, there are waves of proliferation of already differentiated cardiomyocytes. For example, the tubular, primitive heart that is formed in the early stages of cardiac development mostly consists of non-dividing cells. As development progresses, the tube starts looping and cardiomyocytes resume proliferation at the outer curvatures to form the future chambers.24On the contrary, cardiomyocytes at the inner curvature remain resting and will form the conduction system. Thus, proliferation appears to be a common characteristic of the cardiomyocyte, which depends on developmental signals imparted to the cell.
Little is known about the molecular mechanism by which cardiomyocytes stop dividing after birth. It is also unclear whether the signals regulating the exit from cell cycle are intracellular or extracellular. Burton at al. found that the embryonic cardiac myocytes proliferate in culture and in vivo with closely resembling timing, suggesting the existence of an intrinsic timer which might rely, at least in part, on intracellular activation of the cyclin-dependent kinase inhibitors p18 and p27.25
13 On the other hand, various environmental factors could have effects on cardiomyocytes withdraw from the cell cycle. For instance, one of the major changes occurring at birth is the sudden and marked increase in oxygenation. Oxygen tension in the fetal heart is 18 to 28 mmHg,26but, after birth, it rises rapidly in the whole organism to reach approximately 100 mmHg.27 According to some authors, the sudden increase in oxygenation boosts oxygen free radicals (reactive oxygen species, ROS) production, which, in turn, might affect heart cells at birth stopping cardiomyocyte proliferation and favoring their terminal differentiation. This hypothesis is supported by the presence of cardiac stem cell niches in the epicardium, which is characterized by relative hypoxia.28 Epicardial progenitor cells are active during embryonic development, when they contribute to the formation of cardiomyocytes, vascular, endothelial, and smooth muscle cells,29,30 and are reactivated in adulthood after injury, when their contribution to the cardiomyocyte pool can occur after pre-treatment with thymosin beta-4.31 This factor (a peptide also shown to restore vascular potential to adult epicardium-derived progenitor cells with injury32) is able to activate epicardial progenitors by up-regulating anti-oxidative enzymes, thereby reducing anti-oxidative stress.33
Besides hyperoxia, cardiomyocytes are subjected to increased mechanical stress after birth, due to the sudden rise in ventricular workload. Cells of the inner surface of the developing heart display reduced proliferation relative to those of the outer layer due to greater strain, and are the first to exit the cell cycle.34
In addition to the above mentioned environmental events, changes in the composition of the extracellular matrix seem to be important regulatory events controlling the rate of cardiomyocyte proliferation. Cardiomyocyte growth in the developing heart correlates with shifts in the expression of ECM proteins and integrin receptors.35
Another important regulator of cardiomyocyte proliferation that should be considered is the Hippo pathway. It is a signaling pathway, controlling organ size in mammalian organisms through the regulation of cell size, proliferation, apoptosis and survival differentiation. This pathway is conserved in humans. One of the main components of the hippo pathway is the transcriptional co-activator YAP (yes-activated protein; Yorkie in (YKI) in drosophila) which is the only one promoting tissue growth. YAP is deactivated by kinases of the hippo pathway, i.e. the seven
14 growth inhibitory proteins: fat (Mammalian gene; FATJ), merlin (Neurofibromatosis type-; NF2), expanded (FERM-domain-containing-6; FRMD6), hippo (Mammalian sterile-20-like kinases type 1 and type 2 or MST1, MST2), salvador (SAV1), warts (Large tumor suppressor-LATS) and mob as tumor suppressor (Mps-one binder kinase activator-1; MOB1). (figure 2)
Figure 2. Schematic representation of part of the Hippo pathway (see the text)
When the Hippo pathway is activated, MST1/2 phosphorylate SAV1, and together they phosphorylate and activate MOB1A/B and LATS1/2, which then phosphorylate Yes-associated protein. Phosphorylated YAP is sequestered in the cytoplasm by the 14-3-3 protein and subjected to proteosomal degradation. As a consequence, the TEA domain-containing sequence-specific transcription factors (TEADs) associate with the transcription cofactor vestigial-like protein 4 (VGL4) and suppress target
15 gene expression. When the Hippo pathway is silent, MST1, MST2, LATS1 and LATS2 are inactive, so YAP is not phosphorylated and accumulates in the nucleus where it displaces VGL4 and form a complex with TEADs, which promotes the expression of target genes.36 (figure 2)
Some of the important genes targeted by the Hippo pathway are: Ctgf, Birc5, Ankrd1 and Cyr6.
The protein encoded by Ctgf (Connective Tissue Growth Factor), is secreted by vascular endothelial cells. It takes part in chondrocyte proliferation and differentiation, cell adhesion in many cell types, and is related to platelet-derived growth factor.
Cyr61 (Cysteine-rich angiogenic inducer 61) supports cell adhesion, stimulates cell migration, promotes growth factor-induced cell proliferation and differentiation in some cell types, promotes apoptosis in synergy with TNF family cytokines, and induces cellular senescence in fibroblasts.
Birc5 encodes for Survivin, also called baculoviral repeat-containing 5, which is a member of the IAP (inhibitor of apoptosis) family of antiapoptotic proteins. The encoded protein inhibits caspase activation, leading to negative regulation of apoptosis or programmed cell death.
Ankrd1 encodes for Cardiac ankyrin repeat protein (Carp), which is involved in cardiac ventricular chamber differentiation, maturation and morphogenesis. Mice with genetic deletion of the Mst1, WW45, and Lats die postnatally because of cardiomegaly,37whereas transgenic mice overexpressing Lats238 or Mst139 die from dilated cardiomyopathy. Deletion of Yap alleles using Nkx2.5-Cre, or Tnnt2-Cre, expressed during early heart development, caused embryonic mortality due to myocardial hypoplasia. Deletion at a later developmental phase causes dilated cardiomyopathy leading to death after 10 weeks of age. Transgenic overexpression of a constitutively active, nuclear Yap mutant (Yap S112A), caused aggressive cardiomyocyte proliferation and myocardial thickening (studies reviewed in reference23). The above mentioned results indicate that the hippo pathway controls cardiomyocyte proliferation during intra-uterine development and postnatal life. Due to the factors described in this paragraph and probably to other still poorly understood molecular mechanisms, cardiomyocytes reach their terminally differentiated state, hence losing proliferative capacities. Again, they can re-enter
16 cycling after myocardial injury, but to an extent insufficient for effective regeneration.16,17
1.2 Gene therapy to stimulate cardiomyocyte proliferation
Over the last two decades there have been numerous attempts to stimulate cardiomyocyte proliferation. To date, investigators have essentially utilized four strategies to replace the loss of terminally differentiated, functional cardiomyocytes:
transplantation of progenitor cells (deriving from cardiac or other tissues) differentiating as cardiomyocytes or other mature cardiac cell lines 40,41 transplantation of myoblasts deriving from skeletal muscle 42,43
converting non-muscle cells in the injured heart to function as cardiomyocytes (reviewed in reference 44)
induction of dedifferentiation and proliferation of residual adult cardiomyocytes (reviewed in reference 44).
The cornucopia of studies on cardiac delivery of stem cells have already led to clinical applications,45 even though there is little evidence that the transplanted cells are retained in injured hearts. The present project is based on the third strategy listed above, which is presently considered as the most challenging. Several studies focused on fibroblasts reprogramming as cardiomyocyte-like cells,44 since more than 50% of cardiac cells are fibroblasts. On the other hand, stimulating the division of postnatal adult cardiomyocytes has been less popular approach. The reason is that it is rather difficult to activate the division of mature cardiomyocytes. Nonetheless, over the past years, research performed by different laboratories has indicated that activating mature cardiomyocyte proliferation after cardiac damage is a realistic option.23,46 The oldest evidence of atrial cardiomyocyte proliferation in response to ventricular injury was provided in 1983 by a study in newts.47 Other authors found that ventricular damage correlates with cardiomyocyte proliferation in zebrafish, with ensuing cardiac muscle regeneration,48 and newly formed ventricular cardiomyocytes derive from reprogramming and migration of pre-existing atrial cardiomyocytes.49 In neonatal mice, cardiac apex removal induced global activation of DNA replication of the entire ventricle.50 Cardiomyocyte
17 proliferation has been stimulated by manipulation of cell cycle proteins, for example transgenic overexpression of cyclin A2, which is normally silenced in postnatal heart, was reported to stimulate cardiomyocyte proliferation51 and to be beneficial after MI.52 Forced overexpression of the cyclin-dependent kinase 2 also caused an increase in DNA synthesis and proliferating cell nuclear antigen levels in the adult mice transgenic hearts, followed by a transient increase in cardiomyocyte proliferation and the presence of less-differentiated, mononuclear cardiomyocytes.53 Transgenic mice overexpressing the early G1 cyclins showed cyclin D1-revealed sustained DNA synthesis, but abnormal patterns of multinucleation,54 whereas the overexpression of cyclin D2 induced regenerative growth and infarct regression.55,56 These studies in transgenics, considered en masse, suggest that it is possible to achieve postnatal cardiomyocyte DNA synthesis and nuclear division. However, the evidence that karyokinesis is followed by cytokinesis and, most importantly, by cell proliferation, remains insufficient. Another study has reported that the deletion of the homeodomain transcription factor Meis1, a regulator of normal cardiac development, was sufficient to extend the postnatal proliferative window of cardiomyocytes, and to re-activate cardiomyocyte mitosis in the adult heart with no adverse effects on cardiac function.57 Finally, in 2012, Eulalio et al. identified 40 microRNAs able to stimulate DNA duplication and cytokinesis of cultured mouse and rat neonatal cardiomyocytes and successively tested two of them in mouse infarcted hearts.1This seminal study, highlighted by a number of authoritative editorials 58, 59 inaugurated a new strategy for cardiac regeneration based on pro-regenerative molecules rather than stem cell delivery.
1.3 miRNA Biogenesis and mechanism of action
MicroRNAs are small non-coding RNAs (ncRNAs), approximately 20 nucleotides in length, that control gene expression post transcriptionally by regulating mRNA translation or stability in the cytoplasm. In contrast with other small RNAs, miRNAs do not require perfect base pairing, therefore they can regulate a broad network of genes. Each miRNA can target hundreds of different mRNAs, while one mRNA is
18 interfered by multiple miRNAs. It is estimated that miRNAs regulate approximately 30% of the human protein-coding genome.60
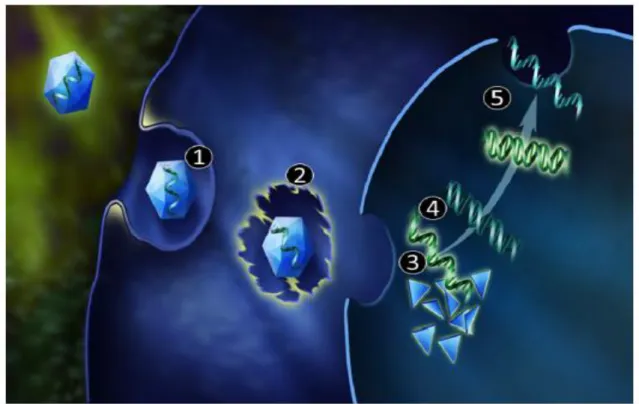
Figure 3: Schematic representation of miRNA biogenesis and mechanisms of action (see text). RISC-RNA- induced silencing complex; ORF – open reading frame
The molecular machinery involving miRNAs can be briefly described as follows. First, the double-stranded miRNA is recognized by protein DGCR8 (DiGeorge
19 syndrome chromosomal region 8, also known as Pasha (figure 3, dark blue). Drosha (figure 3; light blue) associates with DGCR8 to form a microprocessing complex with other cofactors such as RNA helicases (p68 and p72), transcription factors [Smad, p53, estrogen receptor α (ERα)] and RNA-binding proteins (KSRP and hnRNP A1 (figure 3; pink). In the nucleus this complex modulates the catalytic activity of Drosha and processes the primary miRNA transcript into a smaller ∼70-nt hairpin structure, i.e. the precursor miRNA (pre-miRNA). Through the interaction with exportin-5 and Ran-GTP (GTP-binding nuclear protein Ran), the pre-miRNA is carried into cytoplasm, where it undergoes a second step of processing catalyzed by large RNAse protein Dicer (figure 3; violet). Dicer cleaves the loop and a double-stranded ∼22-nt mature miRNA molecule is generated. In the next step, mature miRNA, composed of the guide and the passenger strands, is loaded in RISC (RNA-induced silencing complex; figure 3; orange). The RISC is composed of Argonaute proteins 1–4 (Ago1–4) and several cofactors, such as PACT (protein kinase RNA activator) and TARBP1/2 (transactivation response RNA binding protein). The two strands of the miRNA duplex undergo separation in RISC. The passenger strand is released and often degraded while the guide strand participates in gene silencing.60,61,62 The evidence supports two silencing mechanisms: Slicer-dependent and slicer-independent. The downstream effects, mRNA degradation or translation inhibition, respectively, both ultimately leading to down-regulation of gene expression. One significant difference between the downstream effects is reversibility, mRNA decay is an irreversible process. In contrast, translation inhibition is reversible because stable mRNA can be translated following elimination of translation repression (reviewed in reference63). miRNAs bind the 3’UTR of target mRNAs through a sequence of 2-8 nucleotides in their 5’end. In case of slicer-dependent mechanism, extensive base-pairing between the miRNA guide and mRNA target permit Ago2 mRNA cleavage. Cleavage products are deadenylated to remove the poly (A) tail. Subsequently degradation can occur via the exosome, which is a multi-protein complex with 3’-to-5’ exonuclease activity. Alternatively, the mRNA can undergo decapping by Dcp (Peptidyl-dipeptidase) enzymes which facilitates 5’-to-3’ degradation by the exoribonuclease Xrn1.
20 On the other hand, during translational repression, miRNA can segregate mRNA away from ribosomes to cytoplasmic foci and accelerate deadenylation and decapping processes. Stored mRNA can return to active translation or be targeted for degradation.63
The functional consequence of miRNA-mRNA interaction remains a matter of debate. It was initially thought that miRNAs repress protein output without significantly affecting their mRNA levels in animals (slicer-independent translational repression)64,65 Later, a series of genome-wide studies in cultured mammalian cell lines transfected with miRNA mimics led to the conclusion that miRNAs predominantly induce mRNA degradation (slicer dependent mechanism).66 A follow-up study67 and re-analysis of the previous datasets68 revealed that translational repression precedes mRNA degradation.
Jin et al.69 summarized the relative contribution of translation repression and mRNA degradation to miRNA regulation of targets in miRNA mutant mice. The analyses showed that 48% of target genes were predominantly regulated by translation repression, 29% by mRNA degradation, and 23% by both. Nonetheless, it is still unclear what determines the dominant mode of miRNA mechanism of action.
1.4 microRNAs in cardiovascular disease
1.4.1 microRNAs in cardiovascular disease, historical perspective
Currently it is known that miRNAs regulate the expression of human genes involved in most biologic processes, like metabolism, apoptosis, proliferation, differentiation, metastasis. The importance of miRNAs was documented for the first time in 1993.64,65 miRNAs were initially recognized as development regulators in flies and worms, but over the past decade it became clear that miRNAs have a critical role also in mammalian development and in the maintenance of homeostasis in a wide array of systems, including the cardiovascular .61
The first findings that suggested a possible involvement of miRNAs in human cardiovascular diseases were published in 2005-2006. In 2005, Zhao et al.70 reported that miR-1-1 and miR-1-2 were specifically expressed in cardiac and skeletal muscle precursor cells and miR-1 overexpression during heart development
21 in a mouse model led to a decreased pool of proliferating ventricular cardiomyocytes, suggesting a fine modulation of the expression of critical cardiac regulatory proteins controlling the balance between differentiation and proliferation during cardiogenesis.70 In the same year, Kwon et al. found that miR-1 in Drosophila modulates cardiogenesis.71 Following this discovery, Chen et al. in 2006 reported that miRNA-1 promotes myogenesis, whereas miR-133, clustered together with miR-1 on the mouse chromosome 2, stimulates myoblast proliferation.72 Also in 2006 more than 12 miRNAs were found to be deregulated during cardiac hypertrophy and heart failure.73 Overexpression of miR-195 caused heart failure in mice and hypertrophic growth of cultured rat cardiomyocytes.73,74 Abnormal patterns of miRNAs in cardiac hypertrophy were demonstrated and their roles were defined by several following studies.74
The disruption of miRNA-processing mechanisms at different postnatal ages also determines pathological ventricular remodeling and contractile dysfunction,75,76 indicating that miRNAs are not only required for normal heart development in the embryonic and neonatal period, but are critical for cardiac homeostasis also in adulthood. Recent evidence indicates that specific miRNAs are involved in postnatal cardiomyocyte mitotic arrest. By comparing the miRNA expression profile of postnatal mouse cardiac ventricles at different time points, the miR-15 family was found to be upregulated at day 10, when cardiomyocyteshave already exited the cell cycle.77 Overexpression of miR-15 family members during development caused premature cardiomyocyte cell-cycle arrest, leading to congenital heart hypoplasia, and prevented regeneration following MI in 1 day old mice neonatal hearts.78 Studies in zebrafish revealed a similar role of another miRNA family, namely miR-133, in arresting cardiac cell proliferation and regeneration after apical resection injury.79 Also miR-29a was found to suppress cardiomyocyte proliferation, while its inhibition promoted cardiac cell division conceivably by targeting cyclin D2.80
22 1.4.2 Control of cardiomyocyte proliferation by microRNAs
Eulalio et al. were the first to test the hypothesis that if some miRNAs can force cardiomyocyte exit from the cell cycle, and induce heart failure,80,81 others could promote their proliferation.1They screened the whole human miRNA library, which includes more than 800 miRNA mimics. The miRNA mimic technology is an innovative approach for gene silencing. miRNA mimics are chemically synthesized, double-stranded RNA molecules designed to “mimic” native microRNAs. Once introduced into cells, this RNA fragment, mimicking an endogenous miRNA, can bind specifically to its target gene and produce posttranscriptional repression, more specifically translational inhibition, of the gene. Unlike endogenous miRNAs, miRNA mimics act in a gene-specific fashion.82 By screening a library of 875 available miRNA-mimics Eulalio et al. found that more than 200 miRNAs stimulated neonatal rat cardiomyocyte proliferation while only 40 of them were able to stimulate DNA duplication and cytokinesis also in cultured mice neonatal cardiomyocytes.1 For a more detailed characterization and subsequent tests in vivo, the authors selected the top two best performing candidates, miR-590-3p and miR-199a-3p, that most effectively promoted proliferation in rat and mouse studies, respectively. First they systemically administrated cardiotropic adeno-associated viruses encoding for the above mentioned miRNAs and demonstrated cardiomyocyte proliferation induction in healthy hearts. The analyses of 5-ethynyl-2′-deoxyuridine (Edu) incorporation, a marker of nucleic acid synthesis, revealed a marked increase in the number of positive cells in miR-199-3p or miR-590-3p treated hearts, indicating proliferation (figure 4). Strikingly, these two miRNAs promoted cardiac regeneration in an adult mouse model of MI. The viral vectors carrying miR-199-3p or miR-590-3p were injected in the border zone of the infarcted area shortly after ligation of the left ascending coronary artery. The authors observed a dramatic decrease in subsequent scar extension, as well as impressive left ventricle functional improvement, in comparison with animals treated with a control miRNA (figure 5).
23 Figure 4. miR-590 and miR-199a induce CM proliferation in neonatal rats, in vivo. a, scheme of in vivo protocols using synthetic miRNAs (N=8 per group); b, EdU and α-actinin staining of rat hearts injected with miRNAs. Scale bars, 1 mm (top), 100 mm (bottom); c, percentage of proliferating cells. Figure taken from reference 1
However, controversy remains about the source of new cardiomyocytes during normal turnover as well as post injury. Although it is possible that resident cardiac stem cells also contribute as a source, several studies have demonstrated that the majority of new cardiomyocytes in the adult heart derives from preexisting cardiomyocytes, previously thought to have permanently exited the cell cycle, which means they “return” in more immature, regenerative state.48,8350, 83 These findings are consistent with the neonatal mouse model of cardiac regeneration, as well as the adult zebrafish model, in which cardiac regeneration is believed to occur through proliferation of existing cardiomyocytes.
To determine the genes that could contribute to the increased cardiomyocyte proliferation in response to miR-199a-3p or miR-590-3p overexpression, Eulalio et al. correlated the expression of miRNAs with the expression patterns of mRNAs. Global transcriptome changes identified 1,056 upregulated transcripts (773 for hsa-miR-199a-3p; 283 for has-miR-590-3p) and 697 downregulated transcripts (95 for has -miR-590-3p; 602 for hsa-miR-199a-3p).
24 Figure 5: miR-590-3p and miR-199a-3p induce marked cardiac regeneration after MI in mice. a: Schematics of MI protocol in adult mice. Echo, echocardiography. b–d: LVEF (b), LVFS (c) and LVAW thickness-s (d). Dashed line, non-infarcted animals injected with AAV9-control. N=10–16 per group. e: Masson trichrome staining of heart cross sections. Scale bar, 1mm. f: Infarct size. n56–10 per group. All panels, mean±s.e.m.; *P<0.05, **P<0.01, ***P<0.001 relative to control. Taken from reference 1
Functional analysis of the upregulated genes revealed that most of them belong to the “cell cycle”, “DNA replication, recombination and repair” and “cellular growth and proliferation” categories. Analysis of the downregulated transcripts showed enrichment for genes of the categories: “skeletal and muscular system development and function” and “cellular assembly and organization”.1
25 In order to identify the target genes principally responsible for the observed phenomena, the authors tested the phenotypic consequences of downregulated transcripts. Selective siRNA-based screen of downregulated target genes revealed 45 genes whose downregulation alone was sufficient to reactivate the cell cycle in neonatal cardiomyocytes: 5 siRNAs targeted transcripts were downregulated by has-miR-590-3p; 45 siRNAs targeted transcripts were downregulated by hsa-miR-199a-3p. There was no single candidate gene knockdown as effective as miRNA overexpression in activating cell proliferation, suggesting that the identified miRNAs most probably affect cell-cycle reentry by the combined targeting to multiple genes. Only three of the miR-199a-3p and miR-590-3p target genes overlapped, namely
Homer1, Hopx, and Clic5 (figure 6).
Homer1 encodes for a protein that modulates Ca2+ signaling in the heart by interacting with the Ca2+ release channel ryanodine receptor (RyR). Hopx encodes for a homeodomain protein that inhibits embryonic cardiomyocyte proliferation.
Clic5 is also known as an inhibitor of cell proliferation.44,84
Besides the above mentioned targets, there is evidence that miRNAs may also target the hippo signal transduction pathway, discussed earlier, while the hippo pathway itself regulates miRNA biogenesis in a cell-density-dependent manner. At low cell density, YAP binds and inactivates p72 (RNA helicase, a regulatory component of the miRNA-microprocessor) leading to miRNA suppression in cells. On the contrary, at high cell density, hippo-mediated cytoplasmic retention of YAP facilitates p72 association with the miRNA processing machinery, thus causing widespread miRNA repression (figure 7).85
Several microRNAs control the activity of hippo signaling pathway at least to some extent. Increased miR302-367 expression leads to a marked increase in cardiomyocyte proliferation, partially by repression of the hippo pathway.86 Tian et
al showed that miR-302 targets Mst1, Lats2 and Mob1b, the key kinases of hippo
cascade. The loss of these essential components in the developing mouse heart caused increased proliferation in cardiomyocytes. When active, YAP can translocate in the nucleus and activate the transcription of its target genes Ctgf, Cyr61, Birc5 andAnkrd1. (figure 2).
26 Figure 6: Downregulation of genes by miR-590-3p and miR-199a-3p that causes increased CM proliferation. a. Workflow for miRNA target identification. b-c. Percentage of proliferating mouse CMs after siRNA treatment. Targets increasing proliferation twofold are shown. Red, genes downregulated by both miRNAs. d. EdU staining of mouse CMs treated with selected siRNAs. Scale bar: 100 mm. Figures taken from reference 1
Like most ground-breaking studies, the ones mentioned in this paragraph raise many new questions. Is it possible to control cardiac cell division? What is the mechanism of cardiac cell-cycle reentry? Without scrupulous lineage tracing experiments, can we be sure that the new cardiomyocytes are derived from preexisting ones? Do these findings in rodents translate to the human pathophysiology?
The present study was aimed at addressing such key questions, especially the potential for clinical translation. That is why we needed to utilize an animal model phylogenetically closer to the human organism.
27 Figure 7. Hippo pathway controls miRNA biogenesis in the cell-density dependent manner. (taken from reference 85)
1.5 The need for large animal models to test new therapies
One of the definitions of translational medicine is the transfer of new findings from basic science to clinics. Some years ago, an NHLBI Working Group was gathered to identify the obstacles hindering the translation of experimental findings on post-ischemic myocardial injury and protection into clinical practice practice.87 They observed that, while cell cultures and rodent models remain fundamental for the advancement of biomedicine, there is a pressing need to reconsider animal models that adequately approximate actual clinical circumstances, for instance large animals, as a necessary step before experimental findings can be transferred to the medical practice (figure 8).
The need for clinically relevant tests in large animal models becomes particularly pressing in the era of “biological” therapies, such as gene and stem cell delivery. For instance, a recent meta-analysis of experimental studies on the effects of stem cell
28 therapy in pig ischemic heart has further highlighted the importance of pre-clinical tests in large animal models as a necessary step to direct subsequent clinical trials.87 There is a significant differences in cardiac characteristics such as beating rate, oxygen consumption, adrenergic receptor ratios, and response to loss of regulatory proteins, when mice are contrasted to humans.88,89 Comparing murine to human systems becomes problematic when making interpretations of human MI or heart failure pathophysiology. Therefore, large animal models, which more closely approximate human physiology, function and anatomy, are essential to develop the discoveries from murine models into clinical therapies and interventions for heart failure.90 Several studies and meta-analyses showed that the large animal models
are valid to predict outcome of clinical trials.90–92
It is highly recommended that pre-clinical studies are based on high standards, such as randomized protocol design, blinded functional analysis and accurate follow-up.87 The present project followed those criteria.
29 1.5.1 Pig model of myocardial infarction
Numerous discoveries on myocardial ischemia and infarction have been made in canine models, but several important factors, including coronary artery anatomy, contributed to preference for alternative large animal models such as pigs and sheep. Quantitative evaluation of the anatomy and distribution of swine coronary arteries has demonstrated the presence of sparse collaterals, mainly localized in the subendocardium and mid-myocardium, unlike the extensive epicardial collaterals found in dogs.93 Like man, in pigs the left coronary artery is usually longer and larger in diameter than the right coronary artery. In most of cases, the right coronary artery is dominant, supplying posterior septum and atrioventricular node via the posterior descending coronary artery, although in rare cases balanced blood supply or left coronary artery dominance can be seen. At least half of the left ventricle is supplied by the LAD, which feeds also most of the intraventricular septum. The percent distribution of coronary flow supply of the different areas of the heart in pigs is as follows: (table 2)93
LAD
RCA
Cx
Left Ventricle
49%
25.50%
25.50%
Right Ventricle
27.60%
72.40%
-
IVS
58%
42%
-
Sinoatrial node
-
100%
-
Table 2: Blood supply to different sections of the heart by coronary arteries in pigs.
Cx-Circumflex coronary artery, IVS-intraventricular septum; LAD-Left anterior descending coronary artery; RCA-right coronary artery.
The size and number (from 2 to 9) of the LAD diagonal branches varies inversely with the marginal branches of the circumflex coronary artery, which are occasionally more numerous or reach the apex.93
Overall, swine coronary anatomy and myocardial blood supply described in literature seem very similar to those found in humans. Therefore, pigs have been used as the model of choice to study the pathophysiology of myocardial ischemia
30 and infarction. Experiments assessing maximal coronary artery flow suggested that coronary reserve and collateral circulation were lower than in dogs, but more similar to humans.94 Acute occlusion of the coronary artery in pigs resulted in infarction of most of the tissue in the area at risk.94The lack of the collateral vessels was the reason why two hours occlusion of the coronaries were sufficient to cause transmural infarction.15
Swine models of MI have been used to study scar expansion and LV remodeling in the post-MI setting.95 And, given the above-mentioned characteristics, MI in pigs is currently the preferred large animal model to test the effects of stem cell transplantation on LV remodeling/regeneration and contractile function.96,9797 Another reason for using swine models is that the size of these animals allows morphological and functional assessments based on clinical-standard diagnostic tools, such as echocardiography, magnetic resonance imaging (MRI), computed tomography, positron emission tomography (PET) and single-photon emission computed tomography (SPECT). Studies by our group have shown the remarkable advantages of multi-modal cardiac imaging in the evaluation of post-MI recovery.98,99
1.5.2 miRNA delivery
Despite the promising potential in the area of cardiac regeneration, cardio-selective therapeutic microRNA delivery remains a challenging problem. Clinical applications of this strategy require a series of pre-clinical studies to evaluate not only the therapeutic efficacy, but also the safety of systemic or intramyocardial administration of pro-regenerative miRNA molecules, as there is a potential risk of inducing undesired proliferation in non-target tissues. Other possible limitations that need to be addressed are the miRNA inappropriate biodistribution and disruption or the saturation of endogenous RNA machinery, and finally its instability
in vivo.
There are several options for miRNA delivery to the target. The feasibility of direct transfer in vivo was demonstrated using viral (transduction) and nonviral vehicles (transfection) in several animal species, including pigs, rabbits, and dogs.100–103 For instance, investigators have utilized miRNA mimics as naked nucleic acids or
31 microRNAs carried by liposomal vectors, while miRNA precursor genes can be enclosed into viral vectors with high tropism for cardiomyocytes. The miRNAs, delivered as a miRNA precursor genes or as double stranded molecules then intercepts the cellular RNAi process at different levels (Figure 9).
When a double-stranded synthetic miRNA, delivered as miRNA mimic or conjugated with liposomes, enters the cardiomyocyte, the active strand is incorporated into the RNA-induced silencing complex, leading to its pairing with multiple target mRNAs. The most attractive characteristics of non viral gene delivery vectors are their safety profile and minimal immunogenicity. However, naked gene delivery is more suited for applications that do not require high-density gene transfer, whereas there is a high probability that naked miRNAs are degraded by the abundantly expressed nucleases. Liposomes also have a favorable safety profile for in vivo administration. They are biodegradable, biocompatible, physically stable and easy to produce. Liposomes can protect the carried molecules and be modified for selective targeting, thus permitting to use a lower drug load with less systemic effects. They are approved by the FDA and have been constituents of commercially produced drugs for the last 20 years. Despite these advantages, the efficiency of miRNAs delivery as naked strands or liposome conjugates proves, in many cases, unsatisfactory. Thus, to achieve a sustained, intracellular miRNA production, the best option is to embed the corresponding gene into a viral vector genome. After reaching the nucleus, the vector genome is transcribed and results in a structured miRNA precursor which is processed with the same modalities typical of endogenous miRNAs. Even though gene transfer with recombinant viral vectors is a seasoned approach, it has recently generated new interest, fueled by a number of promising experimental104,105,106,107 and also clinical studies performed in MI and heart failure.108,109,110,111
The most commonly used viral vectors are retroviruses (including the lentiviruses), adenoviruses (ADs) and adeno-associated viruses (AAVs).112 ADs and AAVs have the advantage of transducing nondividing cells. Both of them have proved to transduce the myocardium with acceptable efficiency.
32 Figure 9: Schematic representation of the RNAi machinery inside a cardiomyocyte and the different sites of interception of exogenously delivered miRNAs.
A considerable limitation of AD vectors is their immunogenicity that ultimately limits gene expression days to weeks after gene transfer. On the other hand, AAVs are very little immunogenic, thus permitting a long term expression of the carried therapeutic transgenes.113,114 Several preclinical studies have shown persistent
AAV-33 mediated gene expression years after vector delivery. For example, a clinical trial for the treatment of hemophilia showed consistent gene expression in skeletal muscle one year after AAV injection.113 There are new methods available for the production of the large amounts of AAV vectors necessary for a satisfactory transduction in large animal models. Therefore, in our case AAVs can be considered as the vectors of choice.
1.5.2.1 Adeno associated viral vectors
AAV is a small virus without envelope and with a linear, single stranded DNA belonging to the family Parvoviridae, subfamily Parvovirinae and genus Dependovirus.115 AAV depends on a helper virus such as adenovirus for effective replication. AAV is not known to be associated with any human disease, that is why it is considered as a highly safe vector. Moreover, the only mandatory cis-elements for AAV vectors are the inverted terminal repeats that flank the viral genome. Cis-regulatory sequences and promoters are DNA sequences in the vicinity of the structural portion of a gene that are required for gene expression.116 (in contrast trans elements are usually considered to be proteins that bind to the cis-acting sequences to control gene expression). As a result, transduction by AAV does not lead to the expression of any viral genes, which could be a reason of their low immunogenicity.117 To date, 13 AAV serotypes have been described in literature, although the vectors used for gene transfer are based on serotypes AAV1–9. Together, the AAVs display serotype-specific tissue and cell tropism, likely due to their diverse receptors.115 Regarding the heart, it has been shown that different serotypes display different transduction efficiency in different species. Generally, in all species AAV1, AAV6, AAV8, and AAV9 show stronger cardiotropism than other serotypes. For instance, in Rhesus Macaques, percutaneous, transendocardial injections of AAV6 caused more efficacious myocardial transduction compared to AAV8 and AAV9118, while, according to another study, AAV2 is also able to induce reasonable transduction of cardiomyocytes after intramyocardial injection.119AAV serotypes 6, 8 and 9 transduce canine hearts most effectively when injected in myocardium.120 Recent studies have shown marked cardioprotective effects of gene therapy with AAV9-carried VEGF-B in dogs with heart failure.121 Our knowledge of
34 cardiotropism of specific AAV serotypes in swine is very limited. It has been shown that, after direct injection in myocardium, AAV1 is a more efficient transgene vector than AAV2 in pig hearts.122 Selective retroinfusion of AAV6 in the anterior cardiac vein substantially increased reporter gene expression in the targeted distal LAD territory compared to AAV2 vectors in swine.123
AAV-mediated transduction starts with the binding of the virus to its receptors, followed by endocytosis (Figure 10).120
Figure 10: AAV entry in the cell: 1 indicates receptor binding and endocytosis; 2, escape into cytoplasm; 3, nuclear import; 4, capsid disassembly; 5, double-strand synthesis; and 6, transcription. (taken from reference 120
)
The mechanism of endocytosis can be different for different serotypes of AAV, for instance it can function via the so-called CLIC/GEEC (Clathrin Independent Carriers / GPI-anchored protein Enriched Endocytic Compartment) pathway for AAV2 or both clathrin-coated vesicles or caveolae for AAV5.124,125 It is documented that, at least for these two prototypical AAV serotypes, endocytosis is followed by transport to the Golgi and after entering the cytoplasm AAV is imported into the nucleus as an intact particle, most probably through the nuclear pore. Import of viral particles into the nucleus is followed by capsid removal and viral genome release. Before
35 transcription of the transgene, the single-stranded AAV genome is converted into a double strand genome (Figure 10).120
1.5.3 AAV delivery technique
Various methods have been used to deliver viral vectors to the target organ(s). When considering the heart as the target organ, the general criteria followed to assess the efficacy of gene delivery are:
1. Distribution of myocardial transduction (target organ vs systemic diffusion) 2. Specificity of transgene expression in myocardium (low off target organ
exposure)
3. Efficiency of myocyte transduction (percentage of transduced myocytes = multiplicity of infection)
4. Technical difficulty, side effects, accessibility 5. Delivery-related inflammatory response 126
The following figures and tables explain schematically the most commonly used methods and their advantages and disadvantages.
Anterograde coronary artery infusion (Figure 11) Advantages:
Cardiac-specific
Minimally invasive and relatively safe
Global and homogenous myocardial transduction. Disadvantages:
Inefficient
Large vector leakage to the systemic circulation.
Infusion through the lumen of an inflated angioplasty catheter during short-term occlusion of the coronary artery (Figure 11B) may result in higher gene expression but remains controversial, 127 while the temporary blockage of both coronary vein and artery clearly increased myocardial gene expression but also more frequently caused myocardial injury. 128
36 Figure 11. Anterograde arterial infusion.
A. Coronary artery infusion of the vector without blocking coronary flow. B. Coronary artery infusion of the vector with occlusion of a coronary artery by inflated angioplasty catheter. The vector is injected distal to the occlusion. C. Coronary artery infusion of the vector after simultaneous occlusion of coronary artery and vein by inflated angioplasty catheters. In theory, the vector remains in the coronary circulation until balloons are deflated. (taken from reference 120)
Retrograde coronary venous infusion (Figure 12) Advantages:
Cardiac-specific
Minimally invasive and relatively safe (especially in the clinical setting, for patients with impaired coronary artery circulation, severe atherosclerosis or coronary artery obstruction)
Global and homogenous myocardial transduction Disadvantages:
Higher efficiency compared to anterograde coronary delivery, if coupled with induced short term ischemia (figure 12B), 127but still less efficient compared to other methods
Large vector leakage in the systemic circulation
37 Figure 12. Retrograde venous infusion.
A. The vector is injected into a coronary artery, collected from the coronary sinus and after blood re-oxygenation re-infused into the coronary artery. B. Retrograde coronary venous infusion of the vector after occlusion of coronary artery and vein at once by inflated angioplasty catheters. The vector is infused into a coronary vein and remains in the coronary circulation until balloons are deflated. ( taken from reference 120)
Aortic cross-clamp and infusion in the aortic root or LV cavity
Mainly used in rodents. The vector is injected into the aortic root or the left ventricular cavity after cross clamping aorta and pulmonary artery over a few heart beats.129 Several studies have used this method in piglets, showing selective myocardial uptake and gene expression.130,131
Advantages:
Increased gene transfer efficiency Disadvantages:
Requires chest opening and cardiac arrest High risk of myocardial injury
Non cardiac-specific Intravenous Infusion
Advantages:
The simplest method Disadvantages:
38 Limited cardiac specificity
Uptake by many other organs
A variant of this method, which adds ultrasound-induced microbubble destruction in the target organ, increases the permeability of capillary and cell membranes and slightly improves gene transfer efficiency.132,133
Epicardial Painting
Gene delivery to the atria without affecting ventricular cardiomyocytes. In literature this method has been used for the treatment of atrial fibrillation.134, 135 The pericardium is incised to expose atria. The viral vector solution is painted onto the atria with a specific paintbrush. Each atrium is coated twice, and specific timing between painting coats is necessary to allow adsorption.
Advantages:
Cardiac-specific
Complete atrial transmural penetration High degree of gene transfer
Disadvantages:
Accessibility, as it requires open chest. 135 Electroporation
Electrical fields are used to temporarily destabilize the normally impermeable membranes and let macromolecules into the cytoplasm.136
Advantages:
Enhanced transfer of naked DNA via myocardial injection and retrograde perfusion
Cardiac-specific Disadvantages:
At the appropriate field strengths, the application of these fields to tissues results in little, if any, damage or trauma. However, the electrical pulse must be synchronized with heart rate to avoid the occurrence of ventricular fibrillation.137 The problem with synchronized pulse delivery is that a short pulse length is required in order to be synchronized with the QRS wave. It is
39 not clear at this time when the most optimal delivery time would be and how long they would persist.137
Pericardial Injection (Figure 13)
Intrapericardial delivery is performed surgically in rodents, or with percutaneous approach in larger animals. The percutaneous access to the pericardial space can be achieved via a substernal/xiphoidal puncture.
Advantages:
Safe and practicable with fluoroscopy or intravascular ultrasound Minimally invasive via a substernal/xiphoidal puncture
Minimal, but not absent leakage of the vector towards off target organs, most probably due to pericardial fluid rapid absorption by the lymphatic system. Delivery in a closed compartment potentially favors prolonged vector
persistence.
Figure 13: Percutaneous pericardial injection via a substernal approach. (From reference 120
)
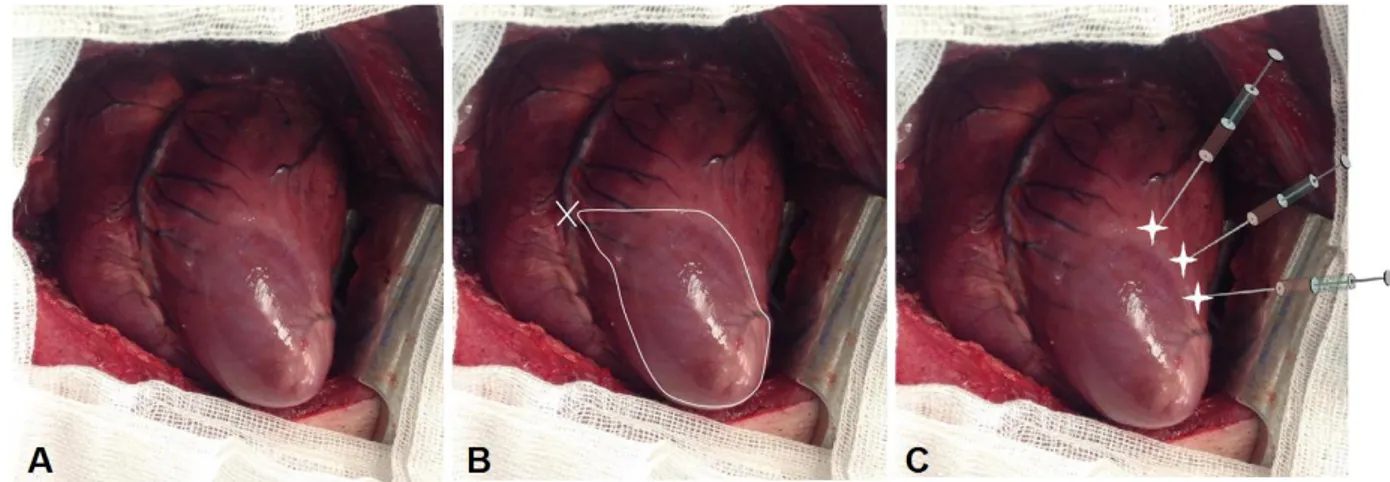
Direct Intramyocardial Injection (Figure 14)
Direct intramyocardial injection is one of the most commonly used for gene transfer, in clinical trials as well as in small or large animal models. The vector can be injected through the endocardium by a catheter inserted percoutaneously, or through the epicardium during open chest surgery. We have chosen this latter method for the
Disadvantages:
tightly joined epicardial cells may prevent vector penetration in myocardium. This limitation can be overcome by administering polyethyleneimine or proteolytic enzymes, which, on the other hand, may cause cardiac toxicity. 195,196
40 present study, principally because it allows a better control of AAV delivery under direct visualization and in clearly identifiable injection sites. The local area around the injection site has a high density of transgenes, however gene expression starts to become negligible 5 to 10 mm from the injection site.126 Multiple injections are needed for adequate coverage of the target area in large animals.
As for the other methods, it also presents advantages and disadvantages. Advantages:
Vector delivery bypasses the endothelial barrier
High density of gene delivery in myocardium (concentration in the injection site) and therefore minimal exposure to the vector of off target organs, although some systemic leakage cannot be completely prevented.126
Simple and relatively safe.
Vector deactivation by circulating DNAses and/or neutralizing antibodies can be limited
Minimal immunological complications
Figure 14. Direct intramyocardial injection. A. The vector is injected through the endocardium by a percutaneously inserted catheter. B. The vector is injected via an epicardial approach requiring open chest surgery (from reference120).
41 Disadvantages:
Small extension of gene expression Multiple injection sites required
Acute inflammatory response and potential scarring of myocardium at the injection sites.
One of the above listed problems is the acute inflammation at the injection sites. The inflammatory response is more likely due to direct trauma caused by injection, rather than stimulation of an immunologic response to the viral vectors, as it has been seen also after injection of plasmid DNA.138,139 Inflammation could be the real problem in humans, but it usually doesn’t prevent efficient gene delivery,138,139 and can be neglected in the case of animal studies.
1.5.4 Direct AAV injection in infarcted hearts
In the specific case of MI, an appropriate target region for direct intramyocardial AAV injections is the so-called border zone. This is a zone adjacent to necrotic or irreversibly damaged myocardium, which is less ischemic and thus potentially viable.140 The main changes present in the border zone of MI are reduced local blood flow (at a level between normal myocardium and the central ischemic zone), mitochondrial swelling with intact cellular architecture, altered tissue ratios of K+/Na+ and for Mg2+/Ca2+, decreased myocardial oxygen tension and Q-T and S-T segment changes.141 According to experimental studies, immediately after MI the border zone is small and its function only moderately impaired. Usually after about two weeks the area of dysfunctional myocardium increases, the border zone deforms and contractility becomes almost as low as some regions of the infarcted myocardium.142 The cause of progressive deterioration in contractility of the border zone is not clearly known, but most probably it can be attributed to the continuous myocyte death combined with the increased wall stress and consequent elevated strain.143
The border zone consists of a mixture of ischemic and viable, functional cardiomyocytes. In adult mice, after experimental MI, the border zone displays a 10-fold increase of cardiomyocyte cycle activity.16 As mentioned above (see section 1.1)