Homeodomain-interacting protein
kinase-2 phosphorylates p53 at
Ser 46 and mediates apoptosis
Gabriella D’Orazi*†, Barbara Cecchinelli*, Tiziana Bruno‡, Isabella Manni*, Yuichiro Higashimoto§, Shin’ichi Saito§, Monica Gostissa¶, Sabrina Coen*, Alessandra Marchetti*, Giannino Del Sal¶#, Giulia Piaggio*, Maurizio Fanciulli‡, Ettore Appella§ and Silvia Soddu*∀ *Molecular Oncogenesis Laboratory and ‡Cell Metabolism and Pharmacokinetics Laboratory, Regina Elena Cancer Institute, Via delle Messi d Oro 156, 00158 Rome, Italy †Department of Oncology and Neurosciences, University ‘G. D’Annunzio’, Via dei Vestini 3, 66013 Chieti, Italy §Laboratory of Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA ¶National Laboratory, Inter-University Consortium for Biotechnologies, AREA Science Park, Padriciano 99, 34012 Trieste, Italy #Department of Biochemistry, Biophysics and Chemistry of Macromolecules, Via Giorgieri 1, 34100 Trieste, Italy ∀e-mail: e-mail: [email protected] Published online: 3 December 2001, DOI: 10.1038/ncb714
Phosphorylation of p53 at Ser 46 was shown to regulate p53 apoptotic activity. Here we demonstrate that
homeodomain-interacting protein kinase-2 (HIPK2), a member of a novel family of nuclear serine/threonine kinases, binds to and activates p53 by directly phosphorylating it at Ser 46. HIPK2 localizes with p53 and PML-3 into the nuclear bodies and is activated after irradiation with ultraviolet. Antisense inhibition of HIPK2 expression reduces the ultraviolet-induced apoptosis. Furthermore, HIPK2 and p53 cooperate in the activation of p53-dependent transcrip-tion and apoptotic pathways. These data define a new functranscrip-tional interactranscrip-tion between p53 and HIPK2 that results in the targeted subcellular localization of p53 and initiation of apoptosis.
T
he p53 tumour suppressor protein is a crucial component of cellular mechanisms that are initiated by a variety of cellular stresses1,2. Under normal conditions, p53 is a short-livedpro-tein that is highly regulated and maintained at low or undetectable levels1. After stress, p53 is activated mostly at the post-translational
level by a complex series of modifications that include the phospho-rylation and acetylation of specific residues in the amino-terminal and carboxy-terminal domains. In addition to post-translational modifications, protein–protein interactions and subcellular relocal-ization also have a role in the activation of p53 (refs 3, 4). The acti-vation of p53 leads to the transcription of several genes whose products trigger different biological outcomes (for example, cell-cycle arrest, apoptosis, DNA repair, replicative senescence or differ-entiation)5. Not all of the pathways involved in these processes are
known but recently phosphorylation at Ser 46 was shown to be involved in the regulation of apoptosis after DNA damage6,7.
However, even in this case a clear role for this modification and the enzyme(s) responsible for it remain to be discovered. To identify the enzyme(s) and any possible cofactor involved in the activation process, we used the murine wild-type p53 lacking the trans-acting domain as bait in a yeast two-hybrid system. The screening for p53-interacting proteins was performed with a cDNA library gen-erated from mouse embryos at day 11, when p53 mRNA expres-sion is most detectable during development8. We found that p53
physically and functionally interacts with homeodomain-interact-ing protein kinase-2 (HIPK2). HIPK2 is a member of a novel fam-ily of nuclear serine/threonine kinases, which also includes HIPK1 and HIPK3 identified through their ability to interact with the Nkx-1.2 homeoprotein9. HIPKs can act as co-repressors for
home-odomain transcription factors9but, unlike other transcriptional
co-repressors, HIPKs have protein kinase activity. The physiological role of these kinases is currently unknown but here we define a function for HIPK2 in p53 activation and apoptosis.
Results
Identification of novel p53-interacting factors. The murine wild-type p53 lacking the trans-acting domain was used as bait in a yeast two-hybrid system to identify p53-interacting proteins. The screen-ing was performed with a cDNA library generated from mouse embryos at day 11, when p53 mRNA is maximally expressed during development8. Of the 13 clones sequenced, 11 encoded
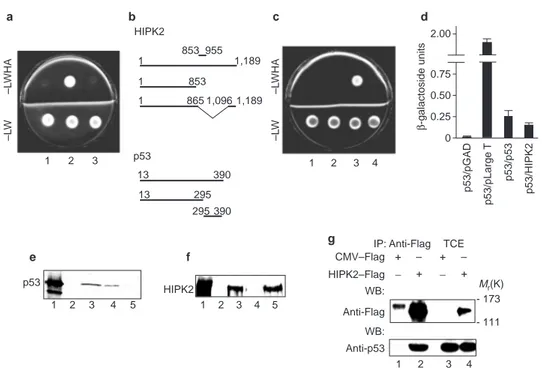
C-terminus-containing portions of p53. The remaining two encoded 101 and 102 amino acid residues encompassing the C terminus (residues 853–955) of a recently identified protein, HIPK2 (Fig. 1a). To exam-ine further the specificity of this interaction, a full-length HIPK2 and a truncated form of HIPK2, consisting of residues 1–853 (Fig. 1b), were matched with the p53 bait. The full-length protein interacted with p53, whereas the truncated mutant HIPK2(1–853) lost this ability (Fig. 1c). The efficiency of this p53–HIPK2 inter-action was comparable to that of p53 homodimers as quantified by
β-galactosidase activity in liquid cultures of yeast10(Fig. 1d).
To determine whether p53 interacts directly with HIPK2 itself, binding in vitro was assayed with 35S-labelled in-vitro-translated
forms of either p53 or HIPK2 along with bacterially produced glu-tathione S-transferase (GST) constructs of HIPK2 or p53, respec-tively. Full-length HIPK2 bound to p53 and also the originally cloned construct, HIPK2(853–955) (Fig. 1e, lanes 4 and 3, respec-tively), whereas deletion of this region abolished the binding (Fig. 1e, lane 5). The reciprocal experiment showed that HIPK2 binds the C-terminal domain of p53 (Fig. 1f, lane 5), but not its DNA-binding domain and N-terminal domain (Fig. 1f, lane 4). Consistent with these results was the observation that HIPK2 can also bind mutant p53, whose mutations reside mostly in the DNA-binding domain11(data not shown).
To test for interaction in vivo between p53 and HIPK2, wild-type p53-expressing HEK 293 cells were transiently transfected with an expression vector encoding Flag ([EYKEEEK]2)-tagged
HIPK2 protein. Total cell extracts (TCEs) were immunoprecipitated with anti-Flag antibodies, and the binding of p53 and HIPK2 was detected by western blotting (Fig. 1g, lane 2). No p53 was present in the immunoprecipitates obtained from the extracts of cytomegalovirus (CMV)–Flag control-transfected HEK 293 cells (Fig. 1g, lane 1). The reciprocal experiment did not co-immunopre-cipitate detectable levels of HIPK2 in HEK 293 cells, probably because of the high p53 expression levels promoted by the adenovirus E1 pro-teins. However, HIPK2 was easily co-immunoprecipitated from p53-transduced H1299 cells (see Fig. 4f). These results indicate that p53–HIPK2 complexes can form in mammalian cells.
HIPK2 localizes together with p53 in the nuclear bodies. HIPK2 is known to localize into nuclear speckles12. Recently, it has been
shown that PML can relocalize p53 in PML-specific nuclear sub-domains, the so-called nuclear bodies (NBs); furthermore, PML promotes p53 acetylation and transactivation13–16. To determine
whether the speckle distribution of HIPK2 is due to the localization into NBs, and whether p53 and HIPK2 localize together in the NBs, indirect immunofluorescence and confocal laser microscopies were performed. p53−/−H1299 cells were transiently transfected with a
haemagglutinin (HA)–HIPK2 expression vector. A partial localiza-tion of the endogenous PML together with the HA–HIPK2 was present in these cells (Fig. 2a), indicating that the HIPK2 speckle structures described originally12are indeed NBs. Next, p53−/−MG63
cells were microinjected with expression vectors encoding wild-type p53 and green fluorescent protein (GFP)–HIPK2 with or without PML-3. p53 and GFP–HIPK2 were found to localize together significantly in the NBs only in the presence of exogenous PML-3 (Fig. 2b, and data not shown). Similar results were obtained
when endogenous PML was activated by arsenic trioxide17(Fig. 2c).
These findings indicate that both exogenous and endogenous PML can facilitate p53–HIPK2 complex formation in the NBs, although HIPK2 seems also to be present in dots that do not contain PML or p53 (Fig. 2a, b, merge).
HIPK2 cooperates with p53 in the activation of p53-dependent transcription. HIPK2 was originally described as a co-repressor for homeodomain transcription factors9, and localization of p53 in the
NBs was recently shown to increase its transcriptional activity14–16.
To test the effect of HIPK2 expression on p53-mediated transcrip-tion, p53−/−H1299 cells were co-transfected with expression vectors
encoding p53 and HIPK2 together with a reporter vector carrying the mdm2 promoter18,19. As shown in Fig. 3a, HIPK2 increased
p53-mediated transactivation in a dose-dependent manner. In contrast, the expression of HIPK2 alone had no effect on the
mdm2 promoter (Fig. 3b). In addition, HIPK2(∆865–1096), which
is unable to bind p53, did not affect mdm2-promoter activity (Fig. 3b), indicating that the interaction of HIPK2 with p53 is essential for the modulation of p53 transcriptional activity. However, this effect was not seen with the p53-responsive p21Waf1 promoter (Fig. 3c), indicating that the modulation of p53 tran-scriptional activity by HIPK2 is promoter-specific for H1299 cells. HIPK2 phosphorylates p53 at Ser 46 in vitro and in vivo. HIPK2 is a serine/threonine kinase9; we therefore tested whether it can
phosphorylate p53 in a kinase assay in vitro. Comparable amounts of in-vitro-translated HIPK2 and a kinase-inactive mutant form, HIPK2-K221R (ref. 9), were incubated with a GST–p53 protein in the presence of [γ-32P]ATP. Only wild-type HIPK2 was able to
phosphorylate p53 in this assay (Fig. 4a). To evaluate whether the
–L WHA –L W –L WHA –L W 1 2 3 1 2 3 4 HIPK2 853 955 1,189 1 1 853 1 865 1,096 1,189 p53 13 13 390 295 295 390 2.00 0.75 0.50 0.25 0 p53/pGAD p53/HIPK2 p53/p53 p53/pLarge T β -galactoside units p53 1 2 3 4 5 1 2 3 4 1 2 3 4 5 HIPK2 _ + + – _ + + – CMV–Flag HIPK2–Flag WB: WB: Anti-Flag Anti-p53 Mr(K) - 173 - 111 a e f g b c d
IP: Anti-Flag TCE
Figure 1 HIPK2 interacts with p53 both in vitro and in vivo. a, HF7c yeast cells were co-transfected with constructs PGBT9LAM–HIPK2(853–955) (lane 1), PGBT9p53–HIPK2(853–955) (lane 2) and PGBT9–HIPK2(853–955) (lane 3) and plat-ed on mplat-edium lacking leucine and tryptophan (−LW) to verify bait and prey plasmid expression, or on medium also lacking histidine and arginine (−LWHA) to examine bait–prey interaction. b, Schematic representation of HIPK2 and p53 proteins, showing the deletions and truncations employed. c, Yeast two-hybrid assay as in
a to test the interaction between p53 and full-length HIPK2 (lane 3) or its truncated
form HIPK2(1–853) (lane 4). Lane 1, constructs PGBT9LAM–HIPK2(1–1189); lane 2, construct PGBT9–HIPK2(1–1189). d, Y817 yeast cells were induced to express
the indicated proteins and assayed for β-galactosidase activity. pGAD is the empty vector used as negative control; Large T is the large T antigen of SV40 used with p53 as strong positive control. e, GST pull-down assays with radiolabelled p53 with the use of the following GST fusion proteins: lane 3, GST–HIPK2(853–955), lane 4, GST–HIPK2(1–1189); lane 5, GST–HIPK2(1–853). Lane 1, input; lane 2, GST con-trol. f, GST pull-down assays with radiolabelled HIPK2 with the use of the following GST fusion proteins: lane 3, GST–p53(13–390); lane 4, GST–p53(13–295); lane 5, GST–p53(295–390). Lane 1, input; lane 2, GST control. g, Co-immunoprecipitation of transfected HIPK2–Flag and endogenous wild-type p53 proteins in HEK 293 cells with anti-Flag antibody. IP, immunoprecipitation.
kinase-inactive HIPK2-K221R was still able to bind p53, HEK 293 cells were transfected with HIPK2–Flag or HIPK2-K221R–Flag and TCEs were immunoprecipitated with anti-Flag antibodies. The pre-cipitated proteins were divided into two fractions. The first fraction was resolved by SDS–PAGE, blotted and immunoreacted for Flag and p53; the second fraction was used in a kinase assay by the addition of [γ-32P]ATP directly to the immunoprecipitates. As shown in
Fig. 4b, the kinase-inactive mutant of HIPK2 can still bind p53 but has no enzymatic activity, whereas the wild-type HIPK2 undergoes autophosphorylation.
HIPK2 binds p53 through its C-terminal domain. To evaluate
the significance of this interaction the full-length
GST–p53(13–390) and the C-terminus-deleted GST–p53(13–295) recombinant proteins were used in a kinase assay with wild-type HIPK2. No phosphorylation of GST–p53(13–295) protein was detectable (Fig. 4c), indicating that the binding between HIPK2 and p53 is required for the kinase reaction.
To identify the site of p53 phosphorylation, chemically synthe-sized p53 peptides (residues 1–39 or 25–65) were employed in immunoblot screening. The use of several specific antisera against p53 phosphorylated on various serine residues20,21showed that, of
the serine residues at the N terminus, only Ser 46 is phosphorylated by HIPK2 (Fig. 4d). The p53 site phosphorylated by HIPK2 was confirmed by a kinase assay in vitro with immunoprecipitated HIPK2–Flag and full-length GST–p53 protein followed by immunoblotting with antisera against p53 phosphorylated on Ser 15 or Ser 46 (Fig. 4e).
To evaluate whether HIPK2 can phosphorylate p53 at Ser 46 in
vivo, p53−/−H1299 cells were simultaneously infected with a p53
recombinant adenovirus (Adp53) and transfected with HIPK2–Flag, HIPK2-K221R–Flag or the vector alone. After 36 h,
cell viability was measured (see below) and TCEs were prepared and immunoprecipitated with anti-p53 (DO-1) antibody. Immunocomplexes were resolved by SDS–PAGE, blotted and immunoreacted with anti-p53-phosphoserine-46 antiserum or anti-Flag antibodies. This last control showed that HIPK2 is co-immunoprecipitated with p53 (Fig. 4f, second panel from the top). Aliquots of the same TCEs were used for western blot analyses to detect the total amount of p53 and Flag chimaeras. As shown in Fig. 4f, HIPK2, but not the HIPK2-K221R kinase mutant, increased the reactivity for the anti-p53-phosphoserine-46 antiserum compared with the vector alone, or with the only infected control (data not PML p53 wt p53 wt HA–HIPK2 GFP–HIPK2 GFP–HIPK2 Merge Merge p53/GFP–HIPK2 Merge p53/GFP–HIPK2 PML3 PML a b c
Figure 2 HIPK2 co-localizes with p53 into PML NBs. a, Immunofluorescence staining of H1299 cells overexpressing HA–HIPK2. Endogenous PML staining by TRITC-conjugated antibodies and HA–HIPK2 staining by FITC-conjugated antibodies are shown. Merging of the two colours results in a yellow signal. b,
Immunofluorescence staining of MG63 cells microinjected with wild-type (wt) p53, GFP–HIPK2 and PML-3. Staining was performed as follows: p53 by TRITC-conjugat-ed antibody, GFP–HIPK2 by means of the intrinsic green fluorescence, and PML by Alexa-conjugated antibodies. Merging of red and green colours is shown.
c, Immunofluorescence staining of MG63 cells microinjected with wild-type p53 and
GFP–HIPK2, then treated with arsenic trioxide. The localizations of overexpressed wild-type p53 and GFP–HIPK2 and endogenous PML were analysed as in b.
800 600 400 200 0 – – – + + + + + 0.5 1 2 5 CMV–p53 HIPK2 (µg) HIPK2 Relativ e lucif er ace activity l.c. a 600 450 300 150 0 – + + + – – – – + – + – – – – + – + CMV–p53 HIPK2 HIPK2 HIPK2 (∆865–1,096) l.c. b – – – + + + + 1 2 5 CMV–p53 HIPK2 (µg) HIPK2 l.c. 400 300 200 100 0 c
Figure 3 Wild-type HIPK2, but not the HIPK2(∆∆865–1096) deletion mutant, is able to increase p53-mediated transcriptional activity. a, Dose–response
analysis of increasing amounts of HIPK2 on p53 activity on the p(mdm2)NA-luc reporter vector, transiently transfected in p53−/−H1299 cells. b, Effect of HIPK2 or
the HIPK2(∆865–1096) deletion mutant on p53 activity on the same reporter vec-tor as in a. c, Dose–response analysis performed as in a on the p21Waf1promoter
transiently transfected in H1299 cells. The CMV–p53 vector coding for the wild-type p53 protein was co-transfected as indicated. All transfections were performed in duplicate and the data are means±s.d. for five independent experiments. Western blot analyses of HIPK2 or HIPK2(∆865–1096) proteins and the related loading controls (l.c.) are shown below each graph.
35S-HA–HIPK2 35S-HA–HIPK2 35S-HA–K221R 35S-HA–K221R GST–p53 γ−32P-p53 γ−32P-GST–p53(13–390) γ−32P-GST–p53(13–390) d.u. 1 5 + + + + + + – – – – + – – CMV–Flag + + – – HIPK2–Flag + – – – – + + + + – – – + + – – + + – – – + – – + – – – + – + + + + – K221R–Flag WB: anti-Flag WB: anti-p53 WB: anti-p53 γ−32P-HIPK2 γ−32P-p53 IP: Anti Flag
IP: Anti-p53 WB: p53Ser 46
WB: Anti-Flag
WB: Anti-p53
WB: Anti-Flag
IP: Anti Flag GST–p53(13–390) GST–p53(13–295) CMV–Flag HIPK2–Flag GST–p53(13–390) GST–p53(13–295) - 80 - 60 Phosphopeptide (positive control) p53 peptide (negative control) p53 peptide with HIPK2 Mr(K) 98 64 50 -GST–p53 p53 1 2 3 4 5 1 2 3 4 5 1 2 3 4 5 Anti-p53Ser 46-P Anti-p53Ser 46-P DO-1 HIPK2–Flag K221R–Flag CMV–Flag Adp53 TCE a d e f b c TCE 1 2 3 4 5 6 7 Mr(K)
Figure 4 HIPK2, but not the HIPK2-K221R kinase-dead mutant,
phosphory-lates p53 in vitro and in vivo. a, HIPK2 or HIPK2-K221R proteins translated in
vitro were analysed by SDS–PAGE and fluorography (upper panel). Densitometric
units (d.u.) were measured to estimate the amounts of proteins translated in vitro to be used with GST–p53 for the kinase assay (lower panel). b, HEK 293 cells were transiently transfected with the indicated HIPK2–Flag vectors. TCEs were immuno-precipitated (IP) with anti-Flag antibody and processed as follows: SDS–PAGE and western blotting (WB) with anti-Flag and anti-p53 (Ab7) antibodies, or incubation in the presence of [γ-32P]ATP, SDS–PAGE and autoradiography. TCEs were also analysed by direct western blotting with anti-p53 (FL393) antibody. c, The indicated GST–p53 constructs were analysed by SDS–PAGE and Coomassie staining (upper panel). Equal amounts of GST fusion proteins were incubated with the indicated immunoprecipitated proteins for the kinase assay (lower panel). d, Immunoblot screening assay on chemically synthesized p53 peptides mixed with
immunoprecipi-tated HIPK2–Flag from transiently transfected H1299 cells. Antibodies specific for p53 phosphorylated at Ser 6 (lane 1), Ser 9 (lane 2), Ser 15 (lane 3), Ser 20 (lane 4), Ser 33 (lane 5) and Ser 37 (lane 6), and for p53(25–65) phosphorylated at Ser 46 (lane 7) were employed. The appropriate phosphopeptide was used as a posi-tive control for each lane. e, Immunoprecipitated HIPK2–Flag obtained as in (d) was incubated in kinase buffer with full-length GST–p53 and resolved by SDS–PAGE. Lanes 1, no added kinase; lanes 2, 0.2µl kinase; lanes 3, 2µl kinase; lanes 4, 10 µl kinase. Membranes were immunoreacted with the indicated antibodies. TCEs from adriamycin-treated cells were used as positive control (lanes 5). f, H1299 cells were infected with Adp53 and transfected with the indicated HIPK2–Flag vec-tors. At 36 h after transfection, equal quantities of TCEs were immunoprecipitated with anti p53 DO1 mAb, resolved by SDS–PAGE and analysed by western blotting with anti-p53Ser 46 and anti-Flag antibodies. Aliquots of the same TCEs were direct-ly anadirect-lysed by western blotting with anti-p53 (FL393) and anti-Flag antibodies.
shown), indicating that HIPK2 can phosphorylate p53 at Ser 46 in
vivo.
Taken together, these results show that HIPK2 can phosphory-late p53 in vitro and in vivo at Ser 46, a recently identified site involved in the regulation of stress-induced apoptosis6,7.
HIPK2 is activated after ultraviolet irradiation. p53 phosphoryla-tion at Ser 46 was originally described in ultraviolet-irradiated apoptotic cells6 and its importance in the regulation of the p53
response to DNA damage in vivo has been demonstrated recently7.
In addition, it has previously been shown that, after high doses of ultraviolet (50 J m−2), p53 from MCF-7 cells is phosphorylated at
Ser 46, leading to the downregulation of p21Waf1and the activation of apoptosis7. To confirm these observations, we analysed Ser 46
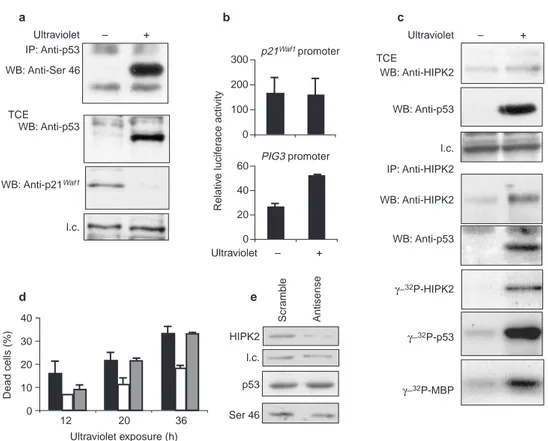
phosphorylation on immunoprecipitated p53, p53 upregulation and p21Waf1downregulation on TCEs (Fig. 5a), and apoptosis (data not shown) 12 h after ultraviolet irradiation (50 J m−2) of MCF-7 cells.
Because we had found no HIPK2–p53 cooperation on the p21Waf1 pro-moter in H1299 cells (Fig. 3c), the activities of this propro-moter and of the p53-responsive, apoptosis-dependent PIG3 promoter were measured in MCF-7 cells 24 h after ultraviolet irradiation. Only the PIG3 pro-moter increased its activity in this pro-apoptotic condition (Fig. 5b).
To evaluate whether these ultraviolet-induced events correlate with the activation of HIPK2, TCEs from control and ultraviolet-irradiated MCF-7 cells were either subjected to western blotting or
immunoprecipitated with anti-HIPK2 antiserum. The western blot analysis of TCEs showed that the expression levels of HIPK2 increased 12 h after irradiation (Fig. 5c). This increase was absent at the earlier time points (that is, 6 h after irradiation; data not shown). The HIPK2 co-precipitated proteins were divided into two fractions. The first fraction was resolved on SDS–PAGE, blotted and immunoreacted for detecting HIPK2 and p53 levels; the sec-ond fraction was used in a kinase assay by the addition of [γ-32P]ATP and the myelin basic protein (MBP) substrate22directly
to the immunocomplexes. As shown in Fig. 5c, the endogenous p53 was co-immunoprecipitated with the endogenous HIPK2 after ultraviolet treatment. Furthermore, HIPK2 underwent autophos-phorylation and efficiently phosphorylated both p53 and MBP after ultraviolet treatment (Fig. 5c). Comparable results were obtained with wild-type p53-carrying RKO cells (data not shown). To determine whether HIPK2 contributes to the ultraviolet-induced, p53-mediated apoptosis, antisense oligonucleotides were used to decrease HIPK2 expression before ultraviolet irradiation. A twofold decrease in HIPK2 protein levels and a 50% decrease in p53 phosphorylation at Ser 46 were found when the cells were treated with the antisense oligonucleotides compared with scramble-oligonucleotide-treated cells (Fig. 5d). A time-course analysis of cell viability showed a decrease in the number of dead cells in the antisense-oligonucleotide-treated population compared with Ultraviolet Ultraviolet – + – + IP: Anti-p53 WB: Anti-Ser 46 WB: Anti-p53 WB: Anti-p21Waf1 TCE l.c. 300 200 100 0 p21Waf1 promoter PIG3 promoter 60 40 20 0 Relativ e lucif er ace activity 40 30 20 10 0 Dead cells (%) 12 20 36 Ultraviolet exposure (h) Scr amb le Antisense HIPK2 l.c. p53 Ser 46 Ultraviolet – + TCE WB: Anti-HIPK2 WB: Anti-HIPK2 IP: Anti-HIPK2 WB: Anti-p53 WB: Anti-p53 γ−32P-HIPK2 γ−32P-p53 γ−32P-MBP l.c. a d e b c
Figure 5 HIPK2 binds and phosphorylates p53 after ultraviolet irradiation
and contributes to ultraviolet-induced apoptosis. a, MCF-7 cells were treated
with ultraviolet at 50 J m−2. Cells were lysed after 12 h and p53 was immunoprecip-tated by DO1 mAb. After SDS–PAGE and transfer to PVDF membrane, western blot-ting was performed with anti-p53Ser 46 antibody. TCEs were also analysed by direct western blotting for p53 detection with anti-p53 (FL393) antibody and p21Waf1
detection with anti-p21Waf1(N-20) antibody Abbreviation: l.c., loading control.
b, MCF-7 cells were transiently transfected with reporter vectors carrying p21Waf1or PIG3 promoter. Luciferase activity was measured 24 h after ultraviolet irradiation.
c, MCF-7 cells were irradiated as in a and processed as follows: (1) TCEs were
analysed by direct western blotting for p53 detection with p53 (FL393)
anti-body and HIPK2 detection with anti-HIPK2 serum; (2) equal amounts of TCEs were immunoprecipitated with anti-HIPK2 antibody and analysed by SDS–PAGE and west-ern blotting with anti-HIPK2 or anti-p53 (Ab7) sera to demonstrate the co-immuno-precipitation of endogenous HIPK2 and p53 proteins; (3) equal amounts of TCEs were immunoprecipitated with anti-HIPK2 antibody and incubated in the presence of [γ-32P]ATP and MBP substrate for kinase assay. d, Percentage of cell death in MCF-7 cells, irradiated as in a and treated with HIPK2-specific mock (black columns), antisense (white columns) and scramble (grey columns) oligonucleotides.
e, Western blot analyses of the same cells as in d lysed at 12 h, for HIPK2 and
untreated or scramble-oligonucleotide-treated cells (Fig. 5d). A TdT-mediated dUTP nick end labelling (TUNEL) assay performed at the 12 h time-point showed the following percentages of positive cells: mock, 16.3±2.8; antisense, 3.6±1.7; scramble-oligonucleotide, 15.3±0.9. This result confirmed the viability data.
These results show that HIPK2 levels, HIPK2–p53 complexes and HIPK2-mediated phosphorylation of p53 at Ser 46 can be reg-ulated by ultraviolet radiation and contribute to ultraviolet-induced apoptosis.
HIPK2 induces apoptosis through p53 phosphorylation. To evalu-ate directly the effects of HIPK2 on the biological activity of p53, wild-type HIPK2 or HIPK2(∆865–1096), which does not bind p53, was transfected in RKO, HEK 293, U373MG and H1299 cells,
which carry wild-type p53 (RKO and HEK 293), mutant p53 (U373MG) and no p53 (H1299), to test for colony formation. In cells containing endogenous wild-type p53 that were transfected with HIPK2, colony formation was strongly decreased, whereas no effect was seen in the same cells transfected with a HIPK2 deletion mutant (Fig. 6a). Furthermore, wild-type HIPK2 did not suppress colony formation in either the p53 mutant or p53-null cells (Fig. 6a). To examine the cause of the suppression of colony for-mation, HEK 293 cells were transiently transfected with the same vectors and analysed for proliferation, viability and DNA content, and subjected to a TUNEL assay. In the HIPK2-transfected cells, a decrease in cell number was associated with decreased viability (Fig. 6b), the appearance of a hypodiploid peak (Fig. 6c) and a positive RKO 293 U373MG H1299 15 10 5 0 100 75 50 25 0 Moc k LxSP HIPK2 HIPK2 ( ∆ 865 – 1,096 ) 293 Mock LxSP HIPK2 HIPK2 (∆865–1,096) Sub-G1 6.8 G1 51.4 S 31.5 G2/M 17.1 Sub-G1 6.0 G1 52.3 S 33.2 G2/M 14.5 Sub-G1 23.2 G1 40.8 S 39.2 G2/M 19.9 Sub-G1 6.8 G1 51.4 S 31.5 G2/M 17.1 30 20 10 0 d170.3 Adp53 HIPK2
Adp53 d170.3 Adp53 HIPK2Adp53
Apoptotic cells (%) Apoptotic cells (%)
40 30 20 10 0 80 60 40 20 0 Bax l.c. 120 80 40 0 HIPK2 l.c. LxSP HIPK2 HIPK2 ( ∆ 865 – 1096) CMV –Flag CMV –p53 HIPK2 CMV –p53 CMV–Flag HIPK2 K221R HIPK2 HIPK2 (∆865–1,096) p53 l.c.
wt p53 S46A S15A Vector
100 80 60 40 20 T unel-positiv e cells (%) LxSP HIPK2 HIPK2 (∆865 – 1,096 ) Viability (%) 10 – 4× cell n umber 293 Relativ e lucif er ase activity Relativ e lucif er ase activity a d g h i e f b c
Figure 6 HIPK2 promotes p53-mediated apoptosis. a, The indicated cells were transfected with pLxSP, pL–HIPK2–SP or pL–HIPK2(∆865–1096)-SP expression vec-tors. Puromycin-resistant colonies were stained 10 d later. b, HEK 293 cells were transiently transfected with the indicated vectors and the cell number and viability were evaluated after 72 h. c, Cell cycle analyses of HEK 293 cells transfected as in
b were performed 48 h after transfection. d, e, Apoptotic death in p53−/−H1299
(d) and p53+/+U87MG (e) cells after infection with Adp53 with or without pL–HIPK2–SP transfection. At 48 h after transduction, cells were stained with pro-pidium iodide for DNA content analyses and evaluation of the hypodiploid picks.
f, Effect of HIPK2 and HIPK2(∆865–1096) deletion mutant of HIPK2 on p53 activity
in HEK 293 cells transiently transfected with Bax (left histogram) and PIG3 (right histogram) reporter vectors. HIPK2 protein expression is shown below with the rela-tive loading control (l.c.). g, Western blot analysis of BAX protein on TCEs from HEK 293 cells transiently co-transfected with pCMV–p53 and the indicated HIPK2–Flag vectors. h, HEK 293 cells were transfected with the indicated vectors. G418-resist-ant colonies were stained 2 weeks later. i, Apoptotic cell death (percentage TUNEL staining) in MCF-7 cells transiently transfected with the indicated plasmids. The expression levels of transduced p53 proteins are shown below with the relative loading control (l.c.). Abbreviation: wt, wild-type.
reaction for TUNEL (data not shown), demonstrating that HIPK2 can induce apoptosis and that this activity requires the binding of p53. Cooperative induction of apoptosis by HIPK2 and p53 was also seen in p53−/− H1299 cells (Fig. 6d) and in p53+/+ U87MG cells
(Fig. 6e).
Because p53-mediated apoptosis relies, at least in part, on the transcriptional activation of several pro-apoptotic genes, the activ-ity of the bax and PIG3 promoters23,24and the expression of Bax
protein were evaluated in HEK 293 cells after transfection with HIPK2 and HIPK2(∆865–1096). Consistent with the previous data was the observation that only wild-type HIPK2 stimulated the transcription of these two genes (Fig. 6f, g).
Finally, we evaluated whether the effect of wild-type HIPK2 on the induction of apoptosis requires both its enzymatic activity and the phosphorylation at Ser 46 of p53. To this end, two different types of experiment were performed. First, the HIPK2-K221R mutant, which does not phosphorylate p53, was used in a colony formation assay. A complete rescue of the colony-forming efficien-cy was found (Fig. 6h), indicating that wild-type HIPK2 mediates p53-dependent apoptosis through its kinase activity. Second, H1299 cells were transiently co-transfected with wild-type HIPK2 or HIPK2(∆865–1096) and with wild-type p53 or the p53 mutants p53-S46A or p53-S15A. All three p53 constructs alone showed a basal level of apoptosis in transient transfection. However, wild-type HIPK2, but not the HIPK2(∆865–1096) construct, significant-ly increased the number of TUNEL-positive cells in co-expression with wild-type p53 and, to a smaller extent, with the p53-S15A mutant, whereas no differences were found when HIPK2 was co-expressed with the p53-S46A mutant (Fig. 6i). These results strong-ly support the involvement of the phosphorylation at Ser 46 of p53 in the induction of apoptosis and indicate that HIPK2 is an upstream regulator of p53-dependent apoptosis.
Discussion
The tumour suppressor protein p53 is known to function in the prevention of genetic instability by controlling cell-cycle check-points and the activation of apoptosis1,2. To mediate these functions,
the p53 protein must be activated by post-translational modifica-tions, which require interaction with specific enzymes3,4. We have
used yeast two-hybrid screening to identify new p53 cofactors. Here we report a novel interaction between HIPK2 and p53 that results in the modulation of p53-dependent apoptosis. In particular, we found that (1) HIPK2 interacts with the C terminus of p53 through its speckle retention signal (SRS); (2) HIPK2 and p53 co-localize with PML-3 into the nuclear bodies and cooperate in the activation of p53-dependent transcription; (3) HIPK2 specifically phosphory-lates p53 at Ser 46, a recently identified site involved in apoptotic reg-ulation6,7; (4) ultraviolet-induced phosphorylation of p53 at Ser 46 is
associated with activation of HIPK2 and stimulation of HIPK2–p53 interaction; and (5) antisense-induced inhibition of HIPK2 expres-sion decreases ultraviolet-induced p53 phosphorylation at Ser 46, and also apoptosis.
HIPK2 is a recently identified member of a new family of co-repressors for homeodomain transcription factors such as
Drosophila NK-1 and NK-3, and mouse Nkx-1.2 and Nkx-2.5
(ref. 9). Besides the homeoprotein-interacting domain, HIPK2 pos-sesses a kinase domain of unknown function and an SRS responsi-ble for the subnuclear localization of the protein (that is, nuclear dots)9,12. In contrast to the homeoproteins, p53 binds HIPK2 in the
SRS-containing region, indicating a different role for the p53–HIPK2 interaction. This inference is supported by the mani-festation of the HIPK2 kinase activity only in the p53–HIPK2 inter-action. Whether these two types of HIPK2 interaction are distinct or whether HIPK2 links p53 to the homeodomain transcription factors remains to be determined.
We found that HIPK2 partly localizes together with PML, a def-inite component of NBs13, indicating that at least part of the
described HIPK2-containing nuclear dots12 are indeed NBs. This
localization seems to be independent of p53 because it can take place in p53−/−H1299 cells, indicating that HIPK2 and p53 reach
the NBs independently. This is consistent with the recent finding that p53 is recruited into the NBs by PML-3 (refs 15, 16). Interestingly, p53 relocalization in these nuclear structures has been observed during oncogenic Ras-induced replicative senescence13,14
or PML-induced pro-apoptotic function15,16, indicating that p53
molecules activated in the same subnuclear compartments can enter different pathways, resulting in different biological outcomes. Our finding that HIPK2 localizes with p53 in the NBs and phos-phorylates p53 at Ser 46 should help in defining the modification process at the NB level that determines which signalling pathway that activated p53 will enter.
Phosphorylation at Ser 46 of p53 was originally observed in apoptotic cells after ultraviolet irradiation and p38 mitogen-acti-vated protein (MAP) kinase was shown to mediate this event in
vitro6. More recently, Oda et al.7showed that Ser 46
phosphoryla-tion specifically regulates p53-induced apoptosis in vivo after severe DNA damage. Indeed, they showed that Ser 46 phosphorylation is a late event after DNA damage and is associated with p21Waf1 down-regulation and the transcription of pro-apoptotic genes including the recently identified p53AIP1 (ref. 7). Furthermore, replacement of Ser 46 on p53 but not p38 MAP kinase inhibitors block this DNA-damage-induced apoptosis7, indicating that one or more
kinases other than p38 MAP kinase could be responsible for this specific modification of p53. We have found that HIPK2 can phos-phorylate p53 at Ser 46 in vitro and in vivo and that HIPK2 overex-pression increases apoptosis when co-expressed with wild-type p53 but not with the p53-S46A mutant. HIPK2 is up-regulated and activated by high doses of ultraviolet radiation that results in HIPK2 autophosphorylation, easily detectable formation of HIPK2–p53 complexes, and p53 or MBP substrate phosphoryla-tion. Moreover, overexpression of HIPK2, but not of its kinase-dead mutant, induces apoptosis in wild-type p53-expressing cells even in those cell types in which p53 overexpression was shown to induce only growth arrest, such as HEK 293 and U87MG cells7,25.
Interestingly, in U87MG cells, the presence of growth arrest and the absence of apoptosis observed upon exogenous wild-type p53 pro-tein expression have recently been correlated with the absence of p53 phosphorylation at Ser 46 (ref. 7). The induction of apoptosis that we observed in these cells upon HIPK2 expression (Fig. 6e) strongly supports these observations that Ser 46 phosphorylation might regulate p53-induced apoptosis. In keeping with these results is also the finding that exogenous wild-type p53 expression directly induces apoptosis in H1299 cells26(Fig. 6d), in which we detected
phophorylation at Ser 46 (Fig. 4e) and high basal expression levels of the endogenous HIPK2 protein (Fig. 3, non-transfected con-trols). Moreover, co-expression of p53 and HIPK2 in these cells increased both the level of phosphorylation at Ser 46 and the rate of apoptosis (Fig. 4e and 6d). Taken together, the findings reported here strongly support HIPK2 as a candidate kinase for the Ser 46-depend-ent activation of p53 and open the way for a greater understanding of the relationship between post-translational modifications of p53, its activation process, and the control of different p53-dependent pathways.
Methods
Yeast two-hybrid assay, HIPK2 cloning and expression constructs
For two-hybrid screening, the GAL4-DNA-binding domain of the pGBT9 vector fused to the murine p53 cDNA (residues 73–390) (Clontech) was used to screen a murine 11-day embryo MATCHMAKER
cDNA library cloned into the pGAD10 vector (Clontech), as described27,28. Murine full-length HIPK2
was cloned from a mouse lung Lambda cDNA library cloned into the Lambda ZAP II vector (Stratagene). The following vectors were employed to construct HIPK2 full-length or deletion mutants: LxSP recombinant retrovirus vector carrying the puromicin resistance gene, pCMV-Tag (Stratagene), pcDNA3.1 (Invitrogen) modified by adding HA or GFP tags, and pGEX (Amersham-Pharmacia). pCMV–HIPK2-K221R–Flag was obtained from the pCMV–HIPK2–Flag vector with the
the pL–HIPK2SP vector by digestion with RsrII and BstEII, repairing overhanging ends and re-ligation. In vitro pull-down and co-immunoprecipitation assays
GST fusion proteins were produced in BL21 bacteria and purified on glutathione–Sepharose resin (Pharmacia). Protein translated in vitro was synthesized with a TNT-coupled reticulocytes lysate
sys-tem (Promega) in the presence of L-[35S]methionine. The protein interaction assay was performed as
described28.
For co-immunoprecipitation in vivo, HEK 293 cells were plated on 60-mm Petri dishes and tran-siently transfected with the bishydroxyethylaminoethanesulphonate (BES)-modified calcium
phos-phate method29. At 36 h after transfection, TCEs were prepared with lysis buffer (50 mM Tris-HCl pH
7.5, 5 mM EDTA, 300 mM NaCl, 150 mM KCl, 1 mM dithiothreitol, 1% Nonidet P40 and a mix of
protease inhibitors (Sigma)) as described12. Equal amounts of proteins were immunoprecipitated for 2
h at 4°C with 10 µl per sample of anti-Flag monoclonal antibody (mAb) (M2; Sigma) pre-adsorbed
on Protein G–agarose beads (Pierce). Immunocomplexes were collected by centrifugation, washed five times with lysis buffer, separated by SDS–PAGE and blotted to nitrocellulose membranes (Bio-Rad). Filters were blocked in TBS–Tween plus 3% dried skimmed milk and incubated with anti-Flag mAb or anti-p53 sheep polyclonal antiserum (Ab 7; Calbiochem).
Cell culture conditions and treatments
Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (HEK 293) or RPMI (MCF-7, RKO, U87MG, U373MG and H1299) medium supplemented with 10% fetal calf serum (Gibco-BRL, Life Technology), glutamine and antibiotics. For DNA damage, subconfluent cells were irradiated with
50 J m−2ultraviolet and 12 h later were lysed for further analyses. Adenoviral infection was performed
as described30by employing the recombinant adenovirus Adp53, carrying the human wild-type p53
under the control of the CMV promoter or the insertless control virus dl70.3 (ref. 31) (kindly provid-ed by S. Bacchetti and F. Graham).
For the inhibition of HIPK2 expression, single-strand oligonucleotides were synthesized in the
anti-sense orientation (5′-GGTGTGAGGGGAGAAAAC-3′) or in a scrambled form (5′
-GAGAAACAGTAGGGGGTG-3′). Cell treatment was performed as described32.
Transactivation assay
Cells were transiently transfected with reporter vectors in which the luciferase gene was driven by the
p53-dependent intronic promoter of the murine mdm2 gene19, the human bax promoter23, the PIG3
promoter24or the p21Waf1promoter (kindly provided by M. Oren and B. Vogelstein) and normalized
with the use of a co-transfected β-galactosidase construct. Transcriptional activity was evaluated after
co-transfection with wild-type HIPK2 and HIPK2 deletion/mutant vectors. Luciferase assay was
per-formed as described previously33.
Anti-HIPK2 serum
Polyclonal antibodies recognizing HIPK2 were prepared by injecting 100 µg of L–HIPK2–SP plasmid
into the leg muscle of 4-week-old BALB/c female mice34. Plasmid injection was repeated after 14 d and,
30 d later, mice were bled and the sera were tested on HIPK2–Flag and HIPK2 proteins. Anti-HIPK2 rabbit antiserum was kindly provided by M. L. Schmitz.
Microinjection and immunofluorescence
Cells grown on coverslips in 35-mm Petri dishes were microinjected with the Automated Injection
System (Zeiss, Oberkochen, Germany) as described16. Immunoflorescence was performed with the
fol-lowing antibodies: anti-p53 rabbit polyclonal16; anti-PML (PGM3, Santa Cruz); anti-HA (12CA5;
Roche Molecular Biochemicals, Monza, Italy), tetramethylrhodamine isothiocyanate (TRITC)-conju-gated goat anti-mouse IgG1, fluorescein isothiocyanate (FITC)-conju(TRITC)-conju-gated anti-mouse IgG2b (Southern Biotechnologies), Alexa-conjugated anti-mouse immunoglobulin (Molecular Bioprobes) and TRITC-conjugated anti-rabbit immunoglobulin (Sigma). Treatment with arsenic trioxide was
per-formed as described previously35. Images were acquired with a Zeiss laser scan microscope (LSM 510).
Kinase assays in vitro
Kinase assays were performed with (1) recombinant, (2) overexpressed and (3) endogenous proteins. For recombinant proteins, HA–HIPK2 and HA–HIPK2-K221R translated in vitro were incubated
with GST–p53 (Santa Cruz) in kinase buffer (20 mM HEPES pH 7.4, 10 mM MgCl2, 200µM sodium
orthovanadate] in the presence of 5µCi [γ-32P]ATP and 50 mM unlabelled ATP for 30 min at 30°C.
Reaction products were resolved by SDS–PAGE and 32P-labelled proteins were detected by
autoradiog-raphy.
Fir overexpressed proteins, cells were transfected with pCMV–HIPK2–Flag, pCMV–HIPK2-K221R–Flag or the empty vector as described for the co-immunoprecipitation assay. Cell lysates were immunoprecipitated with anti-Flag mAb and the immunoprecipitates were subjected to kinase assay as described above.
For endogenous proteins, TCEs were prepared from ultraviolet-irradiated cells and immunoprecip-itated with mouse or rabbit polyclonal anti-HIPK2 serum. Immunocomplexes were used in the kinase assay by the addition of the MBP substrate (Sigma). For immunoblot screening, chemically synthe-sized p53 peptides (residues 1–39 and 25–65) were mixed with HIPK2–Flag proteins immunoprecipi-tated from transiently transfected H1299 cells. Western blotting of kinase reactions was performed
with p53 phosphoserine-specific antibodies20,21.
Western blot analysis
For western blot analysis, TCEs were separated by SDS–PAGE, blotted to nitrocellulose membrane
(Bio-Rad) and incubated with anti-p53 (FL393), anti-Bax (N-20) or anti-p21Waf1(N-20) polyclonal
antibodies (all from Santa Cruz Biotechnology), or with anti-HSP70 mAb (StressGen) or anti-tubulin mAb (Sigma) for loading control. Filters were washed in TBS–Tween then incubated with horseradish-peroxidase-conjugated IgG (Cappel). Antibodies were detected with an enhanced chemiluminescence reaction kit (Amersham Corp.) in accordance with the manufacturer’s instructions. For the detection
of Ser 46 phosphorylation in vivo, equal amounts of TCE were prepared as described20,21and
immuno-precipitated with anti-p53 DO1 mAb (Calbiochem) and western blot analysis was performed with the
anti-p53Ser 46 antibody described for the kinase assay or with anti-p21Waf1antibody (Santa Cruz). For
the detection of endogenous HIPK2 on TCEs, proteins were transferred to PVDF membrane (Millipore) and western blot analyses were performed with rabbit anti-HIPK2 serum.
Cell cycle analysis
Cells were transiently transfected as described above and 48 h after transfection were fixed in
methanol-acetone 4:1 v/v and stained with 20µg ml−1propidium iodide (Sigma) for 30 min at room
temperature in the presence of 40µg ml−1RNAse A (Roche). DNA content was evaluated by
cytofluo-rimetry with an Epics XL analyser (Coulter Corporation).
Colony-formation assay
Cells plated in 60 mm Petri dishes were transfected by using the BES method with the following
plas-mids: pCMV–HIPK2–Flag, pL–HIPK2–SP, pCMV–HIPK2-K221R–Flag, pL–HIPK2(∆865–1096)SP or
the empty vectors pCMV-Flag and pLxSP. At 24 h after transfection, cells were selected with 2µg ml−1
puromycin (for transfections performed with the pLxSP-based vectors) or 1 mg ml−1G418 (for the
pCMV–Flag-based vectors), for 10 d or 2 weeks, respectively, before staining of the surviving colonies with crystal violet.
RECEIVED 30 JANUARY 2001; REVISED 8 AUGUST 2001; ACCEPTED 21 SEPTEMBER 2001; PUBLISHED 3 DECEMBER 2002.
1. Ko, L. J. & Prives, C. p53: puzzle and paradigm. Genes Dev. 10, 1054–1072 (1996). 2. Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307–310 (2000). 3. Giaccia, A. J. & Kastan, M. B. The complexity of modulation: emerging patterns from divergent
signals. Genes Dev. 12, 2973–2983 (1998).
4. Vousden, K. H. p53: death star. Cell 103, 691–694 (2000).
5. Gottlieb, T. M. & Oren, M. p53 in growth control and neoplasia. Biochim. Biophys. Acta 1287, 77–102 (1996).
6. Bulavin, D. V. et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phos-phorylation and apoptosis in response to UV radiation. EMBO J. 18, 6845–6854 (1999). 7. Oda, K. et al. P53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by
Ser-46-phosphorylated p53. Cell 102, 849–862 (2000).
8. Schmid, P., Lorenz, A., Hameister, H. & Montenarh, M. Expression of p53 during mouse embryo-genesis. Development 113, 857–865 (1991).
9. Kim, Y. H., Choi, C. Y., Lee, S.-J., Conti, M. A. & Kim, Y. Homeodomain-interacting protein kinas-es, a novel family of co-repressors for homeodomain transcription factors. J. Biol. Chem. 273, 25875–25879 (1998).
10. Schneider, S., Buchart, M. & Hovens, C. M. An in vitro assay of β-galactosidase from yeast.
Biotechniques 20, 960–962 (1996).
11. Hollstein, M., Sidransky, D., Vogelstein, B. & Harris, C. C. p53 mutations in human cancers.
Science 253, 49–53 (1991).
12. Kim, Y. H., Choi, C. Y. & Kim, Y. Covalent modification of the homeodomain-interacting protein kinase 2 (HIPK2) by the ubiquitin-like protein SUMO-1. Proc. Natl Acad. Sci. USA 96, 12350–12355 (1999).
13. Pearson, M. et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210 (2000).
14. Fereyre, G. et al. PML is induced by oncogenic ras and promotes premature senescence. Genes
Dev. 14, 2015–2027 (2000).
15. Guo, A. et al. The function of PML in p53-dependent apoptosis. Nature Cell Biol. 2, 730–736 (2000).
16. Fogal, V. et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19, 6185–6195 (2000).
17. Muller, S., Matunis, M. J. & Dejean, A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus EMBO J. 17, 61–70 (1998).
18. Wu, X., Bayle, J. H., Olson, D. & Levine, A. J. The mdm-2 autoregulatory feedback loop. Genes
Dev. 7, 1126–1132 (1993).
19. Barak, Y., Juven, T., Haffner. R. & Oren, M. mdm2 expression is induced by wild-type p53 activity.
EMBO J. 12, 461–468 (1993).
20. Sakaguchi, K. et al. Damage-mediated phosphorylation of human p53 threonine 18 through a cas-cade mediated by a casein 1-like kinase. Effect on Mdm2 binding. J. Biol. Chem. 275, 9278–9283 (2000).
21. Higashimoto, Y. et al. Human p53 is phosphorylated on serines 6 and 9 in response to DNA dam-age-inducing agent. J. Biol. Chem. 275, 23199–23203 (2000).
22. Hofmann, T. G., Mincheva, A., Lichter, P., Droge, W. & Schmitz, M. L. Human Homeodomain-interacting protein kinase-2 (HIPK2) is a member of the DYRK family of proteine kinases and maps to chromosome 7q32-q34. Biochimie 82, 1123–1127 (2000).
23. Miyashita, T. & Reed, J. C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299 (1995).
24. Polyak, K., Xia, Y., Zweir J. L., Winzler, K. W. & Vogelstein, B. A model for p53-induced apoptosis.
Nature 389, 300–305 (1997).
25. Matsuzawa, S., Takayama, S., Froesch, B. A., Zapata, J. M. & Reed, J. C. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: suppression by BAG-1.
EMBO J. 15, 2736–2747 (1998).
26. Haupt, Y., Barak, Y. & Oren, M. Cell type-specific inhibition of p53-mediated apoptosis by mdm2.
EMBO J. 15, 1596–1606 (1996).
27. Fanciulli, M. et al. The interacting RNA polymerase II subunits, hRPB11 and hRBP3, are coordi-nately expressed in adult human tissues and down-regulated by doxorubicin. FEBS Lett. 384, 48–52 (1998).
28. Fanciulli, M. et al. Identification of a novel partner of RNA polymerase II subunit 11, Che-1, which interacts with and affects the growth suppression function of Rb. FASEB J. 14, 904–912 (2000).
29. Chen, C. & Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA.
Mol. Cell. Biol. 7, 2754–2752 (1987).
(Humana Press, Clifton, NJ, 1991).
31. Bacchetti, S. & Graham, F. Inhibition of cell proliferation by an adenovirus vector expressing the human wild type p53 protein. Int. J. Oncol. 3, 781–788 (1993).
32. Leonetti, C. et al. Antitumor effect of c-myc antisense phosphorothioate oligodeoxynucleotides on human melanoma cells in vitro and in mice. J. Natl Cancer Inst. 88, 419–429 (1996).
33. Manni, I. et al. NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25c
promoters upon induced G2arrest. J. Biol. Chem. 276, 5570–5576 (2001).
34. Ito, K., Takeuchi, Y., Ito, K. & Kato, S. Strain-dependent antibody response induced by DNA immunization. Immunol. Lett. 74, 245–250 (2000).
35. Lombard, D. B. & Guarente, L. Nijmegen breakage syndrome disease protein and MRE11 at PML
nuclear bodies and meiotic telomeres. Cancer Res. 60, 2331–2334 (2000).
ACKNOWLEDGEMENTS
We thank M. Crescenzi and G. Blandino for helpful advice and stimulating discussions. G.D. and S.S. are particularly grateful to A. Sacchi for her constant availability, support and scientific inspiration. I.M. and M.G. are recipients of fellowships from Fondazione Italiana per la Ricerca sul Cancro. This work was supported by a New Unit Start-Up Grant to S.S. and by Investigator Grants to G.D.S. and G.P. from Associazione Italiana per la Ricerca sul Cancro.