47
3.Synthesis and Characterization of End-Functional P3HT Building Blocks
Poly(3-hexylthiophene)s (P3HTs) constitute an important class of conjugated polymers. Being chemically and thermally stable materials, soluble P3HTs are attractive for exploring their electronic and optical properties. The side chains not only ensure solubility, but are a key parameter for the mesoscopic structure in thin films and; hence, act as a property determining element. Perfection of the primary structure of the polymer, including molecular weight and polydispersity, is a prerequisite to obtain well-defined materials in a reproducible manner. End-group functionalization of these polymers opens the possibility to graft P3HTs onto surfaces, prepare block copolymers, or attach specific electroactive end groups and thereby extend the range of applications of these materials.The hybrid materials consisting of conjugated polymers and quantum dots (QD) are of interest from the standpoint of increased performance relative to either of the non-hybrid counterparts (fully inorganic or fully organic), with many applications envisioned in the areas of photovoltaic (PV) cells1 and LEDs.2 They inherit decent mechanical strength from conjugated polymers and good photostability and high conductivity from QDs. The conjugated polymers and QD nanocomposites are generally prepared by mixing these two components in the presence of solvent or by constructing a conjugated polymer and QD bilayer or conjugated polymers/QD alternating multilayer by either physical or chemical method.3 In the former case it is difficult to control the detailed morphology and dispersion of QDs within conjugated polymers while the latter two procedures are quite complex and unsuitable for high throughput production. The interface between conjugated polymers and QD, accomplished by stabilizing the ligand from QDs during film processing, is not well controlled, thereby reducing the efficiency of the electronic interactions between them. The effective charge transfer, profoundly influenced by the quality of the interface, is crucial for a conjugated polymer/QD composite to be used in the fabrication of photovoltaic cells.4 In particular bicontinuous and nanoscopic phase-separated mixture of conjugated polymer /QD is favorable for charge generation and transport. However, such morphology is currently difficult to realize by using a conventional blending approach.5 In fact, for PV active layer applications, it is important to stabilize QDs in an appropriate electro donating conjugated polymer host. An architecture based on conjugated polymer/QD nanocomposites with well-controlled interfaces provides a means of achieving uniform dispersion of nanoparticles and maximizing the interfacial area, which carries advantages over cases where nanoparticle aggregation dominates. To
48
date, only a few elegant studies have centered on the direct integration of QDs onto conjugated polymers via ligand exchange to replace the insulating surfactants,6 electrostatic interaction, or direct growth from the QDs’ surface7 to achieve a more controlled interface and morphology on a molecular scale.
The synthesis of regioregular P3ATs has been described in the pioneering work of McCullough et al., who reported their first synthesis via a generation of regiodefined 2-bromo-5-(bromomagnesio)-3-alkylthiophene followed by a Kumada-type cross-coupling protocol, resulting in P3ATs with nearly 100 % HT linked repeat units.8 A different route was proposed by Rieke et al. in which the organometallic reactive species is generated utilizing activated “Rieke-zinc” (Zn*) as metalating agent9 (Scheme 3.1). The choice of the catalyst used in the thienyl-thienyl coupling polymerization can either lead to regiorandom with Pd(PPh3)4 or regioregular with Ni(dppe)Cl2, P3ATs.10
Scheme 3.1 The Rieke method producing either regiorandom or HT-P3ATs depending on the type of catalyst
To overcome the drawbacks of these methods (multistep procedure in the case of the McCullough method and use of “Rieke-zinc” in the Rieke method) McCullough et al. developed the Grignard metathesis (GRIM) procedure, which allows the generation of regioregular P3ATs with high molecular weight (Mn up to 40,000 g/mol) and narrow polydispersity in a relatively simple fashion.11 Similar to the Rieke method, the GRIM procedure starts from 2,5-dibromo-3-alkylthiophene, which undergoes a Grignard intermediate when treated with 1 eq. of RMgCl (R = alkyl). The reaction proceeds with a moderate degree of regioselectivity (80:20) at room temperature regardless of the Grignard reagent employed.
In this chapter, we describe the multi-step synthesis of a mono phosphonic acid terminated regioregular P3HT. In the first step, the Grignard metathesis (GRIM) polymerizations was used to prepare asymmetrically terminated rr-P3HTs with a halide atom at one chain end and an allyl at the other, hereafter called mono-capped. 2,5-Dibromo-3-hexylthiophene was chosen as the starting
S S S S S S S Br Br ZnBr Br Br R R ZnBr R Zn* / THF
+
R R n R R n Pd(PPh3)4 Ni(dppp)Cl2 regiorandom head-to-tail (HT) R= C6H1349
material for the preparation of the thienyl Grignard, and bis(diphenylphosphinopropane) dichloride (Ni(dppp)Cl2) as the catalyst that initiates the chain-growth of the polymer. The simple addition of a RMgX (allyl magnesium bromide) to the growing chain gives mono-capped poly(3-hexylthiophene) (P3HT) with an allyl chain-end.12 This chain-end can then be used as a reactive site to introduce new end groups or polymer blocks at the P3HT termination. Using a mono-capped rr-P3HT is critical for the final application of the end-functionalized rr-P3HT as a stabilizer of inorganic nanoparticles. In fact, difunctional P3HT would promote NP aggregation due to bridging of the polymer. Adding functional groups to side chains of a conjugated polymer often requires significant changes in synthetic route, and may disrupt solid state packing of the polymer chains, resulting in non-semiconducting films. In contrast, adding functional end-capping groups such as the strongly coordinating phosphonic acid is a more efficient way for obtaining a uniform dispersion of the CdSe NPs in the polymerization without destroying the polymer crystallinity. Previously, this general approach has been successfully used by Higgins et al13 for pyrrole terminated alkanethiols. Here we synthesized the asymmetrically allyl/phosphonic acid terminated rr-P3HT in a multistep reaction. First we synthesized the allyl/Br terminated rr-P3HT, which was then modified with the phosphonic acid precursor diethyl 4- acetylene benzene phosphonate using the Sonogoshira coupling reaction. After hydrolysis of the phosphonic ester, the phosphonic acid terminated rr-P3HT (P3HT-PA) is used for CdSe NPs stabilization to replace the low MW stabilizer by the ligand exchange. According to the literature the phosphonic acid is in fact a good stabilizer for CdSe NPs.14
In summary, the starting material, regioregular P3HT, was synthesized using the McCullough route.15 This synthesis results in the attachment of bromide groups at one end of 90% of the polymer chain, this end becoming the selected chain termination. The bromide group may then be used for cross-coupling reactions. In our case it was used to attach an alkynyl end group, which could then be further modified to attach a protected phosphonic acid group. Alkynyl functional group in a different procedure, the same asymmetrically allyl/alkynyl terminated P3HT was used as the reagent in a “Click” reaction with an azide-terminated PAN to synthesize the block copolymer P3HT-b-PAN, which is described in chapter 4. The phosphonic acid terminated P3HT on the other hand was used for CdSe NPs stabilization by ligand exchange, which is described in chapter 6.
3.1 Synthesis of asymmetrically allyl/alkynyl terminated rr-P3HT
The synthesis of asymmetrically allyl/alkynyl terminated rr-P3HT was performed in the following steps:
50 3.1.1 Synthesis of 2, 5-dibromo-3-hexylthiophene
In the absence of light, N-bromo succinimide (NBS) (1.98 g, 11.14 mmol) dissolved in DMF (12 mL) was added dropwise under nitrogen to a solution of 3-hexylthiophene (937 mg, 5.57 mmol) in DMF (13 mL) at −20 °C over a 1 hr period. The mixture was stirred overnight at room temperature. Then the reaction mixture was poured into distilled water and extracted with dichloromethane (3 x 15 mL). The combined organic layers were washed with water, dried over magnesium sulphate, and the solvent evaporated to dryness in vacuum to yield the raw product. Further purification was performed using flash silica gel column chromatography (in n-hexane) to give a colorless oil in 93 % yield, (Scheme 3.3).
FT-IR (KBr window): ν = 3095 (aromatic C-H stretching), 2958, 2931, 2856 (aliphatic C-H stretching), 1540, 1465 (aromatic C=C stretching), 1376 (methyl bending), 825 (aromatic C-H out-of plain), 725 (methyl rock), 472 (C-Br stretching) cm-1.
1 H NMR (200 MHz, CDCl3, at RT): δ = 6.79 (s, 1 H, CH-CBr), 2.51 (t, 2 H, C-CH2-CH2), 1.54 (pentet, 2 H, CH2-CH2-CH2), 1.29 (m, 6 H, CH2-CH2-CH3), 0.88 (t, 3 H, CH3) ppm. 13 C {1H} NMR (200 MHz, CDCl3, at RT): δ = 142.98 CBr-C), 130.94 CBr-CH), 110.29 (S-CBr-CH), 107.91(S-CBr-C), 31.56(CH2-CH2-CH3), 29.54(C-CH2-CH2), 29.48 ((CH2)2-CH2 -(CH2)2), 28.78 (C-CH2-CH2), 22.56 (CH2-CH2-CH3), 14.07 (CH3) ppm.
3.1.2 Synthesis of asymmetrically allyl/Br end-capped rr- P3HT
The allyl/Br asymmetrically end capped regioregular poly(3-hexylthiophene) was synthesized as follows: A dry 100 mL three-neck flask was flushed with nitrogen and charged with 2,5-dibromo-3-hexylthiophene (3.04 g, 9.32 mmol) and anhydrous THF (20 mL). A 1.0 M solution of t-butyl magnesium chloride (9.32 mL, 9.32 mmol) in tetrahydrofuran (THF) was added via a syringe, and the reaction mixture was stirred at room temperature. After 2 hr, anhydrous THF (80 mL) was added to the reaction flask, followed by the addition of Ni(dppp)Cl2 (0.084 g, 0.154 mmol). The polymerization was allowed to proceed at room temperature for exactly 10 min followed by the quick addition of a 1.0 M solution of allyl magnesium bromide (2.34 mL, 2.34 mmol) by means of a dry syringe. The reaction mixture was stirred for another 10 min before quenching in excess methanol. The polymer was purified by sequential Soxhlet extractions with methanol, hexane and chloroform. The obtained polymer was isolated from the final chloroform solution and characterized by 1H NMR (yield = 1.10 g, 36.67%). The polymerization degree (DPn=60) was estimated by integrating the resonance peaks of the methylene protons of the hexyl side chain vs those of the allyl protons in the 1H NMR spectrum.
51
FT-IR (KBr window): ν = 3048 (thiophene C-H stretching), 2956, 2925, 2856 (aliphatic C-H stretching), 1513, 1455 (C=C stretching), 1374 (methyl bending), 1093, 1016, 914, 819 (aromatic C-H out-of plain), 724 (methylene rocking), 668, 657, 613, and 511 (C-Br stretching) cm-1.
1
H NMR (300 MHz, CDCl3): δ= 0.9 (t, J = 7 Hz, 3H), 1.35-1.43 (m, 6H), 1.69 (t, J = 7 Hz, 2H), 2.80 (t, J = 7 Hz, 2H), 3.49 (d, J = 7 Hz, 2H), 5.11 (m, 2H), 5.98 (m, 1H) 7.46 (s, 1H) ppm.
GPC: Mn = 6068; PDI = 1.11 (against PS standard). 13
C {1H} NMR (300 MHz, CDCl3): δ = 140.3(S-CBr-C), 134.1(S-C-CH), 132.2 (CH2-CH-CH2), 130.9(S-CBr-CH), 129.0(S-CBr-CH), 115.9(S-CBr-C), 33.3 (CH-CH2-C), 32.2(CH2-CH2-CH3), 31.0(C-CH2-CH2), 29.9((CH2)2-CH2-(CH2)2), 29.7(C-CH2-CH2), 23.1(CH2-CH2-CH3) and 14.6 (CH3) ppm.
3.1.3 Synthesis of allyl/alkynyl terminated rr-P3HT by Sonogoshira Coupling reaction
In schlenk A, allyl/Br terminated P3HT (100 mg, 1x10-5 mol) and trimethylsilyl acetylene (0.17 µL, 1.2x10-5 mol) in 4 mL THF was mixed with bis(triphenylphosphine)palladium(II) dichloride (PdCl2(PPh3)2, 28 mg, 4x10-5 mol). In schlenk B, copper(I) iodide (CuI, 3.81 mg, 2x10-5 mol) was dissolved in 6 mL Et3N, then transferred in schlenk A. The resulting mixture was degassed and filled with nitrogen thrice to remove oxygen, and then it was heated under stirring at 65 °C for 20 hr. At the end of reaction, Et3N was removed at reduced pressure and the polymer was precipitated in methanol, filtered, washed repeatedly on the filter with hexane and methanol to remove any unreacted trimethlysilyl acetylene and collected in chloroform. After the removal of the solvent under vacuum, the resulting product was deprotected as follows: the TMS protected polymer was dissolved in 10 mL THF and the solution was cooled to −20 °C with a brine bath. A 0.20 M solution of TBAF.3H2O (10 equivalent with respect to chain ends, i.e. 32 mg in 0.5 mL THF) was slowly added and the reaction mixture was stirred at this temperature for 30 min and then at room temperature for 4 hr. The polymer was recovered by precipitation in methanol. The asymmetrically allyl/alkynyl terminated rr-P3HTs was purified by a series of reprecipitations from solution into chloroform / methanol. After the final drying 64.7 mg of polymer was obtained, this was characterized by 1H, 13C NMR and FT-IR.
FT-IR (KBr window): ν = 3293, 3054 (thiophene C-H stretching), 2959, 2856 (aliphatic C-H stretching), 2036 (C≡C), 1512, 1455 (C=C stretching), 1377(methyl bending), 1251, 1096, 1017, 845 (aromatic C-H out-of plain), 761(methyl rock), 673, 651 cm-1.
1
H NMR (300 MHz, CDCl3): δ= 0.9 (t, J = 7 Hz, 3H), 1.35-1.43 (m, 6H), 1.69 (t, J = 7 Hz, 2H), 2.80 (t, J = 7 Hz, 2H), 3.49 (d, J = 7 Hz, 2H), 5.11 (m, 2H), 5.98 (m, 1H) 7.46 (s, 1H) ppm.
52 13
C {1H} NMR (300 MHz, CDCl3): δ= thiophene resonances at 142.8, 140.9, 138.0, 138.0, 136.6, 134.8, 134.4, 132.7, 126.3, 126.0, 125.3, 121, 115.9, acetylene carbon resonances at 76.5, 73, hexyl resonances at 32.5, 33.4, 29.6, 27.8, 25.2, 24.7, 23.9, 23.1, 14.0 ppm.
3.2 Asymmetrically end capped rr-P3HT chain end modification by phosphonic group
The allyl/Br terminated rr-P3HT of low molecular weight was prepared by increasing the catalyst amount as described in § 3.1.2 and then phosphonic acid functional group was introduce in two steps.
3.2.1 Synthesis of diethyl 4-bromobenzene phosphonate
In a three necked flask equipped with nitrogen inlet, a solution of p-dibromobenzene (10.0 g, 42.4 mmol) in dry THF (25 mL) was added to magnesium turnings (1.12 g, 46.1 mmol) in dry THF (10 mL). The mixture began spontaneously to reflux within 1 ~ 2 min and an ice bath was used to maintain a mild reflux. When the initially vigorous hexothermal reaction had subsided, the solution was left to stir at room temperature for 4 hr, then diluted with dry THF (80 mL) and cooled to 0 °C. The Grignard reagent was then transferred via cannula to a solution of diethylchlorophosphate (9.50 mL, 63.8 mmol) in dry THF (10 mL) at 0 °C. The resulting mixture was stirred at room temperature overnight, and then 10 mL of aqueous hydrochloric acid solution (1.0 M) was carefully added to remove the unreacted magnesium from the reaction, which reacts to magnesium chloride. The aqueous layer was extracted several times with diethyl ether; the combined ether layers were washed with water, dried over anhydrous magnesium sulfate and then filtered. After removal of the solvent by rotary evaporation, the product was recovered by fractional distillation at 65 °C (1.0 mbar) which afforded 2.74 g (9.35 mmol, 22 %) of a colorless liquid.
FT-IR (KBr window): ν = 3053(aromatic C-H stretching), 2981, 2904 (aliphatic C-H stretching), 1579 (ring C=C stretching), 1387 (O-CH2 wagging vibration), 1250 (P=O stretching), 1127 (aromatic =C-H in-plane deformation vibrations), 1024 (P-O-C stretching vibration), 964 (C-H rocking), 779 (aromatic =C-H out of plain vibration), 582 (C-Br stretching) cm-1.
1
H NMR (300 MHz, CDCl3): δ = 7.67−7.55 (m, benzene, 4H), 4.15 (m, OCH2 4H), 1.29 (t,
OCH2CH3,J = 7.0 Hz, 6H) ppm.
13
C {1H} NMR (300 MHz, CDCl3): δ = 134.5, 134.3, 131.6, 131.4, 129.1, 126.3 (phenyl carbon resonance), 61.9, 61.9, 14.3, 14.3 (phosphonate carbon resonances) ppm.
3.2.2 Synthesis of diethyl 4- acetylenebenzene phosphonate
To a mixture of diethyl bromophenylphosphonate (3 g, 10.24 mmol) and trimethylsilyl acetylene (1.21 g, 12.29 mmol) in 3 mL triethylamine (Et3N) was added a solution of
53
bis(triphenylphosphine)palladium(II)dichloride, (287.2 mg, 0.41 mmol) and copper(I) iodide (39 mg, 0.205 mmol) in 3 mL Et3N. The mixture was thrice degassed and filled nitrogen to remove the residual oxygen, and then the reaction was allowed to proceed under stirring at 75 °C overnight. At the end, Et3N was evaporated under reduced pressure and the residual mixture collected in n-hexane and passed through a short column of silica gel to remove the Pd and Cu catalysts. The TMS protected compound was collected as a colorless transparent liquid after solvent evaporation. This raw product was dissolved in 10 mL methanol and added dropwise with a saturated aqueous solution of KOH (4 mol % of product). The resulting mixture was stirred at room temperature for 4 hr, and then the deionized water was added, the aqueous layer was extracted several times with methylene chloride, the combined organic layers were washed with water 3 times, dried over anhydrous magnesium sulfate and then filtered. The solvent was finally removed by rotary evaporation affording 2.14 g (9.35 mmol, 22 %) of the product as a colorless liquid.
FT-IR (KBr window): ν = 3288, 3198 (aromatic C-H stretching), 2982 (aliphatic C-H stretching), 2100 (C≡C), 1598(ring C=C stretching), 1392 (O-CH2 wagging vibration), 1247 (P=O), 1125 (aromatic =C-H in-plane deformation vibrations), 1027 (P-O-C stretching vibration), 960 (C-H rocking), 792 (aromatic =C-H out of plain vibration), 621 (acetylene C-H bending vibration) cm-1. 1
H NMR (300 MHz, CDCl3): δ= 7.80-7.54 (m, benzene, 4H), 4.12 (m, OCH2 4H), 3.20 (s, C≡CH, 1H), 1.31 (t, OCH2CH3,6H) ppm.
13
C {1H} NMR (300 MHz, CDCl3): δ = 131.9, 131.7, 131.4, 131.4, 129.8, 125.7 (phenyl ring carbon resonances), 84.6, 78.5 (acetylene carbon resonance), 61.9, 61.9, 14.3, 14.3(phosphonate carbon resonance) ppm.
3.2.3 Synthesis phosphonic acid terminated rr-P3HT
Asymmetrically allyl/Br terminated rr-P3HT (0.035 g, 8×10-6 mol) and the previously synthesized diethyl 4- acetylenebenzene phosphonate (2.28 mg, 9.6×10-6 mol) in 3mL THF was mixed with PdCl2(PPh3)2 (3.05 mg, 8×10-6 mmol) and copper(I) iodide (13 mg, 0.068 mmol) in 3 mL Et3N in a schlenk tube, then schlenk tube evacuated and filled thrice with nitrogen to eliminate the influence of oxygen. This reaction mixture was stirred at 65°C for 20 hr. At the end of the reaction, Et3N was removed under reduced pressure and the residue was poured into a large excess of methanol. The precipitated polymer was filtered, washed repeatedly to remove excess of unreacted ligand precursor and collected in chloroform. To hydrolyzed the phosphonate group in phosphonic acid, the allyl/phosphonate terminated P3HT was dissolved in 4 mL THF and trimethylsilyl chloride (1 mL) was added dropwise over 30 min. This reaction mixture was stirred at room temperature for 2 hr and followed by mild reflux at 60 °C for 4 hr. At the end of reaction, the
54
polymer was precipitated in excess of methanol and filtered then dried under vacuum to yield 40 mg of product and characterized by 1H NMR.
FT-IR (KBr window): ν = 3400 (O-H stretching), 3056 (thiophene C-H stretching), 2925, 2855 (aliphatic C-H stretching), 2676 (phosphorus acid), 2195 (C≡C stretching), 1509 (asymmetric C-H ring stretching vibrations), 1455 (symmetric C-H ring stretching vibrations), 1374 (C-H deformation sym.), 1261 (P=O), 1095, 1021, 800(aromatic =C-H out of plain vibration), 705 (P-C stretching) cm-1. 1 H NMR (300 MHz, CDCl3): δ= 0.91 (t, 3H), 1.35-1.43 (m, 6H), 1.69 (t, J = 7 Hz, 2H), 2.78 (t, J = 7 Hz, 2H), 3.49 (d, J = 7 Hz, 2H), 5.11 (m, 2H), 5.98 (m, 1H) 7.46 (s, 1H), 7.60-7.79 (m, 4H) ppm. 13 C {1H} NMR (300 MHz, CDCl3): δ = thiophene resonances at 140.1, 133.9, 133, 130.7, 129.0, 116.3, allyl and hexyl resonances at 84.66, 77.4, 31.9, 30.7, 29.7, 29.1, 23.4, 14.3 and phosphonic resonances at 134.7, 132.2, 129.2, 125.3, 85, 7 ppm.
3.3 Results and Discussion
The chain-growth polymerization of 2,5-dibromo-3-alkylthiophenes by the Grignard metathesis (GRIM) methodology has been reported before,16 and applied to modify 2,5-dibromo-3-alkylthiophenes.17 The 2,5-dibromo-3-hexylthiophene has been synthesized according to scheme 3.2.
Scheme 3.2 Synthesis of 2,5-dibromo-3-hexylthiophene
N-bromo succinimide (NBS) was used for selective bromination by using a twofold excess with
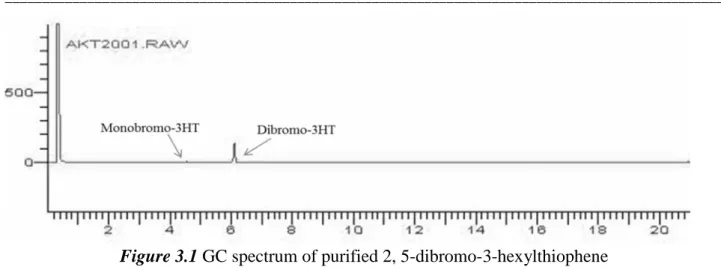
respect to 3-hexylthiophene under mild conditions. A simple distillation afforded the pure product. To obtain the allyl/Br asymmetrically-end capped P3HT, it is very important to start the polymerization with very pure 2,5-dibromo-3-hexylthiophene. The purity of the di-bromo monomer was characterized 99.99 % by GC; remaining the 0.01 % was the mono-bromo compound as shown in figure 3.1.
55
Figure 3.1 GC spectrum of purified 2, 5-dibromo-3-hexylthiophene
After purification, the di-brominated monomer was characterized by 1H NMR and FTIR. In the 1H NMR of figure 3.2, the proton signal of the 4th position of thiophene can be recognized at 6.77 ppm, and there is no residual signal for the proton at the 2nd and 5th position.
1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 f1 (ppm) 3. 45 7. 00 3. 68 2. 26 1. 00 0 .8 9 1 .2 9 1 .5 4 2 .5 0 6 .7 7 7 .2 6 S Br Br CH3 a a b b c c d e f g d,e,f g
Figure 3.2 1H NMR spectrum of 2, 5-dibromo-3-hexylthiophene
Figure 3.3, shows the FTIR spectra of 2,5-dibromo-3-hexylthiophene. The hexyl, moieties shows absorption peaks that are in close correspondence to one another. The ring stretching vibrations in the vicinity of 1520-1450 cm-1, the aromatic C-H stretching vibrations at about 3055 cm-1, and the aromatic C-H out-of-plane vibrations at 820-830 cm-1 are characteristic of 2,5-disubstituted-3-hexylthiophenes.
56 40 00 3500 30 00 2500 2 000 1500 1 000 50 0 5 0 6 0 7 0 8 0 9 0 10 0 11 0 12 0 13 0 % ( T ) W a v e n u m b er (cm-1) 2 9 5 8 2 8 6 4 1 5 3 7 1 4 2 3 1 1 7 7 1 0 0 2 8 2 0
Figure 3.3 FTIR spectrum of 2,5-dibromo-3-hexylthiophene
Obtaining a mono-capped rr-P3HT, that is a P3HT chain with only one allyl-terminated chain end, was crucial to allow further modification of the Br-terminated chain end and thus to prevent the risk of bridging in the subsequent use of the functional P3HT as a stabilizer for CdSe NPs. The bromide in monobromo terminated rr-P3HT is used as reactive functional group for further modification. In this chapter, two different functional modifications are described. In a first route, the brominated chain end was modified to attach an alkynyl group that would be used, as described in chapter 4, for the “Click” reaction with azide−terminated poly(acrylonitrile) (PAN) to obtain a P3HT-PAN block copolymer. The second route was aimed at attaching a phosphonic acid group on one P3HT chain end. Having a phosphonic acid group on only one chain end of P3HT was crucial, since this mono-phosphonic acid P3HT would be subsequently used as a colloidal stabilizer for CdSe nanoparticles; should a difunctional P3HT be present, this would likely cause in such system an unwanted bridging flocculation, as explained in chapter 6. Here, we present the synthesis of asymmetrically allyl/alkynyl and allyl/phosphonic acid terminated rr-P3HTs.
The asymmetrically allyl/Br terminated poly(3-hexylthiophene) was synthesized by GRIM methodology as shown in scheme 3.3. Here, a simplified version of the methodology by McCullough (which involves a multistep procedure) was used, resulting in regioregularity with a
57
very high degree of head to tail HT coupling.18 The mechanism of the polymerization as proposed by McCullough et al., is shown in scheme 3.4.
Scheme 3.3 Synthesis of allyl/Br terminated poly(3-hexylthiophene)
The GRIM method for the synthesis of poly(3-alkylthiophenes) is reported to result in the formation of regioregular polymers with > 98% head-to-tail couplings.19 P3HT with allyl and bromide terminations at the chain ends was prepared as shown in scheme 3.4. 2,5-Dibromo-3-hexylthiophene was synthesized according to the literature20 and was reacted with t-butyl magnesium chloride to yield 2-bromo-5-chloromagnesium-3-hexylthiophene (Scheme 3.5). The addition of Ni(dppp)Cl2 to the reaction mixture is reported to result in the formation of “living” P3HT chains capped by a highly active Ni0 group. The initiation step is the formation of Ni0(dppp) adduct with a tail-to-tail thiophene halide dimer (Scheme 3.4c) and the subsequent oxidative addition of these partners to yield a reactive polymerization center. The ‘tail-first’ insertion of monomers, through a sequence of reductive elimination and oxidative additions, results in a regioregular polymer capped at one end with a tail-to-tail irregularity. At this point, a wide range of rr-polythiophenes bearing a multiplicity of end groups can be obtained by conventional organic reaction. While the polymer was still in solution, either allyl or vinyl end-capping groups were introduced by adding the appropriate second allyl- or vinyl magnesium Grignard reagent to mono capped P3HT. According to previously reported literature,12 the alkenyl Grignard reagent are the most efficient in mono-capping the polymer whereas the others produce a high fraction of symmetrically di-capped polymer. While the reactions with alkenyl Grignard gave as much as 91% mono-capped polymer and negligible amounts of di-capped one,about 14% di-capped P3HT were unexpectedly obtained when using ethynylmagnesium Grignard agent.12
To overcome this problem, a multi-step approach was followed for the synthesis of mono-alkynyl end capped P3HT in this thesis. In particular, to prevent the formation of any symmetrically end-capped polymer, only allyl magnesium bromide was used to obtain allyl/Br asymmetrically terminated rr-P3HT. According to the proposed mechanism, only one tail-to-tail coupling should occur at the initiation stage of the regioregular polymerization of 2,5-dibromo-3-hexylthiophene. Common to chain-growth controlled polymerizations, we found that the molecular weight of P3AT
58
(Figure 3.4) could be controlled by varying the amount of added Ni(dppp)Cl2, and that polydispersities of P3ATs as low as Mw/Mn =1.12 could be obtained.
Scheme 3.4 Mechanism for the synthesis of mono capped rr-P3HT by GRIM method a
c
b
59
12.5 13.0 13.5 14.0 14.5 15.0 15.5 16.0 16.5 17.0
R etention tim e (m in)
Figure 3.4 GPC curve of different molecular weight of P3HT with respect to catalyst amount The structures of the resulting allyl/Br asymmetrically terminated regioregular poly(3-hexylthiophene) (rr-P3HT) was investigate by 1H NMR spectroscopy (Figure 3.5).
60
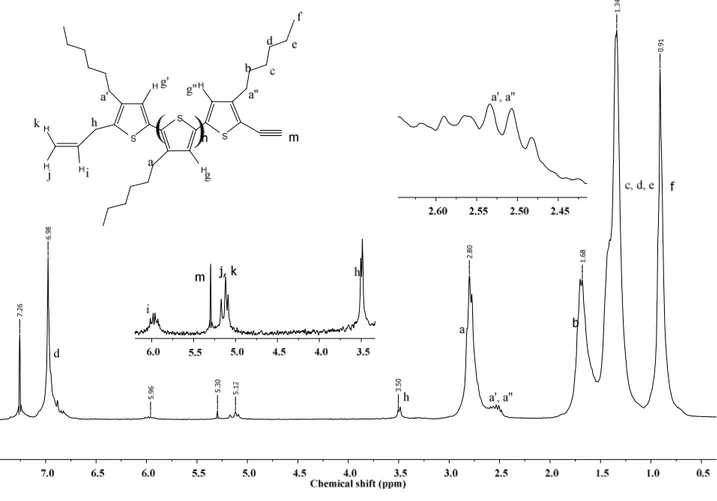
The main absorption signals of rr-P3HT are assigned as shown. Two small triplets at δ ~ 2.5 ppm of the same intensity can be assigned to the protons on the first methylene of the n-hexyl substituent (a′ and a′′) of the two end units of allyl/Br asymmetrically terminated rr-P3HT. Furthermore, the appearance of two small singlets g′ and g″ at different resonance frequencies is due to different chemical environment around g′ and g′′ (Figure 3.5). One end of the chain has an allyl group whereas the other has bromide, which influences the resonance frequency of the proton in position 4 of the thiophene ring. The allyl group at the polymer chain end can be observed by the resonance at 3.5, 5.1 and 5.9 ppm, whereas protons hk and hj show as a structured signal resulting from two overlapping double doublets at 5.1 ppm due to geminal coupling and coupling with proton hi. By NMR analysis one can distinguish between the two different types of chain end. The appearance of the two separate triplet signals at different resonance frequencies is due to different chemical environment around a′ and a′′, and this allow to confirm that allyl/Br terminated rr-P3HT contains only one structural defect per polymer chain. The recorded spectra are in full support of the scheme 3.5, showing that the first step is a tail-to-tail coupling, followed by Ni0 initiated polymerization. From the integrals of the 1H resonances of the first methylene protons (a, a′, and a″) of n-hexyl chain, the polymer was characterized as a regioregular poly(3-hexylthiophene) with at least 98 % HT linkage in the polymer chains. By assuming a well-defined asymmetrically end capped polymer structure, that is without the occurrence of the dead end termination or symmetrically end capped functions, the NMR technique allows a relatively accurate determination of the number average molecular weight from the integration of specific resonances from 3-hexylthiophene end units relative to those of internal units. For instance, DPn for the abovementioned polymer can be determined from the ratio of the integrals of resonance i to g and results in 26 monomer units, corresponding to Mn = 4497 (including end groups). Moreover, the presence of the allyl group is indicated by the resonances at 3.52, 5.11 and 5.98 ppm and n-hexyl chain resonances at 0.9, 1.35-1.43, 1.69, 2.80 and 3.49 ppm.
This allyl/Br terminated P3HT was used to prepare allyl/alkynyl terminated P3HT by the Sonogoshira coupling reaction.21 As described above, direct alkynylation with an alkynyl Grignard agent gives 86 % alkynyl mono capped rr-P3HT12 and 14 % di-capped; the latter is an undesired side product as it may come bridging flocculation where the final phosphonated polymer in used for stabilization of CdSe nanoparticles. To avoid the formation of dialkynyl-terminated polymer, the alkynyl group was attached on allyl/Br asymmetrically terminated rr-P3HT through a multi-step reaction. Firstly, allyl/Br asymmetrically terminated rr-P3HT was reacted with trimethylsilyl
61
acetylene in the presence of PdCl2(PPh3)2 and CuI catalysts. This TMS-protected polymer was deprotected by TBAF·3H2O to yield allyl/alkynyl terminated P3HT as shown in scheme 3.5.
S C6H13 Br n S C6H13 C n C Si CH3 CH3 CH3 TMS-C CH 2. PdCl2(PPh3)2 1. 3. CuI S C6H13 C n CH TBAF.3H2O THF
Scheme 3.5 Synthesis of allyl/alkynyl terminated P3HT
After TMS deprotection, the polymer was purified by soxhlet extraction with methanol and then with n-hexane to remove unreacted alkynyl precursor, collected in chloroform and dried under vacuum. The appearances of the resonance at 5.30 ppm in the 1H NMR spectrum of allyl/alkynyl terminated rr-P3HT confirmed the attachment of alkynyl group, whereas all other resonances are the same as those of the allyl/Br rr-P3HT precursor (Figure 3.6).
62
Figure 3.6 1H NMR spectrum of allyl/alkynyl asymmetrically terminatedrr-P3HT
The FTIR spectrum of allyl/alkynyl terminated rr-P3HT, shown in figure 3.7, shows the C ≡ C stretching absorption at 2100 cm-1, which confirms the presence of the alkynyl group on the chain end.
63
Figure 3.7 FTIR spectrum of allyl/alkynyl asymmetrically terminated rr-P3HT recorded on KBr window
To synthesize the allyl/phosphonic acid terminated rr-P3HT, first the phosphonic acid precursor was prepared separately. Several attempts to achieve direct attachment of phosphonate precursor of phosphonic acid derivatives onto P3HT gave very poor yields of the desired product; the separation of the final reaction mixture was very troublesome. To overcome this problem, introduction of the phosphonic acid functionality on the P3HT chain end was performed using a three-step synthesis according to scheme 3.6. In the first step, diethyl phosphonate derivative was reacted with the mono-Grignard adduct of p-dibromobenzene and the product was purified by column chromatography. The product 4-bromobenzenephosphonate had a 22% yield and the purity was characterized by GC as 99.99 %. The 1H NMR spectrum of figure 3.8 presents the CH3 and CH2 proton resonances of diethyl phosphonate at 1.36 and 4.11 ppm, respectively. The remaining bromine was then replaced with the trimethylsilyl acetylene group by using the Sonogoshira coupling reaction, which yielded 94 % of the product. The TMS protected phosphonate precursor was finally deprotected in its alkynyl side by TBAF.3H2O (product c, scheme 3.6), and the unprotected compound was attached onto allyl/Br terminated P3HT by another Sonogoshira coupling reaction. The phosphonate chain end of the resulting functional polymer was hydrolyzed into phosphonic acid by treatment with trimethylsilyl bromide.
64
65 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 f1 (ppm) 6 .0 3 4 .5 9 4 .0 0 a b c c a, b d d
Figure 3.8 1H NMR spectrum of 4-bromobenzenephosphonate
The structure of the unprotected product C was confirmed by 1H NMR (Figure 3.9). In particular, the resonance at 0.3 ppm due to TMS disappeared after deprotection and was replaced by a new peak at 3.20 ppm due to the alkynyl proton (C≡C-H). The FTIR spectrum of product C is shown in figure 3.10, in which the C≡C stretching absorption can be observed at 2100 cm-1. As already mentioned, product C was coupled to allyl/Br terminated P3HT by a second Sonogoshira coupling reaction to get allyl/ phosphonate asymmetrically terminated rr-P3HT (product D, scheme 3.7). The catalyst was removed easily by reprecipitation of a concentrated THF solution of the polymer in a large excess of methanol. In the 1H NMR spectrum of figure 3.11, the peaks at 3.51 and 1.21 ppm are associated to the CH2 and CH3 resonances, respectively, whereas the phenyl proton signals occur at 7.40 and 7.72 ppm. Hydrolysis of the phosphonate group at the chain end into phosphonic acid was carried out by reacting with trimethylsilyl bromide in THF/TEA mixed solvents (1:1). The resulting phosphonic acid terminated P3HT was purified by reprecipitation.
66 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 f1 (ppm) 6 .3 0 1 .0 0 4 .0 0 2 .5 9 1 .8 6 CH C CH CH C CH P C O O O C H CH2 CH2 CH3 CH3 a c d e a b c d e C D C l 3
Figure 3.9 1H NMR spectrum of phenyl acetylene phosphonate
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0 1 5 0 0 1 0 0 0 5 0 0 8 0 1 0 0 1 2 0 1 4 0 1 6 0 1 3 9 2 7 9 2 1 2 4 7 9 6 0 1 0 2 7 1 5 9 8 2 1 0 0 % T W a v e n u m b e r (c m- 1) 3 1 9 8
67 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 f1 (ppm) 3.5 4.0 4.5 5.0 5.5 6.0 f1 (ppm) n ai a" b c d e f g gi i j k g" a h m n m n f c,d,e a b h S S S C H3 C H3 CH3 H H H H H H P O O O CH3 CH3 p q q p h p g
[
]
j, k j, k i iFigure 3.11 1H NMR spectrum of phosphonic acid terminated P3HT
In the 13C NMR spectrum of allyl/phosphonic acid terminated rr-P3HT presented in figure 3.12, the thiophene ring resonances at 140.1, 133.9, 130.9 and 129 ppm, and these from the hexyl chain at 31.9, 30.7, 29.7, 29.1, 23.4 and 14.3 ppm can be observed. The phenylene ring carbons can be observed at 136.4, 133.2, 132.3, 131.5 and 127.8 ppm and those of the allyl group at 133, 116.3 and 32.3 ppm, respectively. Finally the resonances from C≡C linker resonances occur at 86.09 and 77.4 ppm.
68
Figure 3.12 13C NMR spectrum of allyl/phosphonic acid terminated rr-P3HT
In figures 3.13a and 3.13b are reported the FTIR spectra of phosphonate and phosphonic terminated rr-P3HT, respectively. In figure 3.13a, the successful attachment of phosphonate precursor at rr-P3HT chain end is revealed by the C≡C stretching peak at 2195cm-1. In figure 3.13b, after hydrolysis of the phosphonate group into phosphonic acid, a new broad peak at 3400 cm-1 appears due to the stretching absorption of acidic and very hygroscopic OH group.
69 4000 3500 3000 2500 2000 1500 1000 500
%
T
ra
n
s
m
it
ta
n
c
e
W avenumber (cm
-1)
S C6H1 3 C C P O O O n S C6H1 3 C C P O O H O H n 2195 2195 3400a
b
Figure 3.13 FTIR of (a) allyl/phosphonate and (b) allyl/phosphonic acid terminated rr-P3HT
In conclusion, asymmetrically end-functionalized rr-P3HT was synthesized successfully adopting multi-step procedures to obtain mono end-capped allyl/alkynyl and allyl/phosphonic acid rr-P3HT. In the following chapters will be discussed below the allyl/alkynyl terminated rr-P3HT was used for the preparation of a block copolymer by “click” reaction with an azide−terminated polyacrylonitrile, and the allyl/phosphonic acid terminated rr-P3HT for CdSe NPs stabilization through a ligand exchange procedure, respectively.
70
1
Milliron, D. J.; Alivisatos, A. P.; Pitois, C.; Edder, C.; Frechet, J. M. J. AdV. Mater. 2003, 15, 58. 2
Lee, J.; Sundar, V. C.; Heine, J. R.; Bawendi, M. G.; Jensen, K. F. AdV.Mater. 2000, 12, 1102. 3
J. Xu, J. Wang, M. Mitchell, P.Mukherjee, M. Jeffries-EL, J. W. Petrich, Z. Lin, J. Am.Chem. Soc, 2007, 129, 12828-12833
4
Arango, A. C.; Johnson, L. R.; Bliznyuk, V. N.; Schlesinger, Z.; Carter, S. A.; Ho¨rhold, H. H.
AdV. Mater. 2000, 12, 1689.
5
(a) Huynh, W. U.; Dittmer, J. J.; Libby, W. C.; Whiting, G. L.; Alivisatos, A. P. AdV. Funct.
Mater. 2003, 13, 73 (b) Liang, Z.; Dzienis, K. L.; Xu, J.; Wang, Q. AdV. Funct. Mater. 2006, 16,
542 (c) Gur, I.; Fromer, N. A.; Chen, C. P.; Kanaras, A. G.; Alivisatos, A. P. Nano Lett. 2007, 7, 409.
6
(a) Advincula, R. C. Dalton Trans. 2006, 2778 (b) Liu, J. S.; Tanaka, T.; Sivula, K.; Alivisatos, A. P.; Frechet, J. M. J. J. Am.Chem. Soc. 2004, 126, 6550.
7
(a) Skaff, H.; Sill, K.; Emrick, T. J. Am. Chem. Soc. 2004, 126, 11322. (b) Odoi, M. Y.; Hammer, N. I.; Sill, K.; Emrick, T.; Barnes, M. D. J. Am.Chem. Soc., 2006, 128, 3506.
8
Barbarella, G.; Zambianchi, M.; Bongini, A.; Antolini, L., Adv. Mater. 1994, 6, (7-8), 561. 9
(a) Wu, X. M.; Chen, T. A.; Rieke, R. D., Macromolecules 1995, 28 (6), 2101, (b) Chen, T. A.; Rieke, R. D., J. Am. Chem. Soc. 1992, 114 (25), 10087, (c) Chen, T. A.; Obrien, R. A.; Rieke, R. D., Macromolecules 1993, 26 (13), 3462, (d) Chen, T. A.; Rieke, R. D., Synth. Met. 1993, 60 (2), 175.
10
. Chen, T. A.; Wu, X. M.; Rieke, R. D., J. Am. Chem. Soc. 1995, 117, (1), 233. 11
(a) Iovu, M. C.; Sheina, E. E.; Gil, R. R.; McCullough, R. D., Macromolecules 2005, 38 (21), 8649, (b) Loewe, R. S.; Ewbank, P. C.; Liu, J. S.; Zhai, L.; McCullough, R. D., Macromolecules, 2001, 34, (13), 4324, (c) Loewe, R. S.; Khersonsky, S. M.; McCullough, R. D., Adv. Mater. 1999, 11(3), 250.
12
Malika Jeffries-El, G. Sauve, Richard D. McCullough, Adv. Mater, 2004, 16, 1017. 13
S. J. Higgins, Chem. Soc. Rev., 1997, 26, 247 14
Jordan T. Kopping and Timothy E. Patten, J. Am. Chem. Soc., 2008, 130, 5689. 15
Jinsong Liu and Richard D. McCullough, Macromolecules, 2002, 35, 9882 16
Sheina EE, Liu J, Iova MC, Laird DW, McCullough RD, Macromolecules, 2004, 37, 3526. 17
Sheina EE, Khersonsky SM, Jones EG, McCullough RD, Chem Mater, 2005, 17, 3317. 18
Koeckelberghs G, Vangheluwe M, Samyn C, Persoons A , Verbiest T, Macromolecules, 2005, 38, 5554.
71
19
R. D. McCullough, S. Tristram-Nagle, S. P. Williams, R. D. Lowe, M. Jayaraman, J. Am. Chem.
Soc., 1993, 115 (11), 4910–4911.
20
O. S. Oluwafemi and N. Revaprasadu, New J. Chem., 2008, 10, 1432–1437. 21