How does newness come into world? How is it born? Of what fusions, translations, conjoinings is it made? How does it survive, extreme and dangerous as it is? What compromises, what deals, what betrayals of its secret nature must it take to stave o� the wrecking crew, the exterminating angel, the guillotine?
Is birth always a fall?
Do angels have wings? Can men fly?
— S����� R������, �e Satanic Verses In 1929, Paul Adrien Maurice Dirac, one of the founding fathers of quantum mechanics, stated [1]:
‘‘�e general theory of quantum mechanics is now almost complete, the im-perfections that still remain being in connection with the exact fitting in of the theory with relativity ideas. �ese give rise to di�culties only when high-speed particles are involved, and are therefore of no importance in the consideration of atomic and molecular structure and ordinary chemical reactions, in which it is, indeed usually su�ciently accurate if one neglects relativity variation of mass with velocity and assumes only Coulomb forces between the various electrons and atomic nuclei. �e underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the di�culty is only that the exact application of these laws leads to equations that are too complicated to be soluble. It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of the main features of complex atomic systems without too much computation.’’
�ese words have become somehow an article of faith for modern quantum chemistry. E�orts in the development of theory, algorithms and techniques have made computational quantum chemistry a viable alternative to both theory and experiment. Fi�y years ago this branch was of limited or no use in the understanding of chemical phenomena: it was based on rather approximate models and its predictions could, at best, be qualitative. A giant leap was made possible both by the theoretical refinement of these models and by the evolution of computer hardware. Nowadays, arbitrarily accurate calculations can be made on small systems; whereas a careful choice of approximations, extends the possibilities of computational chemistry towards larger and larger systems, such as proteins. Reaction energies may be evaluated within few kilojoules per mole, spectral data within a few reciprocal centimeters.
�e theory is not, however, free from intrinsic limitations. Some of Dirac’s statements in the quote above have been disproved [2].
In 1905 Albert Einstein publishes a seminal paper containing the ideas of the theory of Special Relativity [3]. �e theory is rooted on two simple assumptions:
P�������� 1: all inertial frames are equivalent;
P�������� 2: Maxwell’s equations have the same form in all inertial frames,
leading to the well-known formulas for the length contraction, time dilation and velocity-dependence of the mass of a body:
l= l0 � 1−v2 c2 t= t0 � 1−v2 c2 m= �m0 1−v2 c2 .
Einstein’s theory of relativity and quantum mechanics are the two great developments of 20th century physics. Dirac’s main contribution was the merging of these two aspects, in the famous equation that bears his name.
Trusting Dirac’s words, most of the successful development in quantum chemistry has been based on nonrelativistic quantum mechanics, although, from a theoretical point of view, it would be more correct to base quantum chemistry on relativistic quantum mechanics. But from the point of view of a chemist, the question arises spontaneously: relativity, why bother? As relativistic corrections depend on the velocity one would tend to think that chemical bonding and structure would be una�ected: valence electrons have small kinetic energies. A�er all, most chemical phenomena take place at energies well below the relativistic domain.¹
(Un)fortunately, this is not completely true. It became clear fairly early in the history of theoretical chemistry, that a nonrelativistic theory could not explain certain trends in observed properties. As recently reviewed by Pyykkö [4], relativistic e�ects may play a prominent role, especially in Inorganic Chemistry. Periodic trends, such as the increase in atomic dimensions and ionization potentials, are not observed for the heaviest elements of the Periodic Table.
Consider the metal-carbon bond length experimentally determined in group 12 dimethyl compounds. An increase in bond length from Zn to Cd is found, but in going from Cd to Hg one observes a decrease in bond length. �e expected trend is a monotonic increase from Zn to Hg; Rao et al. [5], who first determined the bond lengths, concluded that ‘‘this anomalous magnitude of bond lengths for the Cd and Hg compounds is not understood.’’ �e decrease in bond length can be explained in terms of relativistic e�ects.
Another, similar, example of relativistic e�ects on chemical bonding is given by the series of the coinage metals hydrides, CuH, AgH and AuH; which has been repeatedly studied in the literature. Nonrelativistic Hartree–Fock calculations on AuH predict a bond length of183pm, longer than that of AgH. Relativistic calculations predict instead a contraction to157pm, which is shorter than that of AgH and in line with the experimental bond length (152pm) and trend.
1 �e rest energy of an electron (mec2) is half a mega electronvolt, while most chemical processes occur in an
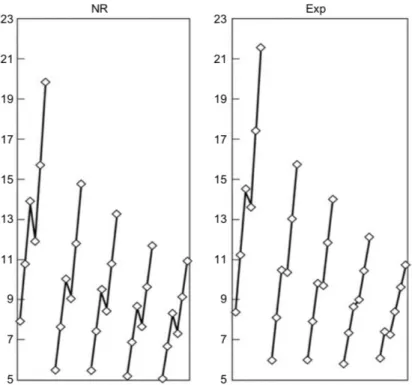
Figure 0.1. Ionization potentials in eV of the p block elements as a function of occupation number for each principal quantum number from n = 2 to n = 6 (le� to right, respectively, in each panel). Each � represents an occupation of the p-shell; e. g. p1the lower one, p6the upper
one. In the le� panel, nonrelativistic results applying Koopmans’ theorem. Experimental results in the right panel. Reproduced from ref. 7.
In Figure 0.1 we report the trends in the ionization potentials of thep-block elements. �e le� panel displays the nonrelativistic results obtained from Koopmans’ theorem [6], in the right panel the observed experimental trend is reported.
From nonrelativistic theory, one would expect a monotonic increase in ionization potential in going from ap1to ap3valence configuration. �is is due to an increase in nuclear charge only partially screened by the other valence electrons. Adding one more electron (p4) results
in a decrease in ionization potential, as pairing electrons causes a loss in exchange energy. Continuing to fill thep-shell again should result in a similar increase as in the beginning of the series.
Apart from the numerical discrepancies, this is indeed the trend observed whennis2, 3and4. Forn = 5there is a marked reduction in the di�erence between thep2 and p3 ionization potentials, while the trend still holds. For the heaviest congeners, the calculated trend is, however, completely wrong. Due to the spin–orbit splitting of thepshell, a decrease in ionization potential is observed in going from ap2to ap3configuration, while there is an increase in going fromp3top4!
As a final example, let us mention the case of gold. Why is gold yellow? Comparisons be-tween relativistic and nonrelativistic calculations were available since the 1960s, showing that
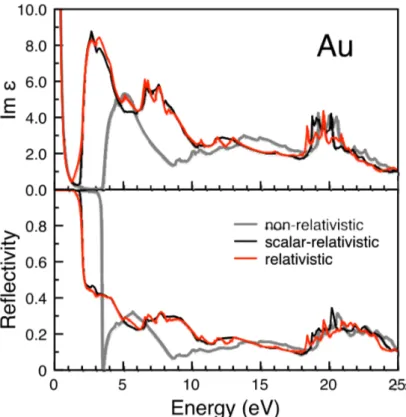
Figure 0.2. Calculated nonrelativistic, scalar relativistic and relativistic dielectric constants for bulk metallic gold. Note the (gray-to-red) relativistic shi� from 3.5 to 2 eV in both curves. �e upper and lower panels give the imaginary and real parts, respectively. Figure reprinted from ref. 8.
the excitation energies from the top of the 5d band to the Fermi level, in the half-filled 6s band, lie in the middle of the visible energy range when relativistic e�ects were included. Without relativistic e�ects, that excitation energy would be much larger, in the UV. Later and recent calculations all confirmed this theoretical observation [8]. As seen in Figure 0.2, a maximum in the optical absorption, near2eV (visible range), is well reproduced. A nonrelativistic calcu-lation, moves the absorption to approximately3.6eV, in the UV. �us nonrelativistic gold is white, like silver, and the yellow color of gold indeed comes from relativity.
�ese examples should convince the reader of the necessity of including relativity into quantum chemical calculations. Whereas the standard methods of nonrelativistic quantum chemistry are widely spread and used also by non-specialists, the applications of relativistic quantum chemistry are still confined among a restricted group of specialists. As will be clear from subsequent discussions in the main body of this thesis, the theory of relativistic quantum chemistry is by far more complicated and involved than its nonrelativistic counterpart. Even if a good number of computational codes is nowadays available to the community, the di�erent theoretical background on which they are based hampers their use as computational blackboxes.
A similar situation in encountered when quantum chemistry deals with the description of complex systems, such as solutions or proteins. Unlike relativity, the inclusion of the environ-ment in the description of a chemical system has been considered from the early stages in the evolution of quantum chemistry. Already in 1936 Onsager [9] had proposed a model to account for the presence of a liquid on point-like electric moments.
As the development of our knowledge of solutions reflects to some extent the development of chemistry itself, this interest is very well justified. Despite the large number of reactions known to happen in the solid state and in gas phase, we can state that almost always chemistry happens in solution [10].
�e theoretical treatment of environment e�ects su�ers from a dimensionality problem, since even the most simplified picture of the system under consideration would require taking into account 500–1000 atoms, at least. Application of the most accurate methods in quantum chemistry is therefore impossible and not even desirable: as always in problems with a high dimensionality, the microscopic detail in the physical description cannot account for the macroscopic behaviour of the system. �is is an important point that will be discussed in greater detail later. Models must be devised to overcome the dimensionality ‘‘disease’’: the ability to account, at least qualitatively, for environment e�ects is rather fundamental in all experimental branches of chemistry.
�is thesis is a tentative attempt to join these two worlds: relativistic molecular electronic-structure theory and the inclusion of the environment e�ects relevant to chemistry.
On the one hand, we will present the theoretical development of the Polarizable Continuum Model [11], a continuum model for solvation, in the framework of 4-component relativistic molecular electronic-structure calculations; a limited region of the great realm called Rel-ativistic Quantum Chemistry [7, 12]. On the other hand, the computational development undertaken to put the ideas and the theory in practice will be exposed in detail. At the best of our knowledge, this is the first attempt ever made to consider environment e�ects in relativistic molecular electronic-structure 4-component calculations.
�e thesis is divided in three Parts:
I the first Part reviews the theory. Chapter 1 reviews the basic ideas of Relativistic Quan-tum Chemistry, with particular emphasis on 4-component methodology. In Chapter 2 Continuum Solvation Models, and the Polarizable Continuum Model in particular, are presented. �e discussion of the theoretical details of the PCM continues in Chapter 3, where the coupling of a quantum description of the solute with the continuum is analyzed in detail. �e Chapters 3 and 4 constitute the core of this thesis: Chapter 3 describes the relativistic formulation of SCF theory, while in Chapter 4 the Linear Response function will be derived.
II in the second Part the implementation in the relativistic codeDIRAC[13] will be
docu-mented. In particular, Chapter 5 will present the principles of the modular programming paradigm adopted in this implementation of the PCM, giving details about the recently developedPCMSolvermodule [14]. A detailed analysis of the algorithm and of the ad
testing procedures adopted for the validation of the new functionality. Some preliminary results obtained on a family of simple chemical species, namely the Group 16 dihydrides, are presented in Chapter 7. Relativistic and nonrelativistic results, both in vacuo and in solvent, for geometries, dipole moments and electric dipole polarizabilities are compared. III in the last Part, the reader will find a large number of the most technical details. A discussion of the mathematical machinery behind the formulation of the Polarizable Continuum Model is given in Appendix A. Details about Second Quantization and mean-field theory are given in Appendix B.
A number of notations and typographic conventions has been adopted in order to maintain consistency throughout. We summarize them here:
A, D, . . . integral operators over a closed subset ofR2 H, Φ, . . . first or second quantizedN-electron operators
r, r�, . . . position vectors inR3
s, s�, . . . position vectors in a closed subset ofR2 v, q, . . . vectors in an arbitrary vector space K, F, . . . matrices in an arbitrary vector space
0N, IN the zero and the identity in anN-dimensional vector space
Summation over repeated indices, both discrete and continuous, is always to be understood: fr = Cκrgκ≡ � κ Cκrgκ, f(r) = U(r, r�)g(r�)≡ � dr�U(r, r�)g(r�).
Further conventions have been adopted for the indices of functions. Lower case Latin letters are used for MO 4-spinors, while lower case Greek letters (κ, λ, µ, . . .) are reserved for one-electron basis functions in 2-spinor or scalar form. Specific ranges of letters are used as follows:
r, s, t, . . . general MO 4-spinor indices i, j, k, . . . occupied MO 4-spinor indices a, b, c, . . . virtual MO 4-spinor indices
Upper case Latin letters always refer to functions in vector spaces other than the two listed above. Complex conjugation will always be shown using a dagger (†) instead of a star (∗).
SI-based atomic units have been used [15]: me= e = �h = 1
4πε0 = 1
the unit of length is the Bohra0, while that of energy is the HartreeEh. �e speed of light is
then:
c= 137.035 999 074 a0Eh�h−1
Some useful conversion constants to SI units are here listed [16]: 1 a0 = 0.529 177 210 92Å= 5.291 772 109 2× 10−11m
1 Eh= 27.211 385 05eV= 2625.499 640 4kJ mol−1 1D= 3.335 641 0× 10−30C m