1
UNIVERSITA’ DEGLI STUDI DI PISA
Facoltà di Farmacia
Corso di laurea specialistica in Farmacia
Tesi di laurea
NUOVI FARMACI NELLA TERAPIA ANTICOAGULANTE
ORALE:
ATTUALITA’ E PROSPETTIVE
Candidato: Relatore:
F. DONDOLINI
M.C BRESCHI
2
1.PARTE GENERALE
1.1 Introduzione
La terapia anticoagulante orale (TAO) costituisce un trattamento di fondamentale importanza nella cura e nella prevenzione delle malattie cardiovascolari e di quelle a rischio tromboembolico in particolare. I pazienti sottoposti a questa terapia sono estremamente numerosi in Italia come in tutto il mondo e tendono ad aumentare costantemente.
Lo scopo principale della terapia è quello di ridurre il potere coagulativo del sangue, in modo da renderlo più “fluido”. Ciò permette di evitare la formazione di coaguli e protegge il paziente da eventi tromboembolici, quali infarto al miocardio, ictus, embolie arteriose periferiche, trombosi venose ed embolie polmonari.
Al fine di minimizzare il rischio emorragico, la riduzione della coagulabilità del sangue deve avvenire in modo controllato e reversibile. Per ottimizzare l’efficacia e garantire contemporaneamente la sicurezza della terapia i pazienti in trattamento devono essere controllati periodicamente dal punto di vista clinico e laboratoristico.
1.2 La malattia trombo embolica venosa
La malattia trombo embolica venosa o tromboembolismo venoso (TEV) è una delle patologie più comuni del sistema circolatorio. Si tratta di una condizione anatomo-clinica costituita da una patologia trombotica a carico del circolo venoso profondo degli arti inferiori (e/o del piccolo bacino) associata o meno ad embolia polmonare. La reale incidenza delle suddetta malattia nella popolazione generale è difficile da determinare perché la maggior parte delle informazioni riguarda pazienti ospedalizzati. Nei paesi occidentali si calcola sia la terza malattia cardiovascolare più comune dopo la cardiopatia ischemica e l’ictus, con un caso ogni 1000 abitanti. Spesso è clinicamente silente e la morte improvvisa per embolia polmonare è la prima ed unica manifestazione. E’ una malattia legata anche all’età, per cui l’invecchiamento della popolazione è destinato ad incrementare nei prossimi anni il
3
numero di casi di tromboembolismo venoso. Le manifestazioni di questa malattia sono la trombosi venosa profonda e l’embolia polmonare.
1.2.1 La trombosi venosa profonda (TVP)
La trombosi venosa profonda (TVP) si manifesta quando all’interno di una vena profonda, generalmente del polpaccio, si forma un trombo, cioè un coagulo di sangue. Questa condizione è estremamente pericolosa perché può frequentemente portare ad embolia polmonare (EP) causata dal distacco del trombo che raggiunge i polmoni, ostacola la circolazione e può causare la morte. Nella grande maggioranza dei casi infatti, l’embolia polmonare è causata dalla trombosi venosa profonda. I trombi venosi sono costituiti in prevalenza da fibrina e globuli rossi con una quota variabile di piastrine e leucociti. La loro più frequente sede di insorgenza sono i sinusoidi venosi o le tasche valvolari (lo spazio fra la cuspide della valvola e la parete vascolare) del polpaccio, ma è possibile anche l’insorgenza nelle vene profonde della coscia o in distretti venosi esposti a traumi diretti.
Ancora oggi si possono ritenere fondamentalmente validi i criteri patogenetici della TVP formulati da Virchow circa 150 anni fa. Virchow sostiene che la formazione di un trombo ha cause riconducibili essenzialmente a tre alterazioni predisponenti principali, descritte dalla cosiddetta triade di Virchow: l’alterazione della parete vasale, l’alterazione del flusso e l’ipercoagulabilità ematica (Virchow 1856). L’ipotesi più largamente accreditata però considera il trombo venoso come il risultato della generazione locale di fibrina in aree di flusso rallentato, mentre la responsabilità patogenetica del danno parietale non è del tutto convincente. Il ruolo della ipercoagulabilità del sangue è riconoscibile nella quasi totalità delle situazioni a rischio di trombosi venosa profonda, sia congenite che acquisite, sottoforma di una alterazione dei meccanismi di regolazione della trombogenesi o di un aumento in circolo di fattori attivati della coagulazione.
Esiste una inequivocabile dimostrazione che pazienti affetti da deficienze o anomalie di alcuni inibitori fisiologici della coagulazione (difetti di antitrombina, proteina C e/o proteina S, resistenza alla proteina C attivata) sono esposti ad un elevato rischio di trombosi, soprattutto nel versante venoso.
4
Una condizione clinica ad alto rischio è data dagli interventi di chirurgia ortopedica maggiore quali protesi d’anca, frattura d’anca, protesi di ginocchio. L’incidenza di TVP e di EP è correlata al tipo di intervento chirurgico o di trauma e, a parità di altre condizioni, è ulteriormente aumentata dall’età del paziente, dalla durata dell’intervento, dalla durata dell’immobilizzazione postoperatoria, dal subentrare di eventuali infezioni postoperatorie e dalla eventuale concomitanza di altri fattori di rischio. Il trattamento preventivo tradizionale con antagonisti della vitamina K ed eparine a basso peso molecolare (EBPM) abbassano sensibilmente l’incidenza di TVP, ma il rischio rimane pur sempre elevato per una patologia che potrebbe essere prevenuta. Per questo i ricercatori hanno messo a punto nuovi anticoagulanti orali di sintesi, più efficaci rispetto alle EBPM e con maggiore tollerabilità, in modo tale da ridurre sensibilmente il rischio di emorragie e addirittura dimezzare il rischio di trombosi venosa nei pazienti sottoposti a chirurgia ortopedica maggiore.
I fattori di rischio per la TVP sono numerosi e comprendono: - Sovrappeso
- Familiarità - Età avanzata
- Ricoveri ospedalieri
- Immobilità prolungata: è certamente uno dei più importanti fattori di rischio per la trombosi venosa ed è infatti la condizione che più comunemente precede un evento tromboembolico. L’incidenza di TVP e/o EP infatti è nettamente superiore nei soggetti confinati a letto per lunghi periodi di tempo. I pazienti operati rimangono a rischio durante tutto il periodo dell’immobilizzazione: infatti eventi trombotici post operatori tardivi non sono prevenuti neppure in coloro che hanno ricevuto una corretta profilassi nelle prima settimana postoperatoria se rimangono allettati o immobilizzati oltre tale periodo di tempo
- Per le donne, sono importanti fattori di rischio l’assunzione della pillola anticoncezionale e la gravidanza (in particolar modo il secondo trimestre e il puerperio). Il rischio trombotico in gravidanza deve essere separato in due fasi distinte, il periodo ante-partum e quello post-partum. I rischi della prima
5
condizione riflettono quelli associati con le modificazioni fisiologiche della gravidanza (ingrandimento dell’utero, che predispone alla stasi venosa; elevazione dei livelli di numerosi fattori della coagulazione e del fibrinopeptide A). Tramite attente osservazioni però si è visto che l’incidenza di trombosi venosa nel periodo ante-partum non differisce più di tanto da quella di donne in età fertile. Il rischio di complicanze tromboemboliche invece appare notevolmente più elevato nel periodo post-partum.
La trombogenicità dei preparati a basso contenuto di estrogeni comunemente impiegati per la contraccezione o per la terapia sostitutiva post-menopausale è tuttora oggetto di dibattito. Recenti indagini dimostrano che un ruolo cruciale è probabilmente svolto dalla componente progestinica associata agli estrogeni: a parità di dosaggio di questi ultimi, infatti, i contraccettivi contenenti progestinici di terza generazione sono associati ad un rischio di complicanze tromboemboliche venose nettamente superiore a quello dei contraccettivi a base di progestinici delle generazioni precedenti (WHO, 1995)
- Interventi chirurgici e traumi
- Pazienti oltre i 40 anni che subiscono un intervento di chirurgia addominale - Pazienti sottoposti a protesi all’anca
- Politraumatizzati (incidenti stradali) - Pazienti con fratture agli arti inferiori
- Pazienti sottoposti ad interventi chirurgici di ortopedia
- La presenza di altri eventi cardiovascolari gravi quali ictus cerebrale e infarto Un’elevata incidenza di malattia tromboembolica venosa si registra in pazienti con scompenso cardiaco congestizio o con infarto miocardico acuto. Il rischio è particolarmente alto quando le due condizioni sono associate. Recenti indagini hanno dimostrato che l’insufficienza cardiaca rappresenta un fattore associato indipendentemente con lo sviluppo di TVP in pazienti ambulatoriali (Cogo et al., 1994)
- Problemi di stasi del sangue, che tende a non scorrere normalmente - Ipercoagulabilità (anomalie dei fattori di coagulazione)
6
- Neoplasie maligne. Studi recenti hanno dimostrato che i pazienti con neoplasie sono particolarmente esposti al rischio di complicanze tromboemboliche durante o immediatamente dopo l’esecuzione dei cicli di polichemioterapia. Tra le ipotesi che sono state formulate per spiegare il meccanismo delle trombosi da chemioterapia figurano la tossicità diretta di alcuni chemioterapici per i vasi sanguigni, la riduzione dei livelli degli inibitori circolanti e la riduzione dell’attività fibrinolitica. La frequente associazione tra neoplasie maligne e malattia tromboembolica venosa, ha spinto i ricercatori ad indagare sull’eventuale predittività di eventi tromboembolici venosi ai fini dello sviluppo di neoplasie in pazienti che apparentemente non ne sono portatori al tempo dell’osservazione. Tale associazione ha risultati contraddittori ma ha avuto recentemente una potente conferma (Prandoni et al., 1992)
In questa patologia occorre distinguere tre differenti categorie di pazienti:
- Pazienti con il sospetto del primo episodio di TVP: essi giungono all’osservazione con il sospetto clinico del primo episodio di TVP. In circa il 90% di essi la trombosi coinvolge già i distretti prossimali al tempo della diagnosi. I trombi sono per lo più estesi e comportano un alto rischio di embolia polmonare
- Pazienti con il sospetto di recidiva di TVP: si tratta di coloro che, a seguito di un episodio di TVP, giungono all’osservazione con il sospetto di recidiva flebo trombotica. La scelta del metodo di diagnosi adatto a riconoscere i pazienti con recidiva di TVP è difficile, perché in molti casi i test di diagnosi persistono alterati a seguito del primo episodio, precludendo la possibilità di identificare un nuovo episodio
- Pazienti asintomatici ad alto rischio di TVP: questo gruppo riguarda i candidati ad intervento chirurgico maggiore e coloro che restano immobilizzati per lungo tempo. Nonostante una profilassi adeguata infatti, dal 10 al 25% di questi pazienti sviluppano un episodio di TVP. I trombi in questo caso sono abitualmente piccoli e non occlusivi
7
I metodi di diagnosi dovrebbero quindi identificare i trombi, anche se piccoli, e dovrebbero essere facili da eseguire e ben tollerati. Purtroppo però la diagnosi clinica della TVP è priva di specificità per cui le più comuni manifestazioni cliniche, quali dolore, tensione, edema, crampi notturni, rossore, aumento di temperatura cutanea e formazione di cordone venoso palpabile, costituiscono l’indicazione per l’esecuzione dei test obiettivi.
La diagnosi strumentale consiste nella flebografia degli arti inferiori, ritenuta l’unica metodica in grado di identificare con accuratezza la trombosi isolata delle vene muscolari del polpaccio, dove ha quasi sempre insorgenza la TVP. Essa è tuttavia una metodica invasiva, a volte mal tollerata dal paziente e con potenziali complicazioni.
Anche l’ecotomografia real-time è considerato un approccio collaudato e praticabile ai pazienti con sospetta TVP e consente la rapida identificazione dei casi in cui la trombosi venosa interessa il distretto prossimale (coinvolgente cioè la vena poplitea e/o vene profonde più prossimali).
Anche il color doppler è una metodica diagnostica di più recente introduzione. In questo caso la possibilità di visualizzare più facilmente grazie al colore, le strutture vascolari, consente di esaminare con maggiore accuratezza anche le vene della gamba.
1.2.2 Tromboembolia polmonare (EP)
La diagnosi dell’embolia polmonare rimane ancora oggi uno dei problemi clinici più comuni e al tempo stesso difficile da risolvere. La mortalità per embolia polmonare rimane molto elevata in assenza di un adeguato trattamento. Data la natura della EP e le differenti modalità cliniche di presentazione, la diagnosi richiede un alto indice di sospetto e l’avvio dell’appropriato iter diagnostico per consentire una sicura definizione. In circa il 90-95% dei casi gli emboli polmonari originano da una TVP degli arti inferiori o del bacino.
La sintomatologia della EP è molto variabile ed aspecifica, essendo correlata sia con l’entità delle alterazioni emodinamiche, a loro volta dipendenti dal numero e dal
8
volume degli emboli, sia con le preesistenti condizioni dell’apparato cardiorespiratorio. Nei soggetti senza precedenti affezioni cardiache o polmonari gli emboli di piccole dimensioni sono nella maggior parte dei casi clinicamente del tutto silenti. L’inizio delle sintomatologia è rappresentato per lo più da dispnea, dolore toracico, tachipnea e tachicardia. La brusca comparsa di questi sintomi in pazienti a rischio, che si trovano già in condizioni predisponenti alla TVP, deve prontamente far sospettare l’embolia polmonare. Nei pazienti anziani, soprattutto se affetti da insufficienza cardiorespiratoria, la diagnosi di EP è molto difficile. In alcuni casi l’embolia può manifestarsi come improvviso aggravamento delle condizioni cliniche, come aumento della dispnea, la comparsa di un’insufficienza ventricolare sinistra o di una fibrillazione atriale. La diagnosi di tale temibile affezione richiede pertanto un alto indice di sospetto e l’avvio di un appropriato iter diagnostico ogni qualvolta i rilievi clinici lo suggeriscano.
Purtroppo nessun esame di laboratorio è dotato di sensibilità e specificità sufficienti per avere valore diagnostico. A causa della ridotta perfusione polmonare, la pressione arteriosa risulta spesso diminuita nell’EP e la sua misurazione è stata perciò considerata un elemento diagnostico indiretto di grande importanza. Inoltre nei pazienti con embolia polmonare, l’elettrocardiogramma (ECG) è pressoché costantemente alterato (95% dei casi), ma le alterazioni presenti sono in larga misura aspecifiche perché osservate anche in pazienti con cardiopatie di altra natura. L’ECG è comunque un accertamento indispensabile per escludere la diagnosi di infarto miocardico. Nell’iter diagnostico possono essere utili i test strumentali che confermano la presenza di una flebo trombosi usati anche per la diagnosi della TVP. Gli emboli però possono originare anche da sedi diverse dalle vene profonde degli arti inferiori, per cui se uno dei test risulta negativo purtroppo non si può escludere l’EP. L’arteriografia polmonare è l’unico metodo in grado di identificare con sicurezza emboli polmonari anche di piccole dimensioni. Essa però non può essere impiegata come metodo di screening sia per la sua invasività che per motivi di costo, di praticità e di disponibilità. E’ pertanto necessario selezionare accuratamente i pazienti candidati a tale indagine.
9 1.2.3 Terapia della trombosi venosa profonda
Il cardine della terapia della trombosi venosa profonda è rappresentato dai farmaci antitrombotici, mentre un ruolo del tutto secondario compete ai farmaci sintomatici dell’edema, della flogosi e dello stato doloroso e febbrile.
Gli obiettivi che si prefigge il trattamento della TVP sono i seguenti:
1. Il blocco della fibrinoformazione, così da ottenere l’arresto dell’estensione del trombo, prevenendo il rischio di embolia polmonare e quello di recidive flebo trombotiche nelle settimane successive all’episodio iniziale;
2. Il ripristino della canalizzazione vascolare, così da ridurre il rischio dell’incompetenza valvolare profonda e di conseguenza l’incidenza e la gravità delle manifestazioni della sindrome post-tromboflebitica.
Il primo obiettivo è raggiunto con la terapia anticoagulante mentre il secondo è considerato appannaggio della terapia trombolitica. La prevenzione delle recidive è ottenuta da un uso congruo dell’eparina e dei farmaci anticoagulanti, che pertanto rivestono un ruolo cruciale per il raggiungimento di tutti gli obiettivi terapeutici delle TVP.
1.2.4 Terapia dell’embolia polmonare
Le finalità che si prefigge la terapia dell’EP sono le seguenti:
1. Arresto immediato della fibrinoformazione e prevenzione delle recidive emboliche: ciò si ottiene con l’eparina e con gli anticoagulanti
2. Conservazione delle funzioni primarie dell’organismo: a tale scopo sono utili l’ossigenoterapia, l’infusione di liquidi, farmaci cardioattivi, vasopressori ed antiaritmici; in alcuni casi solo l’embolectomia d’urgenza può salvare la vita ad un paziente
3. Rapida lisi dei tromboemboli in pazienti selezionati, con il duplice intento di migliorare l’emodinamica circolatoria in fase acuta e di proteggere dallo sviluppo di danni funzionali respiratori a lungo termine: ciò si ottiene con i farmaci trombolitici
10 1.3 Fibrillazione atriale (FA)
In certe condizioni il muscolo cardiaco si contrae in maniera irregolare e assolutamente inefficace per la propulsione del sangue. Questa aritmia è denominata fibrillazione e può interessare sia gli atri sia i ventricoli.
La fibrillazione atriale è l’aritmia cardiaca più diffusa nella popolazione generale, una persona su 4 la sviluppa prima o poi nel corso della vita e in Italia i pazienti sono circa un milione. Le probabilità di sviluppare tale condizione aumentano con l’avanzare dell’età. La fibrillazione atriale consiste in un’alterazione del ritmo cardiaco che origina nelle camere cardiache superiori, gli atri, impedendo loro di funzionare correttamente (disorganizzazione dell’attività atriale). In tali circostanze gli atri non sono più in grado di espellere tutto il sangue, che rimarrà in parte all’interno delle camere con il rischio di formazione di coaguli, uno dei pericoli più gravi per chi soffre di fibrillazione atriale. I coaguli di sangue infatti, se non vengono “sciolti”, possono trasferirsi nel letto vascolare polmonare o sistemico; possono andare ad occludere i vasi cerebrali provocando un ictus, una delle complicanze più temibili e frequenti di questa aritmia. Si constata che la fibrillazione atriale aumenta fino a 3-5 volte il rischio di incorrere in attacchi ischemici transitori (TIA) o ictus cerebrale, inoltre complica malattie dell’apparato cardiovascolare avviandole verso lo scompenso cardiaco con relativo incremento di mortalità. Per questo è importante diagnosticarla correttamente, e dove possibile curarla.
Nel normale ritmo cardiaco, l’impulso generato dal nodo seno atriale (centralina elettrica del cuore), causa la contrazione del muscolo cardiaco e permette la circolazione del sangue. Nella fibrillazione atriale invece, si attivano più impulsi elettrici negli atri, dando origine a contrazioni disorganizzate e frammentarie che a loro volta si trasmettono in modo irregolare ai ventricoli determinando il battito irregolare e accelerato. L’attività elettrica degli atri completamente disorganizzata non corrisponde ad un’attività meccanica efficace. Le onde di depolarizzazione atriale, o onde f, sono di piccola ampiezza e hanno una frequenza molto elevata. In queste condizioni il nodo atrioventricolare riceve dall’atrio molti più impulsi di quanti sia in grado di condurne, esercitando quindi una funzione di filtro che trasmette ai ventricoli un numero di battiti non eccessivamente elevati: numerosi
11
impulsi penetrano, infatti, solo parzialmente nel nodo atrioventricolare e si bloccano al suo interno. Questa variabilità della conduzione atrioventricolare fa sì che i ventricoli si contraggano in maniera irregolare. Pazienti con sintomi sospetti, come palpitazioni irregolari, possono essere sottoposti ad un’attività di monitoraggio del ritmo, come ad esempio l’esame holter al fine di documentare l’aritmia, inoltre l’elettrocardiogramma è l’esame diagnostico più usato per evidenziare la presenza di fibrillazione atriale. Gli aspetti elettrocardiograficamente salienti della FA sono la scomparsa delle onde p, che vengono rimpiazzate dalle fluttuazioni di potenziale continuate e irregolari, chiamate onde f, e l’irregolarità dei battiti.
Dal punto di vista clinico la fibrillazione atriale si suddivide in base al modo di presentazione in:
- Parossistica: quando gli episodi si presentano e si risolvono spontaneamente in un tempo inferiore ad una settimana, talvolta durano meno di 48 ore. E’ associata a sintomi transitori, di durata variabile da minuti ad ore ma che comunque sia si risolvono da soli
- Persistente: quando l’episodio aritmico dura oltre i 7 giorni e non si interrompe spontaneamente ma solo a seguito di interventi terapeutici esterni - Permanente: quando non siano ritenuti opportuni tentativi di cardioversione,
o gli interventi terapeutici si siano dimostrati inefficaci
Le cause della fibrillazione atriale possono essere molteplici e includono: - Difetti delle valvole cardiache
- Difetti cardiaci congeniti - Enfisema e altre pneumopatie
- Esposizione a sostanze stimolanti, quali ad esempio farmaci, caffeina o tabacco, o consumo di alcol
- Insufficienza cardiaca - Attacchi di cuore - Ipertensione
- Ipertiroidismo o altri squilibri metabolici - Un precedente intervento di cardiochirurgia
12
- Malattia del nodo del seno (quando il pacemaker naturale del cuore smette di funzionare correttamente)
- Apnea notturna
- Stress dovuto a polmonite, intervento chirurgico o altra malattia - Infezioni virali
Nella maggior parte dei casi la fibrillazione atriale è la conseguenza di una malattia cardiovascolare, ma può verificarsi anche in soggetti che non soffrono di alcuna cardiopatia. In tal caso si parla di fibrillazione atriale isolata.
I fattori di rischio sono quindi per lo più: la presenza di malattia cardiovascolare, inclusi coloro che hanno subito un intervento di cardiochirurgia, l’avanzare dell’età, problemi di salute cronici o una storia familiare di fibrillazione atriale.
Alcuni individui affetti da tale aritmia non mostrano alcun sintomo per cui ignorano la loro condizione fino a che questa non viene rilevata dal medico durante un esame obiettivo. Coloro che invece manifestano un quadro sintomatologico possono lamentare:
- Palpitazioni e sensazione di cuore in gola, battito cardiaco irregolare o anomalo o tuffo al cuore
- Debolezza
- Stordimento e confusione - Difficoltà respiratorie - Dolore al torace
Diagnosticare una condizione di fibrillazione atriale è importante per far si che questo problema cardiaco non contribuisca allo sviluppo di ulteriori complicanze. La diagnosi, tuttavia, può rivelarsi difficile in quanto si tratta di un evento imprevedibile e i sintomi non sono sempre evidenti. Per questo la collaborazione del paziente, nel fornire indicazioni dettagliate in merito ai sintomi, al proprio medico risulta fondamentale.
La fibrillazione atriale può essere trattata farmacologicamente o con la cardioversione o con l’ablazione chirurgica mininvasiva.
13
La terapia farmacologica prevede l’utilizzo di farmaci antiaritmici o di farmaci di controllo della frequenza ventricolare in associazione a farmaci anticoagulanti. La terapia anticoagulante deve durare almeno 3-4 settimane, inoltre in base a eventuali recidive o alla presenza di cardiopatia, si può intraprendere una profilassi farmacologica antiaritmica.
La cardioversione è utile per garantire un sollievo dai sintomi e ripristinare la funzione cardiaca nei pazienti ai primi stadi della patologia. La cardioversione può essere farmacologica o elettrica e consiste in una procedura in grado di interrompere l’aritmia con una sorta di “reset” del battito.
Nei casi di inefficacia della cardioversione, in base ai sintomi, all’età e al contesto clinico generale, si può valutare l’eventuale passaggio a metodiche terapeutiche invasive quale l’ablazione.
L’ablazione del nodo atrio-ventricolare e l’impianto di un pacemaker permanente elimina l’irregolare contrazione ventricolare associata alla patologia. Questo trattamento è riservato ai pazienti altamente sintomatici.
Le procedure di ablazione con catetere trans venoso hanno lo scopo di bruciare piccole aree nell’atrio, distruggendo le cellule miocardiche che contribuiscono all’aritmia. Si creano lunghe lesioni lineari per prevenire i circuiti di rientro degli impulsi elettrici associati con la fibrillazione atriale.
Le procedure chirurgiche sono un’opzione efficace per il trattamento di questa patologia. L’obiettivo chirurgico è isolare o interrompere i circuiti di rientro responsabili della fibrillazione atriale creando lesioni lineari nell’atrio, preservando la funzione atriale.
1.4 Emostasi
L’emostasi consiste in una serie di reazioni biochimiche e cellulari sequenziali e sinergiche, che hanno lo scopo di impedire la perdita di sangue dai vasi. E’ un meccanismo di difesa, finalizzato al mantenimento dell’integrità dei vasi sanguigni e della fluidità del sangue. Ogni qualvolta un vaso sanguigno viene leso o reciso, l’emostasi viene messa in atto per mezzo di alcuni meccanismi comprendenti la
14
vasocostrizione o spasmo vascolare, l’aggregazione piastrinica, la coagulazione del sangue e lo sviluppo di tessuto fibroso in seno al coagulo sanguigno per chiudere definitivamente l’apertura del vaso.
Figura 1. http://e-learning.med.unifi.it/ “Fasi del processo emostatico”
- Vasocostrizione: Non appena un vaso sanguigno si rompe la sua parete si contrae, riducendo così immediatamente il lume vascolare, il flusso e la fuoriuscita di sangue. La contrazione deriva da riflessi nervosi, spasmo miogeno locale, e fattori umorali locali liberati dai tessuti traumatizzati e dalle piastrine del sangue. Anche i vasi adiacenti sono coinvolti nel meccanismo di vasocostrizione. Per i vasi più piccoli, le piastrine sono responsabili di gran parte della vasocostrizione, liberando la sostanza vasocostrittrice Trombossano A2. Quanto maggiore è il trauma subito, tanto più intenso è lo spasmo. Ciò vuol dire che, in genere, un vaso reciso nettamente tende a sanguinare molto di più di un vaso rotto in seguito a schiacciamento traumatico. Lo spasmo vascolare locale dura parecchi minuti o anche ore, periodo di tempo in cui i processi successivi di formazione del tappo piastrinico e della coagulazione sanguigna, hanno modo di attuarsi.
- Aggregazione piastrinica: la lesione dell’endotelio dei vasi sanguigni induce le piastrine ad aderire alla sede della lesione. A contatto della superficie vasale danneggiata, e in particolare delle fibre di collagene della parete
15
vascolare, le piastrine modificano immediatamente e drasticamente le loro caratteristiche. Iniziano a rigonfiarsi, assumono forme irregolari ed emettono prolungamenti che protrudono dalla loro superficie; liberano granuli contenenti molteplici fattori attivi, diventano adesive e si attaccano alle fibre di collagene. Cominciano a secernere notevoli quantità di adenosina difosfato (ADP) e trombossano A2, che a loro volta agiscono sulle piastrine circostanti attivandole e promuovendo l’adesione di altre piastrine. L’adesività delle piastrine così attivate fa si che esse aderiscano a quelle originariamente attivate. Perciò, la parete vascolare lesa o i tessuti extravasali provocano un ciclo di reazioni che va attivando via via un numero sempre più grande di piastrine; queste, ammassandosi, formano un tappo piastrinico. Le piastrine rilasciano anche serotonina (5-idrossitriptamina) che aumenta la vasocostrizione, e tromboplastina, che accelera la coagulazione del sangue. Se la lesione della parete in un vaso è piccola, il tappo piastrinico da solo è in grado di arrestare completamente la perdita di sangue, mentre nel caso di una grossa apertura, per arrestare l’emorragia è necessario che, oltre al tappo piastrinico, si formi un coagulo sanguigno. Il meccanismo di tamponamento da parte delle piastrine è estremamente importante per chiudere piccolissime rotture che si verificano a carico di piccoli vasi sanguigni anche numerose volte al giorno.
- Coagulazione del sangue: Se il trauma della parete vascolare è di grave entità si instaura il terzo meccanismo dell’emostasi che consiste nella formazione del coagulo sanguigno. Si tratta di un processo complesso composto dall’attivazione sequenziale di diversi fattori presenti nel sangue. Il coagulo comincia a formarsi in 15-20 secondi in caso di lesioni gravi, mentre ci vorranno 1-2 minuti per traumi di minore entità. L’inizio del processo di coagulazione è dovuto a sostanze attivatrici provenienti sia dalla parete vascolare traumatizzata, sia da piastrine e da proteine sanguigne che ad essa aderiscono. Entro 3-6 minuti dalla rottura di un vaso, se l’apertura non è troppo ampia, la lacerazione o estremità recisa, viene riempita per intero dal coagulo. Da 20 minuti ad 1 ora dopo, il coagulo si retrae determinando così
16
un ulteriore restringimento del lume vascolare ed un avvicinamento dei lembi dei piccoli vasi sanguigni sezionati. In questa fase di retrazione del coagulo le piastrine svolgono un ruolo importante.
La coagulazione del sangue deve avvenire rapidamente e restare circoscritta alla zona vasale danneggiata. Consiste in una serie di reazioni che coinvolgono alcune sostanze, dette fattori della coagulazione, identificati con un numero romano o con il nome dello scopritore, e costituiti da enzimi attivati attraverso un meccanismo proteolitico. Normalmente il sangue non coagula all’interno dei vasi per la prevalenza di sostanze anticoagulanti, ma laddove si determina la rottura di un vaso sanguigno l’attività dei pro coagulanti nella sede della lesione aumenta in misura tale da portare alla formazione del coagulo.
I prodotti proteici che sono coinvolti nell’innesco della fase plasmatica o coagulativa vengono prodotti inizialmente in forma inattivata. Nel fegato sono sintetizzati diversi fattori, come la vitamina K, essenziale per la sintesi di fattori di coagulazione di origine epatica. La fase fondamentale per la coagulazione del sangue è la conversione del fibrinogeno in fibrina da parte della trombina. Il coagulo formato da questa reazione si compone di una fitta rete di filamenti di fibrina in cui rimangono intrappolati i globuli rossi e il plasma. Le due vie di coagulazione del sangue, la via estrinseca e la via intrinseca, sia attuano attraverso meccanismi differenti, ma hanno in comune alcuni attivatori (ioni calcio e fosfolipidi) e inoltre convergono sugli eventi fondamentali finali della coagulazione, quali la trasformazione della protrombina in trombina e del fibrinogeno in fibrina.
17
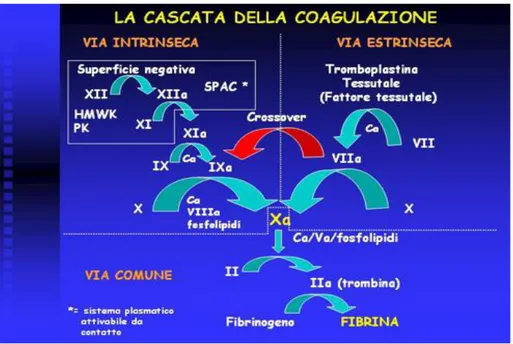
Figura 2. http://e-learning.med.unifi.it/ “La cascata della coagulazione”
Meccanismo di coagulazione estrinseco: vi partecipano fattori che non sono presenti nel sangue ma che sono rilasciati dai tessuti. Esso viene attivato dal contatto del sangue con vasi sanguigni che abbiano subìto una lesione, oppure con tessuti danneggiati. Le membrane cellulari danneggiate liberano sostanze, dette nel complesso tromboplastina tissutale (o fattore III), che innescano una serie di reazioni calcio-dipendenti, determinanti la produzione di attivatore protrombinico. Il complesso lipoproteico del fattore tissutale si unisce a costituire un nuovo complesso con il fattore VII della coagulazione, e in presenza di ioni calcio, agisce enzimaticamente sul fattore X (o fattore di Stuart), trasformandolo in fattore Xa. Il fattore Xa si unisce immediatamente con i fosfolipidi disponibili che fanno parte della tromboplastina tissutale o che vengono liberati dalle piastrine, nonché con la proaccelerina (o fattore V), formando il complesso designato come attivatore della protrombina. L’attivatore protrombinico trasforma la protrombina (fattore II: alfa-globulina prodotta dal fegato e normalmente presente nel plasma) in trombina (fattore IIa). L’azione proteolitica della trombina attiva il fattore V che diventa un ulteriore forte acceleratore dell’attivazione della protrombina. La trombina agisce come un enzima, provocando la precipitazione del fibrinogeno solubile (fattore I) in fibrina, cioè il distacco di piccole porzioni delle molecole di fibrinogeno, che, ridotte
18
di dimensioni (fibrinogeno attivato), si uniscono per formare lunghi filamenti di fibrina.
I filamenti di fibrina si ammassano sulla superficie danneggiata dei vasi sanguigni, formando un coagulo ematico, cioè una massa di consistenza gelatinosa costituita da un reticolo di fibrina , in cui rimangono incluse cellule ematiche e piastrine. Il coagulo provoca quindi l’occlusione della zona vascolare lesa. La formazione del coagulo ematico stimola la produzione di ulteriori coaguli, in quanto la trombina agisce direttamente su fattori di coagulazione diversi dal fibrinogeno e può stimolare la formazione di ulteriore trombina dalla protrombina. La formazione di ulteriori coaguli ematici si limita al sangue che defluisce dal vaso danneggiato e cessa dove il coagulo entra in contatto con il sangue circolante, grazie all’allontanamento della trombina in eccesso e all’azione dell’antitrombina, globulina presente nel sangue e nella membrana plasmatica delle cellule endoteliali, che limita la formazione della trombina (Pasqualino e Panattoni, 2007).
Meccanismo di coagulazione intrinseco: vi partecipano fattori presenti nel sangue e inizia quando quest’ultimo viene in contatto con una superficie estranea, come il collagene o il vetro. Viene dapprima attivato il fattore di Hageman (fattore XII) che, in presenza di ioni calcio, determina una cascata di eventi che coinvolgono numerosi fattori della coagulazione, provocando la formazione di un attivatore protrombinico. Simultaneamente, il trauma sul sangue altera anche le piastrine che conseguentemente liberano fosfolipidi, tra cui la lipoproteina chiamata fattore piastrinico III, che pure ha un ruolo nelle reazioni successive. Il fattore XII attivato agisce enzimaticamente e attiva il fattore XI (o fattore antecedente plasmatico della tromboplastina), Questa reazione richiede anche chininogeno ad alto peso molecolare, e viene accelerata dalla precallicreina. Il fattore XI attivato agisce successivamente con meccanismo enzimatico sul fattore IX (o fattore di Christmas), che viene così attivato a sua volta. Questa reazione richiede ioni calcio. Sempre in presenza di ioni calcio, il fattore IX attivato, agendo di concerto con il fattore VIII, con i fosfolipidi piastrinici e con il fattore III liberato dalle piastrine traumatizzate, attiva il fattore X (o fattore di Stuart). Quindi in caso di scarsa disponibilità del fattore VIII o di piastrine, questo stadio viene meno. Il fattore VIII è quello che
19
manca nei soggetti affetti da emofilia classica, viene perciò detto fattore antiemofilico. A questi passaggi segue la formazione dell’attivatore della protrombina. Questo stadio nel meccanismo intrinseco è uguale all’ultimo stadio del meccanismo estrinseco. Ossia, il fattore X attivato si unisce al fattore V ed ai fosfolipidi piastrinici o tissutali formando il complesso noto come attivatore della protrombina, che nel giro di pochi secondi, determina la scissione della trombina dalla protrombina ed avvia il processo della coagulazione, come precedentemente descritto (Pasqualino e Panattoni, 2007).
Salvo che nei primi due stadi del meccanismo intrinseco, gli ioni calcio sono necessari per lo svolgimento di tutte le reazioni, perciò in assenza di ioni calcio la coagulazione del sangue non ha luogo. Nell’organismo vivente la concentrazione di ioni calcio raramente cade a livelli tanto bassi da influire in maniera significativa sulla cinetica dell’emocoagulazione. Però il sangue prelevato da un individuo può essere reso incoagulabile riducendo la concentrazione di ioni calcio al di sotto del livello minimo richiesto per la coagulazione. Ciò si può ottenere provocando sia la deionizzazione del calcio, facendolo reagire con ioni come il citrato, sia la sua precipitazione con sostanze come lo ione ossalato.
Le piastrine svolgono un ruolo chiave nella conversione della protrombina in trombina poiché la maggior parte delle molecole di protrombina si legano ai propri recettori presenti sulla superficie delle piastrine legate al tessuto danneggiato. Questo legame accelera la trasformazione di protrombina in trombina, che avviene proprio nel tessuto dove è richiesta la formazione del coagulo. Il fattore che limita la velocità del processo della coagulazione, è di solito, proprio la formazione dell’attivatore della protrombina e non le reazioni che ad essa fanno seguito, poiché queste si svolgono assai rapidamente fino alla formazione del coagulo. La trombina agisce sul fibrinogeno (o fattore I), trasformandolo in filamenti di fibrina (o fattore Ia).
Una differenza di grande rilievo fra il meccanismo intrinseco e quello estrinseco sta nel fatto che quest’ultimo ha un carattere esplosivo; una volta avviato, la velocità di evoluzione è limitata soltanto dalla quantità di tromboplastina liberata dai tessuti traumatizzati e da quella dei fattori X, VII e V presenti nel sangue. Se il trauma
20
tissutale è grave, la coagulazione può avvenire in appena 15 secondi. Al contrario, il meccanismo intrinseco è molto più lento e richiede, di solito, da 1 a 6 minuti prima di determinare la coagulazione.
- Organizzazione fibrosa o dissoluzione del coagulo. Una volta formato il coagulo può andare incontro a due destini diversi: può essere invaso da fibroblasti che poi formano tessuto connettivo che si estende attraverso tutto il coagulo, o può subire una lisi completa. Il destino abituale del coagulo che occlude una piccola apertura di una parete vasale è l’invasione da parte di fibroblasti. Ciò è promosso almeno in parte dal fattore di crescita secreto dalle piastrine. Il fenomeno inizia entro poche ore dalla formazione del coagulo e continua fino alla completa organizzazione di quest’ultimo in tessuto fibroso nel giro di circa 1-2 settimane (Levy et al., 2006).
Quando invece si forma un grosso coagulo sanguigno, vengono attivate all’interno del coagulo stesso speciali sostanze ad attività enzimatica che provocano la sua dissoluzione. Più specificatamente, una notevole quantità di plasminogeno (o profibrinolisina) viene ad incorporarsi nel coagulo insieme ad altre plasmaproteine. I tessuti e l’endotelio vascolare lesi liberano un potente attivatore, detto attivatore tissutale del plasminogeno (t-PA), che in un giorno circa, dopo che il coagulo ha arrestato l’emorragia, finisce col trasformare il plasminogeno in plasmina (o fibrinolisina) e rimuove il coagulo. La plasmina è un enzima proteolitico che digerisce i filamenti di fibrina ed anche altre sostanze presenti nel sangue, come il fibrinogeno, il fattore V, il fattore VIII, la protrombina e il fattore XII. Pertanto, quando in un coagulo sanguigno viene a formarsi plasmina, essa è capace di indurre la lisi del coagulo oltre che la distruzione di molti dei fattori della coagulazione, per cui rende il sangue meno coagulabile. E’ possibile infine, affermare che l’eliminazione dei coaguli avviene in diverse fasi: alla formazione del coagulo fa seguito la retrazione del coagulo stesso; la retrazione provoca la spremitura dal coagulo del siero, liquido costituito essenzialmente dal plasma privo del fibrinogeno, della fibrina e di altri fattori della coagulazione. La riparazione delle pareti vasali danneggiate avviene per produzione, all’interno del coagulo di tessuto connettivo
21
fibroso da parte dei fibroblasti stimolati dal fattore di crescita di derivazione piastrinica (Platelet-Derived Growth Factor, PDGF). La dissoluzione dei coaguli, cioè la fibrinolisi, avviene in quanto il plasminogeno (profibrinolisina), proteina plasmatica adsorbita sui filamenti di fibrina, viene trasformato in plasmina dall’attivatore del plasminogeno liberato dai lisosomi delle cellule dei tessuti danneggiati. La plasmina, come enzima proteolitico, digerisce sia il fibrinogeno che i filamenti di fibrina.
La formazione spontanea di coaguli ematici all’interno di vasi non danneggiati viene fisiologicamente impedita da vari meccanismi. I filamenti di fibrina adsorbono la trombina, ostacolando il perpetuarsi di un eventuale processo di coagulazione spontaneamente iniziato; dall’altra parte, l’antitrombina, legandosi alla trombina, ne impedisce l’azione sul fibrinogeno.
L’eparina, secreta dai granulociti basofili e dai mastociti del connettivo periva scolare, esercita una potente azione anticoagulante, in quanto inibisce la formazione dell’attivatore protrombinico, previene l’azione della trombina sul fibrinogeno, favorisce l’azione dell’antitrombina e l’adsorbimento della fibrina. Le cellule secernenti eparina sono abbondanti attorno ai capillari epatici e polmonari, in cui il sangue venoso circola lentamente, favorendo l’eventuale formazione di piccoli coaguli spontanei.
L’endotelio, che riveste la parete interna dei vasi, essendo liscio e dotato di carica elettrica negativa, respinge le piastrine e numerosi fattori della coagulazione, e produce la prostaciclina (PGI2), prostaglandina inibente l’adesione delle piastrine alla superficie endoteliale.
Viceversa, la coagulazione intravasale può avvenire qualora esista un’alterazione dell’endotelio che ne renda la superficie irregolare (come avviene nell’arteriosclerosi), poiché le piastrine sono in grado di aderire a superfici ruvide. In tali casi, all’interno del vaso si forma un trombo, piccola massa solida, costituita da un accumulo di piastrine, circondato da un coagulo di fibrina con eritrociti e leucociti; il trombo può ostruire il lume vasale con gravi conseguenze per i tessuti irrorati da quel vaso sanguigno, oppure può distaccarsi parzialmente o totalmente
22
dalla parete del vaso, dando origine a un embolo, che, trascinato a distanza dalla corrente ematica, provoca fenomeni occlusivi in altri distretti vascolari (trombo-embolie). La coagulazione intravasale spontanea può inoltre avvenire nel sangue che scorra troppo lentamente, in quanto i fattori favorenti la coagulazione non vengono rimossi velocemente e aumentano localmente fino a determinare la formazione di un trombo (come avviene nelle trombosi venose).
I test di laboratorio più comunemente utilizzati per valutare i meccanismi della coagulazione ematica sono il tempo di protrombina (Prothrombin Time, PT) e il tempo di tromboplastina parziale (Partial Thromboplastin Time, PTT), che valutano il tempo necessario per la formazione di filamenti di fibrina in un campione di plasma, in riferimento, rispettivamente, al meccanismo di coagulazione estrinseco e al meccanismo di coagulazione intrinseco.
1.5 La terapia anticoagulante
I sistemi fisiologici che controllano la fluidità del sangue, come abbiamo visto, sono estremamente complessi. Il sangue deve rimanere fluido all‘interno del sistema vascolare ma allo stesso tempo deve coagulare in fretta quando esposto a superfici non endoteliali nei siti di rottura. Nel momento in cui si forma un trombo intravascolare, un sistema di fibrinolisi viene attivato allo scopo di ripristinare la fluidità. In condizioni fisiologiche quindi, un delicato equilibrio previene sia la trombosi che l‘emorragia.
I farmaci anticoagulanti hanno meccanismi d’azione diversi, ma tutti alterano l‘equilibrio tra pro-coagulazione e anticoagulazione. Con questi farmaci tossicità ed efficacia sono purtroppo necessariamente intrecciate, poiché l‘effetto terapeutico desiderato dell‘anticoagulante, non può essere scisso dall‘effetto tossico dovuto alla somministrazione di una dose eccessiva di farmaco.
23
Una condotta terapeutica ottimale, capace cioè di garantire la massima efficacia riducendo al minimo i rischi, si basa sulla presenza contemporanea dei seguenti elementi:
Medico esperto. Il medico che prescrive la terapia anticoagulante orale deve conoscere perfettamente non solo le indicazioni, ma anche le controindicazioni assolute e quelle relative a questo tipo di trattamento. Il curante quindi, prima di prescrivere tale regime terapeutico deve eseguire sempre un’accurata visita medica e valutare attentamente nel singolo paziente il rapporto rischio/beneficio.
Paziente collaborante. Il paziente deve collaborare attivamente con il medico al fine di minimizzare i rischi e di ottenere i migliori risultati dal regime terapeutico intrapreso. Il paziente deve essere perciò accuratamente informato ed educato sugli scopi e sui rischi, sulle modalità di assunzione e di controllo degli anticoagulanti orali e sul corretto comportamento da adottare in alcune situazioni specifiche.
Laboratorio affidabile. Il laboratorio presso il quale si eseguono i controlli della coagulazione, deve garantire i necessari standard di qualità (Coli et al., 2004).
1.5.1 INR (International Normalised Ratio)
L’INR (o rapporto internazionale normalizzato ) è l’indice della coagulabilità del sangue. Esprime il rapporto tra il tempo di protrombina (PT o tempo di Quick) del paziente e quello della media dei valori di PT del laboratorio che esegue l’analisi, elevato alla potenza del valore ISI (International Sensitivity Index) per il sistema analitico utilizzato. Il risultato per un tempo di protrombina effettuato su un individuo normale varierà a seconda del tipo di sistema analitico utilizzato in quello specifico laboratorio. Ciò è dovuto alle variazioni esistenti tra lotti commerciali differenti di fattore tissutale usate nel reagente per eseguire il test. L’INR è concepito proprio per standardizzare i risultati e renderli paragonabili indipendentemente dal laboratorio che esegue l’esame. Ogni produttore assegna un valore ISI per ogni fattore tissutale che viene fabbricato ed immesso in commercio. Il valore ISI indica un particolare lotto di fattore tissutale confrontato ad un fattore tissutale internazionale di riferimento.
24
L’INR è un mezzo utilizzato per monitorare l’efficacia della terapia anticoagulante orale.
I valori normali dell’INR sono generalmente compresi tra 0,9 e 1,2. I valori terapeutici di INR, generalmente compresi tra 2 e 3, dipendono dalla patologia di base per cui è richiesta la terapia anticoagulante. In particolari pazienti il target di INR che si vuole raggiungere può essere maggiore, per esempio nel caso di pazienti con valvola cardiaca meccanica il cui valore di INR deve essere compreso tra 3 e 4 (Coli et al., 2004).
1.5.2 Indicazioni terapeutiche
Alcune indicazioni della terapia con anticoagulanti orali sono ormai consolidate mentre altre nuove indicazioni sono state poste negli anni più recenti. L’argomento quindi risulta ben definito nelle sue linee essenziali ma è soggetto ad aggiustamenti e modifiche per il gran numero di studi e ricerche ancora in corso volti ad ottimizzare i regimi terapeutici.
Gli anticoagulanti orali vengono somministrati in caso di: - Protesi valvolari cardiache
Il trattamento cronico con anticoagulanti orali riduce significativamente il rischio di embolie in portatori di protesi valvolari cardiache.
In caso di protesi meccaniche è raccomandato un trattamento con anticoagulanti orali sine die. Viene comunque tenuta in considerazione l’età del paziente, se ha già avuto o meno precedenti embolici ed anche il tipo di valvola impiantata in modo da agire terapeuticamente in modo prudente e corretto.
Nel caso di pazienti con protesi biologiche il trattamento con anticoagulanti orali viene in genere consigliato solo per i primi tre mesi dall’intervento, periodo nel quale è massima l’incidenza di fenomeni embolici. La terapia viene proseguita nei pazienti che soffrono di fibrillazione atriale cronica.
25
- Malattie valvolari cardiache
Trombosi cardiaca endocavitaria: in caso di trombosi delle cavità cardiache la terapia anticoagulante orale (TAO) è indicata per tutto il tempo in cui la trombosi è rilevabile.
- Fibrillazione atriale (FA)
Nella fibrillazione atriale associata a valvulopatia la terapia anticoagulante orale è considerata obbligatoria, e se si verificano episodi embolici durante il trattamento è indicata l’associazione con aspirina (100mg/die) o dipiridamolo (400mg/die) in caso d’intolleranza all’aspirina.
Nel paziente con fibrillazione atriale non valvolare tra 65 e 75 anni è indicata la TAO con INR 2-3 in assenza di rischi emorragici. Nei soggetti con età superiore ai 75 anni con fattori aggiuntivi di rischio trombo embolico (diabete, ipertensione arteriosa, scompenso cardiaco, dilatazione atriale sinistra, disfunzione sistolica ventricolare sinistra) è indicata la TAO con INR 2-3.
Bisogna tenere conto che il trattamento nell’anziano può associarsi ad una più elevata frequenza e gravità di complicanze emorragiche, per cui si tende ad effettuare un’attenta valutazione del singolo caso.
E’ stato inoltre valutato che non ci sono sostanziali differenze fra il rischio di ictus in pazienti con FA parossistica e FA cronica, per questo motivo vengono utilizzate le stesse indicazioni per le sopracitate casistiche.
- Infarto miocardico acuto
I pazienti con infarto al miocardio dovrebbero ricevere terapia anticoagulante per almeno tre mesi, con prosecuzione sine die nella FA cronica.
- Tromboembolismo arterioso
Per le condizioni di tromboembolismo arterioso viene in genere suggerito un alto livello di anticoagulazione ( INR 3-4,5) a tempo indefinito.
26
- Prevenzione della trombosi venosa profonda
La profilassi con anticoagulanti orali è generalmente da riservare ai pazienti ad altissimo rischio (pregressa trombosi venosa profonda/embolia polmonare, interventi di chirurgia ortopedica maggiore).
- Trattamento della trombosi venosa profonda e dell’embolia polmonare: profilassi delle recidive
L’utilità del trattamento anticoagulante orale a lungo termine nella trombosi venosa profonda (TVP) e nell’embolia polmonare (EP) è stata dimostrata inequivocabilmente in diversi studi clinici (Finazzi et al., 2000). La durata della terapia rimane ancora oggi non completamente definita: di sicuro le recidive tromboemboliche sarebbero ridotte se la terapia anticoagulante fosse condotta senza interruzione per tutti i pazienti, ma molti di questi sarebbero inutilmente esposti al rischio emorragico e ai costi che comunque sia sono associati alla terapia anticoagulante. Viene generalmente raccomandato un periodo di trattamento di 3-6 mesi per i pazienti senza importanti fattori di rischio tromboembolico, e un periodo più lungo (o indefinito) nei casi considerati più a rischio.
Nell’ipertensione polmonare primitiva studi autoptici e bioptici hanno dimostrato la presenza di trombi occludenti le venule e le arteriole polmonari. L’uso della TAO in questi pazienti induce un miglioramento della prognosi e pertanto viene raccomandato da diversi esperti (Finazzi et al., 2000).
- Ictus
I pazienti con ictus tromboembolico e con lesione piccola o moderata, nei quali una TAC eseguita dopo l’insorgenza dei sintomi esclude una emorragia intracranica, devono essere trattati con eparina seguita dalla TAO. Nei pazienti ipertesi o con focolaio ischemico esteso è bene attendere due settimane prima dell’inizio del trattamento anticoagulante.
27
Nei casi in cui la fibrillazione atriale non valvolare è considerata la causa presumibile dell’ictus è indicata l’instaurazione della terapia anticoagulante direttamente dopo la TAC eseguita a 48 ore.
- Arteropatie periferiche
Un ulteriore campo di impiego della terapia anticoagulante orale è nella chirurgia ricostruttiva vascolare.
1.5.3 Esami di laboratorio preliminari
Prima di iniziare la TAO bisogna eseguire e far valutare i seguenti accertamenti: - Test coagulativi di base (PT, APTT);
- Esame emocromocitometrico completo;
- Transaminasi, γ–GT, bilirubina, colinesterasi (al fine di valutare la funzionalità epatica);
- Creatinina, glicemia, uricemia, colesterolo, trigliceridi; - Test di gravidanza in tutte le donne in età fertile
1.5.4 Condizioni a rischio
Le condizioni generali in cui la somministrazione dei farmaci anticoagulanti orali è rischiosa e di difficile gestione sono rappresentate da:
- Mancata collaborazione da parte del paziente a causa di ridotte capacità intellettive;
- Difficoltà di controllo per motivi di lavoro; - Lavoro ad alto rischio traumatico;
- Gravi irregolarità dietetiche; - Alcolismo cronico;
28 1.5.5 Controindicazioni assolute
Nelle seguenti circostanze il trattamento anticoagulante orale non deve essere adottato:
- Gravidanza
Gli anticoagulanti orali non devono essere somministrati durante il primo trimestre di gravidanza per le note malformazioni fetali che possono indurre, e nelle ultime 4-6 settimane, per il rischio emorragico nel neonato dovuto al fatto che l’anticoagulante attraversa la placenta.
- Recente emorragia maggiore, specie se a rischio vitale
In caso di insorgenza di emorragia maggiore, specie se essa può generare un rischio per la vita, è opportuno non somministrare la TAO per un adeguato periodo (almeno un mese).
Esistono, inoltre, numerose condizioni, sia di carattere generale che per presenza di specifiche patologie, nelle quali la TAO deve essere considerata come un trattamento ad alto rischio.
- Età avanzata
L’età avanzata costituisce un fattore di rischio emorragico importante, ma al tempo stesso coincide con l’epoca di insorgenza di un aumentato rischio tromboembolico legato a malattie cardiovascolari. Questo rende necessaria un’attenta sorveglianza clinica ed un’accurata valutazione del rapporto beneficio/rischio, ma non costituisce di per sé una controindicazione.
- Malattie cardiovascolari
L’ipertensione grave rappresenta un fattore di rischio emorragico importante per il paziente in TAO. Si ritiene però che il paziente iperteso, specie se affetto da ipertensione moderata, possa essere sottoposto alla TAO qualora esistano le indicazioni, purchè venga effettuato un adeguato controllo farmacologico dei valori pressori.
29
Altri rischi sono rappresentati dall’endocardite batterica (rischio di disseminazione di emboli settici), dalla pericardite, dall’insufficienza cardiaca grave (rischio emorragico grave per alterato metabolismo degli anticoagulanti).
- Malattie renali
L’insufficienza renale grave rappresenta una condizione a rischio emorragico elevato a causa dell’alterato metabolismo dell’anticoagulante orale. In caso di esecuzione di biopsia renale è necessario evitare di somministrare TAO nelle due settimane successive per il rischio di emoperitoneo.
- Malattie gastrointestinali
La presenza di ulcera-emorragica e ulcera gastro-duodenale attiva (accertate con endoscopia o radiografia dello stomaco-duodeno) sono note e classiche controindicazioni.
In assenza di lesioni sanguinanti, l’ernia iatale e la diverticolosi del colon vengono considerate controindicazioni relative.
Situazioni di malnutrizione, malattie biliari, steatorrea e diete ipocaloriche, possono alterare l’equilibrio della vitamina K e quello degli anticoagulanti orali.
- Malattie epatiche
L’insufficienza epatica grave e l’ittero colestatico aumentano il rischio emorragico.
- Malattie ematologiche
Diverse malattie ematologiche sono ovvie controindicazioni alla TAO. In particolare i soggetti con piastrinopenia severa o piastrinopatia sono ad alto rischio emorragico. Inoltre si ricordi che l’associazione di farmaci antipiastrinici alla TAO è da evitare e meno che non sia prevista dal protocollo terapeutico.
30
- Miscellanea
Alcune manovre invasive come la puntura lombare e l’incannulamento di arterie possono essere estremamente pericolose. I caso di necessità di eseguire tali manovre in pazienti già in TAO, si consiglia di ridurre i dosaggi. Anche le iniezioni intramuscolari vanno evitate e per le vaccinazioni è consigliabile praticare iniezioni sottocutanee o nel muscolo deltoide.
Anche la tireotossicosi ed il mixedema possono alterare il metabolismo di questi farmaci.
Le retinopatie e le malattie infiammatorie intestinali sono controindicazioni relative per le quali è necessaria una sorveglianza clinica particolarmente accurata. Questo vale anche in pazienti che hanno subito un recente intervento chirurgico (Finazzi et al., 2000).
1.6 Anticoagulanti parenterali
1.6.1 Eparina e storia
L’eparina è un polisaccaride solfato naturalmente prodotto nei granuli dei mastociti e basofili a partire da precursori formati da uridindifosfo-(UDP)-zucchero. L’eparina presenta una struttura costituita da polimeri di residui degli acidi D-glucuronico e N-acetil-D-glucosammina alternati e contenenti molti gruppi solforici. Dalle 10 alle 15 catene del glucosamminoglicano, ciascuna contenente da 200 a 300 unità monosaccaridi, sono legate ad un cuore proteico formando un proteoglicano con peso molecolare che varia da 750 Kda a 1.000 Kda. Il glucosamminoglicano subisce successivamente una serie di modifiche che includono: deacetilazione ed N-solforazione dei residui glucosamminici, epimerizzazione dell‘acido D-glucuronico ad L-iduronico, O-solforazione dei residui acidi iduronico e glucuronico nella posizione del carbonio C2, e O-solforazione dei residui glucosamminici nelle posizioni del carbonio C3 e C6. Dopo che l‘eparina proteoglicano è stata trasportata ai granuli dei mastociti, una endo-β-D-glucuronidasi degrada le catene del glucosamminoglicano in frammenti che variano dai 5 Kda ai 30 Kda.
31
L’eparina è un anticoagulante in grado di inibire vari fattori coagulativi, in particolar modo la trombina e il fattore Xa; le sue molecole interagiscono con l’antitrombina in circolo assicurando una difesa antitrombotica naturale.
L’eparina è uno dei farmaci più antichi ancora attualmente in uso. La sua scoperta avvenne nel 1916 da parte di uno studente di medicina, Jay McLean, il quale stava lavorando, sotto la supervisione di William Henry Howell, sugli effetti degli agenti anticoagulanti (Marcum, 2000). L’eparina fu originariamente isolata dalle cellule del fegato canino e nel 1918 fu proprio Howell a coniare il termine “eparina”, dal greco “epar”, che significa per l’appunto fegato. Nel 1920 Howell isolò un anticoagulante solubile in acqua che decise di nominare eparina, per quanto la sua struttura fosse fondamentalmente diversa da quella precedentemente isolata. Dieci anni dopo un gruppo di ricercatori svedesi di cui faceva parte Eric Jorpes, del famoso Karolinska Institute, iniziarono una esaustiva ricerca sull’eparina, pubblicando i risultati ottenuti nel 1935. La società farmaceutica AB Vitrum iniziò la produzione di eparina per uso endovenoso un anno dopo. All’incirca nello stesso periodo gli scienziati canadesi di Toronto Labs Connaught perfezionarono la formulazione dell’eparina, rendendola il più possibile sicura per i pazienti, tramite la sperimentazione di una soluzione salina. In confronto l’eparina svedese fu considerata tossica ed eccessivamente costosa. Nel 1937 i canadesi ricevettero il via libera per commercializzare l’eparina ad uso umano. I trial clinici condotti in quelle circostanze ebbero un grande successo e dimostrarono l’efficacia della nuova eparina nel prevenire la formazione dei coaguli di sangue nei pazienti (Keeling, Wardrop, 2008).
Eparansolfato. L’eparansolfato, così come l’eparina è sintetizzato dallo stesso
precursore disaccaride (acido D-glucuronico legato alla N-acetil-D-glucosammina). Esso però subisce un minor numero di modificazioni del polimero rispetto all’eparina, inoltre contiene più porzioni di acido-glucuronico e di N-acetil-glucosammina e pochi gruppi solfato. Per ottenere un meccanismo antitrombotico naturale, l’eparansolfato interagisce con l’antitrombina circolante sulla superficie delle cellule endoteliali vascolari o nella matrice extracellulare del sub-endotelio. Nei pazienti affetti da neoplasie si può verificare un sanguinamento legato alla circolazione di eparansolfato o di glucosamminoglicano, che provengono probabilmente dalla lisi delle cellule tumorali.
32 1.6.2 Farmacocinetica e farmacodinamica
L’eparina standard o eparina non frazionata (ENF) è costituita da glicosaminoglicani e il suo peso molecolare varia da 5000 a 30 000 dalton con un valore medio di 15 000 dalton. Viene in genere estratta dalla mucosa intestinale del maiale o dal polmone dei bovini, e le preparazioni possono contenere piccole quantità di altri glucosamminoglicani. Nonostante le differenze nella composizione, i preparati commerciali hanno tutti lo stesso meccanismo d’azione. Le eparine a basso peso molecolare (EBPM) sono ottenute da quella standard mediante frazionamento con metodi chimici ed enzimatici e differiscono dall’eparina classica per le proprietà farmacocinetiche e per il meccanismo d’azione. Le eparine a basso peso molecolare hanno peso molecolare variabile tra 1000 e 10 000 dalton, con un valore medio di 4000-6000 dalton. Nel processo della coagulazione del sangue il fattore X attivato (Xa), legandosi a ioni calcio e fosfolipidi, catalizza l’attivazione della protrombina (fattore II) in trombina (fattore IIa), che a sua volta trasforma il fibrinogeno in fibrina con formazione del coagulo. L’attività dei fattori della coagulazione è modulata da inibitori naturali: antitrombina (AT), proteina C e proteina S. La proprietà anticoagulante dell’eparina viene definita indiretta, in quanto dipende dalla presenza nella sua struttura molecolare, di una specifica sequenza pentasaccaridica per mezzo della quale si lega con alta affinità all’antitrombina. Il complesso eparina-antitrombina determina una modificazione conformazionale dell’eparina-antitrombina ed un marcato potenziamento della sua attività inibitoria nei confronti di molti enzimi della coagulazione fra cui la trombina, i fattori Xa e IXa e in modesta parte anche il fattore VIIa (Lindahl et al., 1979). L’inattivazione della trombina richiede la formazione di un complesso ternario: trombina-antitrombina-eparina. L’eparina catalizza l’inibizione di molte proteasi della coagulazione attraverso l’antitrombina. L’inibizione di cui sopra avviene quando la proteasi attacca una sequenza specifica arginina-serina presente nel sito attivo dell’antitrombina e viene intrappolata in un complesso stabile in rapporto 1:1. L’eparina fa aumentare la velocità della reazione trombina-antitrombina di circa un migliaio di volte. Inoltre il legame dell’eparina induce un cambio conformazionale nell’antitrombina che rende il sito reattivo più accessibile alle proteasi. Così una volta che la trombina si è legata all’antitrombina,
33
la molecola di eparina si distacca dal complesso. Il sito di legame per l’antitrombina sulla molecola di eparina è una specifica sequenza pentasaccaridica che contiene un residuo glucosammino 3-O-solfato. E’ necessario che l’eparina contenga almeno 18 unità monosaccaridi per legare simultaneamente la trombina e l’antitrombina e permettere la formazione del complesso ternario (Lane et al., 1984). Per cui le molecole di eparina che contengono un numero inferiore di 18 unità monosaccaride (<5,4 Kda) non catalizzano l’inibizione della trombina da parte dell’antitrombina. Le eparine a basso peso molecolare quindi, producono il loro effetto anticoagulante essenzialmente attraverso l’inibizione del fattore Xa ad opera dell’antitrombina, perché la maggior parte di queste molecole non sono sufficientemente lunghe per catalizzare l’inibizione della trombina. In conclusione, mentre l’eparina standard ha un’attività inibitoria sostanzialmente equivalente sia verso il fattore IIa che Xa, in tutte le eparine a basso peso molecolare invece, l’attività anti-Xa supera quella antitrombinica. Il rapporto tra attività anti-Xa e anti-IIa , sostanzialmente uguale ad 1 nell’eparina classica, presenta nelle eparine a basso peso molecolare una variabilità compresa circa tra 1,6 e 4,2 (Baglin et al. 2006).
Il complesso eparina-antitrombina, inattiva non solo la trombina, ma anche i fattori Xa, IXa, XIa, e XIIa. La trombina è circa 10 volte più sensibile all’inibizione rispetto al fattore Xa. Inattivando la trombina l’eparina non solo previene la formazione di fibrina, ma inibisce anche l’attivazione delle piastrine, dei fattori V e VIII indotta dalla trombina (Hirsh et al., 2001).
L’attività dei prodotti a base di eparina è espressa in Unità Internazionali (UI), calcolate secondo il metodo della Farmacopea Europea, che si basa sul prolungamento del tempo di tromboplastina parziale attivata (aPTT) di plasma di pecora.
1.6.3 Uso clinico
L’eparina viene in genere utilizzata per il trattamento delle trombosi venose ed embolie polmonari. Le eparine a basso peso molecolare sono indicate nella prevenzione e nel trattamento della trombosi venosa e delle sindromi coronariche acute in sostituzione all’eparina standard. I criteri che vengono per lo più utilizzati per la scelta del tipo e della dose di eparina sono:
34
- Il rischio trombotico ed emorragico intrinseco del paziente o correlato alle condizioni cliniche o alle eventuali procedure (rischio del paziente);
- L’efficacia relativa delle differenti preparazioni e dosi di eparina ed il relativo rischio emorragico ad esse correlato (rischio eparinico).
L’eparina viene utilizzata nei casi in cui è richiesta una rapida correzione della coagulazione. Di solito, nella fase iniziale della terapia con un anticoagulante orale viene utilizzata allo stesso tempo l’eparina per almeno 4 o 5 giorni, in modo tale da permettere all’anticoagulante di raggiungere il suo massimo effetto terapeutico. I pazienti con tromboembolismo ricorrente, oltre ad un adeguato anticoagulante orale, possono essere trattati anche con eparina. Quest’ultima inoltre è somministrata in pazienti con angina instabile, infarto miocardico acuto, durante e dopo angioplastica coronarica o inserimento di stent, oppure durante interventi chirurgici che richiedono un bypass cardiopolmonare. Inoltre l’eparina è utilizzata per trattare i pazienti con coagulazione intravascolare disseminata. L’eparina può essere somministrata per infusione continua e monitorata con l’aPTT mantenuto a valori pari a 1,5-2,5 volte quello normale.
Sono stati suggeriti diversi schemi terapeutici: ad esempio le dosi raccomandate dall’American College of Chest Physician sono le seguenti:
- TVP: 80 UI/Kg in bolo, seguita da 18 UI/Kg/ora in infusione continua;
- Angina instabile ed infarto dl miocardio: 60-70 UI/Kg in bolo, seguita da 12-15 UI/Kg/ora in infusione continua;
- Interventi coronarici percutanei: 70 UI/Kg in bolo seguita da dosi regolate per mantenere il tempo di coagulazione attivato > 200sec.