85
86
2.1 Studio del trascritto del gene LEAFY
COTYLEDON1-LIKE nei diversi organi e tessuti di
Helianthus annuus
2.1.1 Materiale vegetale utilizzato
Il materiale vegetale utilizzato è costituito dalla linea pura HOR di Helianthus
annus costituita nel Dipartimento di Biologia delle Piante Agrarie, Sezione di
Genetica.
2.1.2 Estrazione dell’RNA
L’estrazione degli RNA è stata condotta, in accordo con il protocollo del “TriPure Isolation Reagent Kit” (Roche), su embrioni immaturi, cotiledoni, foglie giovani (2 cm di lunghezza), foglie espanse (15 cm di lunghezza), radici, fusto ed infiorescenze immature di H. annuus (HOR). I campioni appena prelevati sono stati congelati mediante immersione in azoto liquido, quindi 300 mg di materiale sono stati polverizzati in mortaio mantenendoli ghiacciati sempre con azoto liquido. La polvere ottenuta è stata trasferita in una provetta da 2 ml ed incubata per 5 minuti a temperatura ambiente dopo l’aggiunta di 1 ml di Trizol. Successivamente, sono stati addizionati 200 µl di cloroformio e la provetta è stata agitata vigorosamente per 15 secondi, quindi lasciata a temperatura ambiente per 5 minuti ed infine centrifugata a
87 9000 rpm per 25 minuti in rotore Kroton A 8.24 a 4 °C. La fase superiore acquosa è stata recuperata e trasferita in un’altra provetta, dove sono stati aggiunti 500 µl d’isopropanolo ed il tutto è stato mescolato delicatamente. La soluzione è stata incubata a temperatura ambiente per 10 minuti, per favorire la precipitazione del RNA, quindi centrifugata a 9.000 rpm in rotore Kroton A 8.24 per 40 minuti a 4 °C. Il surnatante è stato scartato, mentre il precipitato formatosi è stato sottoposto a due lavaggi successivi per eliminare eventuali residui di cloroformio con 1 ml d’etanolo al 75%, centrifugando a 9.000 rpm in rotore Kroton A 8.24 per 20 minuti a 4 °C . Il surnatante è stato scartato e l’RNA precipitato è stato fatto asciugare all’aria per eliminare i residui d’etanolo. L’RNA precipitato è stato poi sciolto in acqua trattata con DEPC e conservato a –80 °C.
88
2.1.3 Corsa elettroforetica su gel di agarosio in condizioni
denaturanti
Il gel è stato preparato sciogliendo l’agarosio in H2O DEPC con aggiunta di
formaldeide e MOPS 10X preriscaldati a 65 °C. La miscela è stata versata sul supporto e lasciata polimerizzare. Il gel polimerizzato è stato posto nel tampone di corsa (MOPS 1X) ed è stata effettuata una precorsa di 10 minuti a 60 mA. I campioni sono stati preparati aggiungendo 3 volumi di soluzione denaturante, incubandoli a 65 °C per 15 minuti. I campioni, così denaturati, sono stati tenuti in ghiaccio per 5 minuti e poi è stato aggiunto il tampone di caricamento in quantità pari a 1/5 del volume finale. Caricati i campioni, è stata effettuata una corsa per circa 2 ore a 50 mA, per verificare l’integrità dell’RNA estratto.
SOLUZIONI UTILIZZATE:
GEL AGAROSIO 1% (70 ml) SOLUZIONE DENATURANTE Agarosio 0,7g 50 ml MOPS10X 52,5 ml H2O DEPC 75 ml Formaldeide 37%
10,5 ml Formaldeide 37% 250 ml Formammide deionizzata 7 ml MOPS 10X
MOPS 10X TAMPONE DI CARICAMENTO
0,2 M MOPS pH 7 Saccarosio 30%
Acetato di sodio 80 mM Blu di bromofenolo q.b 10 mM Na2 -EDTA
89
2.1.4 RT-PCR relativa
Con questa tecnica è stata valutata l’espressione del gene LEAFY
COTYLEDON1-LIKE in diversi organi e/o tessuti di H. annuus. L’RT-PCR Relativa consiste nella
co-amplificazione del cDNA di un gene espresso costitutivamente, che rappresenta un controllo interno, e del cDNA di interesse, utilizzando due coppie di oligonucleotidi specifiche nella stessa reazione di PCR.
2.1.4a SCELTA DEGLI OLIGONUCLEOTIDI PER LA ß-ACTINA
L’actina è una proteina espressa costitutivamente nelle piante e l’mRNA ad essa relativo costituisce lo standard interno con cui sono stati confrontati gli mRNA per l’HaL1L. Oligonucleotidi per l’amplificazione di un frammento del cDNA della ß-actina di lunghezza opportuna, sono stati scelti sulla base dell’allineamento di sequenze di specie vegetali vicine ad H. annuus disponibili in rete.
2.1.4b AMPLIFICAZIONE DELLE SEQUENZE I campioni di RNA usati provengono da:
1. embrioni zigotici di H. annuus a diversi stadi di sviluppo (5, 10, 21, 28 giorni dall’impollinazione);
2. foglie cotiledonari di H. annuus di semi germinati da 10 giorni; 3. foglie giovani di H. annuus (2 cm di lunghezza);
90 4. foglie espanse di H. annuus (15 cm di lunghezza);
5. fusto di piante di H. annuus allo stadio di bottone fiorale; 6. radici di piante di H. annuus allo stadio di bottone fiorale;
7. infiorescenze immature di H. annuus (capolini a 15 giorni dall’antesi);
I) Retrotrascrizione:
La miscela per la retrotrascrizione era costituita da: 1 µl Oligo dT (500 µg/ml)
X µl RNA (1 µg) 1 µl dNTP (10 mM)
H2O DEPC q.b fino a 12 µl
La miscela è stata incubata a 65 °C per 5 minuti quindi messa immediatamente in ghiaccio per 5 minuti. Successivamente, dopo aver centrifugato brevemente la miscela, sono stati aggiunti:
4 µl First Strand Buffer (5X) 2 µl DTT (0,1M)
1 µl Inibitore delle Ribonucleasi
La miscela è stata incubata a 42 °C per 2 minuti, quindi è stato aggiunto 1 µl di SuperscriptTM II (Invitrogen) (200 U/µl) e il tutto è stato mantenuto a 42 °C per
ulteriori 50 minuti. La reazione è stata, infine, bloccata ponendo la provetta a 70 °C per 15 minuti.
91 II) Amplificazione del cDNA mediante PCR
La miscela di reazione della retrotrascrizione (MRT), contenente il cDNA sia per la
β-actina sia per l’HaL1L, è stata utilizzata come stampo nella reazione di
amplificazione condotta con il seguente protocollo. 5 µl MRT 4 µl MgCl2 (25mM) 1,5 µl Buffer 10X 0,2 µl dNTP (10mM) 0,1 µM Primer ß-actina 0,1 µM Primer HaL1L 0,15 µl TaqPolimerasi (5U\µl) q.b a 15 µl H2O
L’amplificazione è stata effettuata tramite PCR utilizzando contemporaneamente entrambe le coppie di oligonucleotidi che, per evitare artefatti, non devono presentare regioni complementari. Le coppie di oligonucleotidi impiegate sono CHI-REL/CHITcDNA per HaL1L e ACT3/ACT5 per la ß-actina (Tab. 2.2). In questo tipo di analisi è di fondamentale importanza conoscere il numero di cicli di PCR in cui i due amplificati raggiungono la saturazione. Ogni PCR, infatti, raggiunge il punto di saturazione quando l’aumento del prodotto di amplificazione non è più esponenziale, ovvero non è più proporzionale alla quantità di stampo iniziale poiché sono utilizzati come stampo anche i prodotti amplificati nei cicli precedenti. Per conoscere il punto di saturazione è stata condotta una PCR dalla quale, al termine di cicli successivi, sono state sottratte aliquote di campione immediatamente trasferite in una adiacente macchina per PCR per subire la fase di estensione finale di 7 minuti a 72 °C. I prodotti di amplificazione, ottenuti nell’intervallo compreso tra 22 e 32 cicli,
92 sono stati analizzati su di un gel di Agarosio ed è stato quindi deciso che il numero di cicli da utilizzare era compreso tra 26 e 30.
CICLI UTILIZZATI: 95° 94° 53° 72° 72° 3’ 30’’ 30’’ 25’’ 7’
93
2.1.5 Corsa elettroforetica su gel di agarosio
I campioni amplificati sono stati sottoposti a corsa elettroforetica su gel di agarosio. L’agarosio era stato disciolto, a concentrazioni variabili tra l’1% ed il 2%, a seconda dell’ampiezza attesa per la banda amplificata, in un tampone Tris-acetato (TAE) 1X, contenente 0,5 µg/ml di bromuro d’etidio. Per il caricamento sono stati aggiunti ad ogni campione 0,2 volumi di tampone di caricamento.
SOLUZIONI UTILIZZATE:
TAMPONE DI CARICAMENTO: Saccarosio 60% (p\v)
Orange G 0,1% (p\v) 25 mM Na2EDTA pH 8,
TAMPONE TRIS-ACETATO 10X (TAE) Tris acetato 0,4 M
0,01 mM Na2 EDTA pH 8,
94
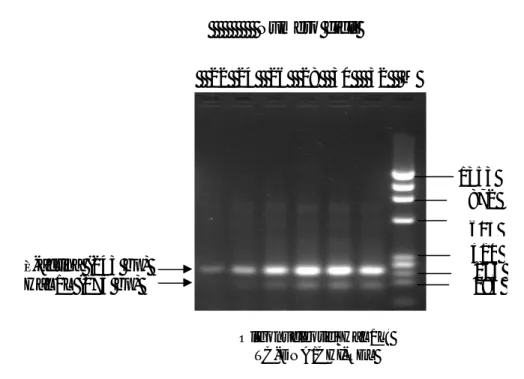
Fig. 2.1: Visualizzazione su gel di Agarosio al 2% dei prodotti di amplificazione ottenuti nell’intervallo compreso tra 22 e 32 cicli.
Dalla Fig. 2.1 si vede che a 26 cicli la PCR è ancora nella fase esponenziale, mentre a 30 cicli il segnale di amplificazione della β-actina è a saturazione. Le quantità relative di ogni prodotto di amplificazione sono state quantificate mediante la scansione diretta del gel di Agarosio al 2% con il densitometro UVP Image Store 5000 (Ultra Violet Product Ltd, Cambridge, England) equipaggiato con l’UVP GelBase-GelBlot TM Windows Software. Per normalizzare i campioni relativamente alla quantità di RNA totale utilizzato e alla efficienza della sintesi di cDNA, le intensità delle bande di amplificazione del frammento di HaL1L sono state rapportate ai prodotti di amplificazione della β-actina. Un valore arbitrario di 1 è stato assegnato in ogni campione al livello della β-actina. È stata poi effettuata l’analisi statistica della varianza e le medie sono state separate mediante il test di Tukey (con P = 0.05). Per quest’analisi sono stati utilizzati tutti i diversi RNA per ciascun tipo di campione.
Numero cicli 22 24 26 28 30 32 M 1353 872 310 194 603 HaL1L (175 bp) ß-actina (243 bp) Oligonucleotidi HaL1L: TC-DNA/CHI-REL 234
95
2.2 Localizzazione del trascritto del gene HaL1L
in H. annuus
2.2.1 Ibridazione in situ RNA:RNA
L'ibridazione in situ (ISH) è una tecnica molto potente e versatile che consente di individuare sequenze di DNA o di RNA, che hanno localizzazioni spesso specifiche, sia nelle cellule sia nei tessuti. Informazioni relative a quest’aspetto non sono, viceversa, deducibili da ibridazioni su Southern o Northern-blot che richiedono l’estrazione, mediante omogeneizzazione, e il trasferimento su membrana degli acidi nucleici. Dal punto di vista metodologico, la tecnica ISH comprende quattro fasi principali:
1. Preparazione della sonda 2. Marcatura della sonda
3. Preparazione dei campioni e dei vetrini (fissazione, allestimento delle sezioni, trattamenti di pre-ibridazione)
4. Ibridazione
5. Visualizzazione e localizzazione del sito d’ibridazione (post-ibridazione) Nel corso di queste tappe deve essere evitata la degradazione degli mRNA da parte delle RNasi. A tale scopo è necessario far uso di guanti, contenitori di plastica sterili e la vetreria deve essere sterilizzata a 180 °C per 2 ore in stufa a secco o autoclavata a 121 °C per 20 minuti. L’acqua, utilizzata per la preparazione di tutte le
96 soluzioni deve essere “Rnasi-free” e viene preparata trattandola con DEPC (dietil-pirocarbonato) allo 0.1%, che è un forte, sebbene non totale, inibitore delle RNasi. L'acqua è poi autoclavata o bollita per inattivare il DEPC. Nei nostri esperimenti il materiale sperimentale utilizzato comprende: embrioni di H. annuus (linea pura HOR) a diversi stati di sviluppo.
2.2.1a FASE1: PREPARAZIONE DELLE SONDE
Gli oligonucleotidi CHI-SR e CHI-SF sono stati utilizzati per l’amplificazione di una regione al 3’UTR per la preparazione di una sonda in grado di distinguere il gene L1L dal gene LEC1 in esperimenti d’ibridazione in situ. Questa regione è inserita nel clone HaL1L-Sonda. Il DNA plasmidico contenente la porzione del cDNA codificante per la sequenza delimitata dai due oligonucleotidi specifici, CHI-SR e CHI-SF, del gene HaL1L è stato amplificato per preparare una sonda molecolare da utilizzare in esperimenti di ibridazione in situ. Dal clone HaL1L-Sonda, conservato a –80 °C, con ansa sterile è stata raccolta un’aliquota di cellule che è stata inoculata in una provetta da 1,5 ml contenente 1 ml di substrato LB ed ampicillina. La coltura è stata lasciata crescere su un agitatore per 14-16 ore a 37 °C. Una diluizione opportuna della crescita è stata utilizzata come stampo per le reazioni di PCR. Con il sequenziamento del DNA plasmidico contenente il cDNA e l’analisi della sequenza è stato possibile capire l’orientamento dell’inserto e stabilire i giusti accoppiamenti oligonucleotidi specifici/ M13F o M13R. Le reazioni d’amplificazione sono state condotte come precedentemente descritto.
Oligonucleotidi utilizzati nelle amplificazioni per la sonda a RNA usata per l’ibridazione in situ:
97 2.2.1b FASE 2: MARCATURA DELLA SONDA A RNA
Il metodo non radioattivo del “DIG-RNA Labeling Kit Nonradioactive” (Roche) è stato utilizzato, secondo protocollo fornito dalla ditta, per realizzare la sonda a RNA complementare al cDNA codificante per HaL1L, in grado di ibridarsi con il relativo mRNA.
MISCELA DI REAZIONE PER LA MARCATURA: Antisenso Senso (CHI-SF/M13F) (CHI-SR/M13R) prodotto di PCR purificato: 200 ng 200 ng 10X transcription buffer: 2 µl 2 µl 10X NTP labelling mixture: 2 µl 2 µl inibitore delle RNasi (20 unità/ µ l): 1 µl 1 µl RNA polimerasi SP6/T7 (20 u/ µl): T7: 2 µl SP6: 2 µl
H2O DEPC fino a 20 µl fino a 20 µl
Il campione è stato incubato per 2 ore a 37 °C. Il DNA stampo è stato rimosso dalla miscela aggiungendovi 2 µl (20 unità) di DNasiI ed incubando 15 minuti a 37 °C. La trascrizione è stata bloccata aggiungendo 2 µl di EDTA 0,2 M DEPC a pH 8 e il trascritto ottenuto è stato precipitato con 2,5 µl di LiCl 4 M DEPC e 75 µl di etanolo assoluto preraffreddato, con incubazione o.n. a –20 °C. Dopo una centrifugazione di 25 minuti a 13.000 rpm, il precipitato è stato lavato con etanolo al 70% DEPC, asciugato a temperatura ambiente e solubilizzato in 30 µl di H2O DEPC, incubando a
37 °C per 15 minuti. La trascrizione effettuata con la T7 polimerasi ha permesso di ottenere la sonda antisenso, la trascizione operata dalla SP6 polimerasi ha invece consentito di ottenere la sonda senso, costituente il controllo negativo.
98 2.2.1c FASE 3: PREPARAZIONE DI CAMPIONI E VETRINI
In questa fase vengono preparati sia i campioni che i vetrini per la successiva ibridazione.
Fissazione dei campioni
.
Entro 5-10 minuti dal suo isolamento, sono stati arrestati i processi biologici del campione, per conservare sia la morfologia del campione sia gli RNA presenti in natura. Questo scopo è stato raggiunto attraverso la fissazione in paraformaldeide al 4% in tampone 1X PBS pH 7.0. Per favorire la penetrazione del fissativo, è stata usata una pompa da vuoto per qualche minuto; quindi, dopo un cambio con fissativo fresco, il materiale è stata posto a 4 °C in lenta agitazione per tutta la notte. Il materiale è stato quindi disidratato, per prepararlo all'inclusione in paraffina, attraverso immersioni in una serie di alcoli a gradazione crescente, a partire dall'etanolo al 30% fino all'etanolo assoluto, mantenendolo a 4 °C e in agitazione. Per l'inclusione, l'etanolo è stato sostituito gradualmente con lo xilolo e quest’ultimo con paraffina in crescenti concentrazioni. Dopo diversi cambi con paraffina pura, questa è stata fatta polimerizzare a temperatura ambiente. I blocchetti di paraffina sono stati tagliati al microtomo ottenendo sezioni spesse 7-8 µm. Durante l'ibridazione le sezioni sono sottoposte ad una serie di lavaggi e trattamenti che ne causano spesso il distacco dai vetrini. Per evitare questo, i vetrini sono stati preparati, prima della deposizione delle sezioni di materiale sperimentale, secondo il seguente protocollo: sono stati accuratamente puliti con un lavaggio in acido nitrico concentrato per 30 minuti, sciacquati in acqua deionizzata per circa 2 ore e poi lasciati asciugare all'aria. Dopo un passaggio in acetone di 15 minuti, i vetrini sono stati asciugati in stufa a 180 °C per 2 ore. Su ogni vetrino è stata, quindi, posta una goccia di poly-L-lysina (PM=300.000, 1 mg/ml in acqua “RNasi-free”), che è stata distribuita su tutta la
99 superficie, usando un coprioggetto, in modo da costituire un film omogeneo. Infine i vetrini sono lasciati seccare in stufa a 40 °C, per tutta la notte, a questo punto erano pronti per essere utilizzati. Dopo aver disposto le sezioni sui vetrini, questi sono stati messi ad asciugare su una piastra riscaldata a 37 °C, per tutta la notte. Tale fase è molto importante perché permette alle sezioni di aderire bene al vetrino e di resistere ai lavaggi della post-ibridazione. La preibridazione consta di una serie di passaggi che permette di: i) incrementare la capacità di accesso della sonda alle sequenze bersaglio; ii) ridurre le probabilità della sonda di ibridarsi specificamente; iii) minimizzare le interazioni con proteine o altre molecole che possono legare la sonda. In particolare, le sezioni incluse in paraffina sono state prima deparaffinate secondo il seguente protocollo: 2X 10 minuti xilene 2X 1-2 minuti 100% EtOH 1-2 minuti 95% EtOH 1-2 minuti 70% EtOH 1-2 minuti 40% EtOH 1-2 minuti H2O
Segue una incubazione in 2X SSC a temperatura ambiente per 15-20 minuti.
Quindi, è stata effettuata la permeabilizzazione che richiede un trattamento con Proteinasi K: il buffer TE 5X (50 mM Tris HCl, pH 8,5, 5 mM EDTA) è stato preriscaldato a 37 °C; a questo è stato aggiunta proteinasi K (10 mg/ml) fino a concentrazione 1 µg/ml, con lo scopo di rimuovere le proteine che potrebbero interferire con la sonda e aumentare il segnale di fondo. La digestione con proteinasi K in TE 5X a 37 °C ha una durata di 30 minuti al termine dei quali viene effettuato un trattamento con glicina 2 mg/ml in PBS a temperatura ambiente per 2 minuti per inattivare la proteinasi K. Successivamente sono state eseguite una breve
100 postfissazione con paraformaldeide al 4% in 2xPBS per 5 minuti e poi l’acetilazione mediante immersione, in agitazione, dei vetrini in una soluzione di trietanolamina 0.1M a pH 8.0, a cui è stata aggiunta anidride acetica fino ad ottenere una concentrazione finale dello 0.5%. Dopo la fase di acetilazione i vetrini sono lavati in PBS 2 volte per 5 minuti a temperatura ambiente e quindi nuovamente disidratati con le seguenti soluzioni di etanolo:
30 sec. 40% EtOH 30 sec. 70% EtOH 30 sec. 95% EtOH 2x 30 sec. 100% EtOH
Le sezioni sono state lasciate asciugare all’aria per 1 ora. SOLUZIONI UTILIZZATE: PBS 10X NaCl 1.3 M Na2HPO4 70 mM NaH2PO4 30 mM TE 5X 50 mM Tris HCl, 5 mM EDTA pH 8,5 2.2.1d FASE 4: IBRIDAZIONE
20 µl delle sonde, senso o antisenso, (100 ng di RNA in 50% formammide demonizzata) sono state denaturate per 2 minuti ad 80 °C, poste in ghiaccio e centrifugati prima di essere aggiunti a 80 µl di miscela di ibridazione. A questo punto
101 vengono posti 100 l della miscela di ibridazione, contenente la sonda ad RNA, nel centro di un vetrino coprendo col parafilm e incubando in camera umida. L’ibridazione avviene per una notte a temperature comprese tra i 48 e i 50 °C.
SOLUZIONE IBRIDAZIONE PER OGNI VETRINO: 7 µl H2O DEPC
40 µl formammide deionizzata 20 µl destran solfato 50% 2 µl Denhardt’s solution 50X 1 µl tRNA (100 mg\ml) 10 µl Sali per in situ 10X 80 µl Volume totale
Questa soluzione è assai viscosa a causa del destran solfato, si è quindi riscaldata prima dell’utilizzo. E’ stata eseguita esegue una serie di lavaggi successivi all’ibridazione:
§ è stato rimosso il parafilm immergendo i vetrini in 0.2X SSC preriscaldato a 55 °C. § 30 minuti per 2 volte in 0.2X SSC preriscaldato a 55 °C agitando delicatamente. § 5 minuti per 2 volte in NTE preriscaldato a 37 °C agitando delicatamente.
§ 30 minuti in RNAsi (20 µg/ml RNasi A in NTE), che digerisce solo l'RNA a singolo filamento e quindi non ibridato, a 37 °C agitando delicatamente..
§ 5 minuti per 2 volte in NTE 37 °C agitando delicatamente. § 60 minuti in 0.2X SSC a 55 °C agitando delicatamente. § 5 minuti in PBS a temperatura ambiente.
SOLUZIONI UTILIZZATE:
5X NTE 20X SSC:
NaCl 2,5 M NaCl 3 M
Tris, pH 8.0 50 mM Citrato di sodio 0,3 M
102 2.2.1e FASE 5: RIVELAZIONE
I vetrini sono stati incubati per 45 minuti con 1% “Roche block” in 100 mM Tris pH 8, 150 mM NaCl.
La soluzione precedente è stata sostituita con 1% BSA in 100 mM Tris 7.5, 150 mM NaCl, 0.3% Triton X-100 per 45 minuti.
L’anticorpo anti-dig è stato diluito nella soluzione BSA/Tris/NaCl/Triton (1:1250) Si coprono i vetrini con 100 µl della soluzione anticorpo anti-Dig e si lascia ad incubare per 2 ore a temperatura ambiente in camera umida.
Sono stati effettuati 4 lavaggi successivi di 10 minuti ciascuno in una soluzione BSA/Tris/NaCl/Triton, a temperatura ambiente agitando moderatamente. Sono stati effettuati 2 lavaggi successivi di 10 minuti ciascuno in una soluzione 100 mM Tris 9.5, 100 mM NaCl, 50 mM MgCl2 a temperatura ambiente agitando moderatamente.
I vetrini sono stati coperti con 100 µl della soluzione NBT-BCIP (200 µl di Roche NBT-BCIP in 10 ml della soluzione Tris 9.5/NaCl/MgCl2) e lasciati ad incubare con
parafilm, in un contenitore in camera umida, in completa oscurità per 1-3 giorni. Il parafilm è stato rimosso e i vetrini sono stati posti in TE per fermare la reazione. I vetrini sono stati disidratati immergendoli per 5 secondi nelle seguenti soluzioni:
40% EtOH 70% EtOH 95% EtOH 2X 100% EtOH 2X xilene
103 I vetrini sono stati analizzati al microscopio ottico (Axioscop, Zeiss) e le immagini sono state acquisite con una videocamera digitale (Laica DC 100 versione 4.1.8.0).
SOLUZIONI UTILIZZATE: 10X PBS pH 7 5X NTE 1.3 M NaCl 2.5 M NaCl 70 mM Na2HPO4 50 mM Tris pH 8 30 mM NaH2PO4 5 mM EDTA pH 8 20X SSC STE 3 M NaCl 0,1 M NaCl 300 mM Na Citrato 10 mM Tris pH 8 1 mM EDTA pH 8 10X PBS / Glicina a 4 °C pH 7 RNasi A a 4 °C 1.3 M NaCl 20 mg/ml 70 mM Na2HPO4 30 mM NaH2PO4 20 mg/ml Glicina
104
2.3 Isolamento e sequenziamento del gene
LEAFY COTYLEDON1-LIKE
2.3.1 Estrazione del DNA
L’estrazione del DNA è stata condotta, in accordo con il protocollo del kit “Genomic DNA from plant” (Macherey-Nagel) su embrioni allo stadio di 5 giorni e foglie giovani (2 cm di lunghezza) di H. annuus, linea pura HOR. I campioni appena prelevati sono stati congelati mediante immersione in azoto liquido, quindi 300 mg di materiale sono stati polverizzati mantenendoli ghiacciati sempre con azoto liquido, in mortaio o tramite sferette di zirconio poste in una provetta con il materiale ghiacciato. La polvere ottenuta è stata sospesa in 400 µl di tampone di lisi e con 10 µl di Rnasi A ed incubata per 1 ora a 65 °C. Il liquido è stato raccolto in una colonna filtrante, provvista nel kit, e centrifugato per 2 minuti a 11.000 x g, è stato recuperato il liquido e posto in una nuova provetta in cui sono stati aggiunti 450 µl di Binding
buffer. Il liquido raccolto viene trasferito in un'altra colonna filtrante e centrifugato 1
minuto a 11.000 x g. A questo punto il DNA è legato al filtro della membrana che viene lavata con tre lavaggi con Wash buffer: 400 µl per 1 minuto e centrifugato a 11.000 x g, 700 µl per 1 minuto, centrifugato a 11.000 x g, ed infine con 200 µl per 2 minuto a 11.000 x g. Alla fine il DNA viene eluito dalla membrana con 50 µl di
Eluition buffer (pre- riscaldato per 5 minuti a 70 °C) tramite centrifugazione per 1
105
2.3.2 Southern blot
La tecnica del Southern Blot è stata utilizzata per identificare enzimi in grado di tagliare il DNA genomico in frammenti, contenenti rispettivamente l’estremità 5’ e 3’ del gene, da utilizzare per il Genome walking.
2.3.2a TAGLIO DEL DNA CON ENZIMI DI RESTRIZIONE
Il DNA genomico è stato sottoposto a digestione enzimatica con il seguente protocollo:
DNA 15 µg
Buffer 10X 1/10 del volume finale Enzima 5 U/µg
H2O x µl fino a volume finale
La quantità totale dell’enzima non deve superare 1/10-20 del volume finale per evitare fenomeni di riduzione dell’efficienza di taglio dovuti alla presenza del glicerolo nella soluzione stock dell’enzima.
La digestione condotta per una notte ad una temperatura di 37 °C, viene bloccata aggiungendo il 20% di stopper (saccarosio 25%, Na2EDTA pH 8, 25 mM, orange-G
0,5%).
Sui campioni di DNA genomico sono state effettuate delle digestione con differenti enzimi di restrizione: PvuI, DraI, EcoRV, StuI, SspI, StyI, RsaI, HpaI.
I prodotti della digestione sono stati caricati su gel d’agarosio (1%) e la corsa elettroforetica è stata effettuata per alcune ore a 20mV.
106 Successivamente il gel è stato sottoposto alla seguente procedura per la preparazione al trasferimento del DNA su membrana:
Depurinazione: il gel è sommerso per 10’ nella soluzione depurinante. La depurinazione del DNA, che facilita il passaggio attraverso le maglie del gel. Breve lavaggio in H2O.
Denaturazione: il gel è sommerso due volte per 15’ nella soluzione denaturante. Breve lavaggio in H2O.
Neutralizzazione: il gel è sommerso due volte per 15’ nella soluzione neutralizzante.
Il trasferimento del DNA su membrana di Nylon carica positivamente (Roche), è effettuato per capillarità, con 20xSSC, per tutta la notte. Questo tipo di trasferimento è stato scelto, sebbene più lungo e laborioso di altri, poiché ha una maggiore efficienza.
La membrana viene quindi lavata con SSC2X, appoggiata su carta da filtro bagnata in SSC2X quindi per fissare il DNA alla membrana questa è posta in UV Stratalinker programmato a 120 mJ, esponendo entrambi i lati.
SOLUZIONI UTILIZZATE:
SSC 20X: Soluzione neutralizzante: NaCl 3 M NaCl 3 M
Sodio-citrato 0,3 M Tris-HCl 0,5 M Portare a pH 7 con acido citrico pH 7.5
e autoclavare
Soluzione denaturante: Soluzione depurinante NaOH 0,5 M HCl 0,250 M
NaCl 1,5 M
107 Sonde utilizzate
Dal clone HaK-3 contenente tutto il cDNA dell’L1L, sono stati amplificati tramite PCR due frammenti che sono stati purificati su gel, marcati con la marcatura non radioattiva, e quindi sono state usati come sonde in prove d’ibridazione su Southern
blot.
1) Sonda1: regione compresa tra i primers SON1F-SON1R posizionati al 5’ della sequenza codificante l’L1L. Lunghezza: 143 pb
2) Sonda3: regione compresa tra i primers SON3F-SON3R posizionati al 3’ della sequenza codificante l’L1L. Lunghezza: 222 pb
Marcatura non radioattiva della sonda
Le sonde sono state marcate utilizzando “DIG-DNA Labelling Kit Non radioactive” (Roche), seguendo le istruzioni fornite dalla ditta.
Si denaturano a 100 °C per 10 minuti circa 0,1-1 µg di DNA in 15 µl di acqua e, dopo aver rapidamente raffreddato la soluzione in ghiaccio, si aggiungono 2 µl di esanucleotidi, 2 µl di dNTP (1 mM dATP, 1 mM dGTP, 1 mM dCTP, 0,65 mM dTTP e 0,35 mM DIG-dUTP) ed 1 µl di Klenow.
Dopo incubazione per una notte a 37 °C, si interrompe la reazione polimerasica con 2 µl di Na2EDTA 0,2 M (pH 8) e il DNA si precipita con 2,5 µl di LiCl 4 M e 75 µl
di etanolo assoluto freddo (-20 °C), lasciandolo per 1 ora a –80°C.
Successivamente si centrifuga il campione a 12000 rpm per 30 minuti a 4 °C, ed il
pellet recuperato si lava due volte in etanolo al 70% e poi, quando è asciutto, si
108 2.3.2c IBRIDAZIONE SU FILTRO
Si pone la membrana nel tubo di ibridazione e si procede ad una preibridazione della durata di circa 1 ora nell’apposito termostato a 39 °C (20 ml di soluzione di preibridazione per circa 100 cm² di membrana).
Si denatura la sonda marcata mediante bollitura per 10 minuti e la si raffredda immediatamente in ghiaccio. Si procede poi con l’ibridazione ponendo nel tubo la soluzione di ibridazione (soluzione di preibridazione contenente sonda marcata alla concentrazione di 20 ng/ml) per una notte, sempre a 39 °C.
Le soluzioni di preibridazione e di ibridazione sono preriscaldate a 68 °C. SOLUZIONI UTILIZZATE:
Buffer 1
NaCl 0,15 M
Acido Maleico 0,10 M, pH 7,5
Blocking stock solution
Sciogliere il blocking reagent fornito dalla ditta in Buffer 1.
Soluzione di preibridazione DIG-Easy Buffer
Soluzione di ibridazione
109 2.3.2d LAVAGGI DELLE MEMBRANE E RIVELAZIONE
Si recupera la sonda e si sottopongono le membrane ad una serie di lavaggi con crescente potere stringente.
I lavaggi, che vengono effettuati sempre nel tubo di ibridazione, sono: § SSC 2X (SDS 0,1%)
A temperatura ambiente, 5 minuti, per due volte § SSC 0,5X o SSC 0,1X (SDS 0,1%)
A 65 °C, 15 minuti, per due volte
A questo punto si procede alla visualizzazione della sonda ibridata utilizzando il “Dig Luminescent Detection Kit” (Roche).
Si lava la membrana in Washing buffer per 5 minuti e si tiene in lenta agitazione in Buffer 2 per 30 minuti.
Successivamente la membrana viene incubata con anticorpo anti-digossigenina legato alla fosfatasi alcalina, diluito 1:20000 in Buffer 2, per 30 minuti in agitazione.
Si effettuano, di seguito, 2 lavaggi di 15 e 20 minuti rispettivamente in Washing buffer e poi si equilibra la membrana per 5 minuti in Buffer 3.
Per visualizzare la chemioluminescenza della sonda ibridata, si procede in camera oscura cospargendo la membrana, adagiata su una pellicola trasparente, di CSPD STAR (circa 1 ml per membrane di 100 cm²) e incubando per 5 minuti a temperatura ambiente.
Successivamente, dopo aver tolto la soluzione in eccedenza (senza seccare la membrana), si effettua ancora un’incubazione per 5 minuti a temperatura ambiente. Si espone infine su lastra Hyperfilm MP (Amersham) per 5’-15’, e successivamente si sviluppa e si fissa con le soluzioni Kodak (seguendo le istruzioni fornite dalla ditta).
110 SOLUZIONI UTILIZZATE Buffer 1 NaCl 0,15 M Acido Maleico 0,1 M, pH 7,5 Washing buffer
Buffer 1 contenente 0,3% Tween 20
Buffer 2
Blocking stock solution diluita 1:10 in Buffer 1
Buffer 3
Tris-HCl 100 mM, pH 9,5 NaCl 100 mM
111
2.3.3 GENOME WALKING 5’ e 3’ RAGE
La tecnica della 5’ e 3’ RAGE è stata utilizzatam nel Genome Walking per ottenere la sequenza completa dell’intero gene dell’L1L. A questo scopo è stato utilizzato il kit “GenomeWalker Universal Kit” (Clontech).
2.3.3 a DIGESTIONE
Il DNA genomico viene digerito con i diversi enzimi di restrizioni scelti grazie alle informazioni derivate dal Southern Blot, ovvero DraI e SspI, per ottenere il frammento a monte del gene, mentre DraI ,SspI e HpaI sono stati scelti per il frammento a valle.
Per ogni reazione sono state utilizzati differenti provette da 1,5 ml contenenti: 25 µl di DNA genomico (0,1 µg\µl)
8 µl di enzima di restrizione (10 U\µl) 10 µl buffer enzima di restrizione (10X) 57 µl H2O deionizzata
In un volume totale di 100 µl
La soluzione è stata incubata 2 ore a 37 °C e successivamente agitata vigorosamente per 10 secondi e posta di nuovo a 37 °C per tutta la notte.
2.3.3b PURIFICAZIONE
Alla fine della digestione, il DNA è stato purificato aggiungendo 100 µl di fenolo, agitando e centrifugando per 3 minuti a 13.000 x g per separare le due fasi organica e
112 acquosa. Alla fase acquosa recuperata è stata aggiunto un ugual volume di cloroformio e le due fasi separate di nuovo centrifugando per 3 minuti a 13.000 x g. La fase acquosa è stata trasferita in una nuova provetta e sono stati aggiunti 2 volumi di etanolo al 95%, 1\10 di volume di 3M sodio acetato (pH 4,5) e 1 µl di glicogeno, il DNA digerito è lasciato precipitare a -20 oC per tutta la notte. Il DNA precipitato è
stato raccolto con una centrifugazione a 10.000 xg, per 25 minuti a 4 °C, lavato per due volte con etanolo freddo all’80% e lasciato ad asciugare all’aria. Il DNA è stato infine solubilizzato in 15 µl di tampone Tris-EDTA (10M\0,1M pH 7,5). A questo punto 1 µl di campione viene visualizzato su gel di Agarosio (1,5%) per quantizzare il DNA recuperato e controllare la qualità della digestione.
2.3.3c LEGAME DEGLI ADATTATORI
Ad ogni campione sono stati legati particolari adattatori caratterizzati da un gruppo amminico all’estremità 3’ in grado di bloccare l’estensione dell’estremità per prevenire la formazione di un sito di attacco del primer AP1, inoltre se l’adattatore viene esteso al 5’ non si crea il sito di legame con il primer AP1 e la reazione si blocca, infine la lunghezza inferiore dei primers che si legano all’adattatore consentono una PCR soppressiva, prevenendo l’amplificazione di stampi con estremità 3’ estesa contenenti il sito per il legame con il primer AP1.
113 Miscela di legame:
X µl contentenenti 0,4 µg di DNA digerito e purificato per ogni digestione 1,9 µl GenomeWalker Adaptor (25µM)
1,6 µl Ligation Buffer 10X 0,5 µl T4 DNA Ligasi (6 U\µl) X µl di H2O fino a 8 µl
La miscela è stata incubata per 16 ore e la reazione fermata tramite incubazione a 70 °C, sono stati quindi aggiunti 72 µl di Tris-EDTA (10M\1M, pH 7,5) e la miscela agitata vigorosamente per 15 secondi.
2.3.3d AMPLIFICAZIONI
Sono state effettuate due amplificazioni successive utilizzando come stampo rispettivamente il DNA legato agli adattatori e la miscela della I PCR diluita 1:50.
Miscela I PCR Cicli utilizzati: 1 µl Template
0,5 µl dNTP 20 mM 5 µl Buffer 10X
1 µl Adaptor Primer1 (10µM)
x µl Primer specifico1 (In7 per 5’ RAGE, 3RAGE3 per 3’RAGE) 1 µl Advantage 2 Polymerase Mix 50X
40,25 µl H2O
94° 72° 94 ° 67° 67° 25’’ 3’ 25’’ 3’ 7’ 7 cicli 32 cicli
114 Miscela II PCR Cicli utilizzati:
1 µl Template
0,5 µl dNTP (20 mM) 5 µl Buffer (10X)
1 µl Adaptor Primer2 (10µM)
1 µl Advantage 2 Polymerase mix 50X
x µl Primer Specifico2 (1RAGE CDS per 5’ RAGE, 3RAGE2 per 3’RAGE) 40,06 µl H2O
I prodotti delle amplificazioni sono stati controllati su gel di agarosio (1,5%) e i frammenti di dimensione attesa sono stati clonati e sequenziati.
94° 72° 94 ° 67° 67° 25’’ 3’ 25’’ 3’ 7’ 5 cicli 20 cicli
115
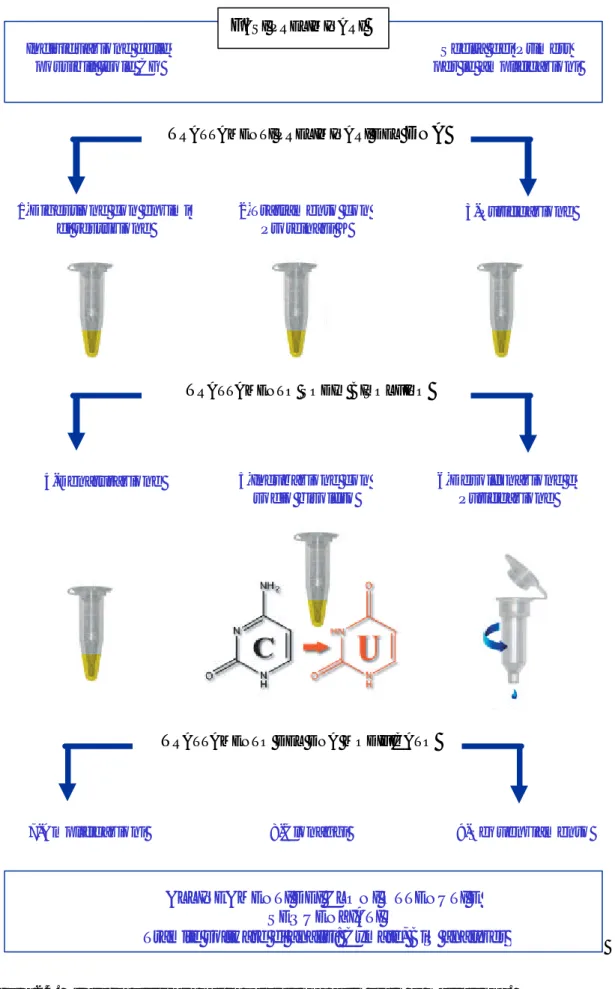
2.4 Analisi delllo stato di metilazione
Due zone indicate come Isola1 (Is1) e Isola2 (Is2) potenzialmente contenenti residui di citosina passibili di metilazione differenziale sono state individuate con il programma “CpGfinder” disponibile al sito www.softberry.com.
Per riconoscere in una sequenza di DNA i residui di citosina, modificati biochimicamente per l’aggiunta in posizione 5 dell’anello purinico di un gruppo metilico, è utilizzabile un trattamento con sodio bisolfito, seguito da una reazione di desolfonazione, che è in grado di convertire in uracile solo le citosine che non hanno subito la metilazione.
Fig.2.3. Rappresentazione della reazione di conversione effettuata dal sodio bisolfito.
Sono stati usati due diversi kit commerciali in accordo con i protocolli degli stessi kit oppure con alcune modifiche.
116
2.4.1 Preparazione dei campioni
La denaturazione del DNA deve essere completa affinchè tutte le citosine non metilate siano effettivamente esposte alla rezione di solfonazione. Il DNA utilizzato era quindi “nativo” o sottoposto a opportuna digestione per ottenere frammenti più facilmente denaturabili.
Il DNA è stato digerito con l’endonucleasi SspI, che genera un frammento di 2024 pb contenente entrambe le Is1 e Is2, incubando per una notte a 37 °C come segue:
10 µl DNA genomico (1µg) 4 µl Buffer 1x
1 µl enzima SspI (10U/µl) H2O q.b fino a 40 µl
Il DNA digerito è stato deproteinizzato con Proteinasi K (600 U\ml), per 1 ora a 37 °C e purificato con il kit “Wizard SV gel and PCR clean-up system” (Promega).
117
Figura 2.4. Strategia utilizzata per la valutazione dello stato di metilazione. ALLINEAMENTI DEI CLONI OTTENUTI E
SEQUENZIATI
Tramite software di analisi: Cymate, BiQ analyzer
Scelta dei Primers per le amplificazioni Individuazione delle
possibili Isole CG
FASI PRELIMINARI
7-Amplificazioni 8-Clonaggi 9-Sequenziamento TRATTAMENTO DEL DNA MODIFICATO
4-Denaturazione 5-Incubazione con sodio bisolfito
6-Desolfonazione e Purificazione TRATTAMENTO SODIO BISOLFITO
1-Digestione con enzimi di restrizione
2-Trattamento con Proteinasi K
3-Purificazione TRATTAMENTI PRELIMINARI DEL DNA
118 2.4.1a TRATTAMENTO CON SODIO BISOLFITO IN ACCORDO CON IL
PROTOCOLLO CHEMICON
Il kit CpGGenome™ FastDNA (Chemicon) prevede l’aggiunta di 550 µl di DNA
Modification Reagent contenente il sodio bisolfito. La soluzione deve essere portata a
pH 7 con l’aggiunta di NaOH. L’incubazione viene protratta a 55 °C per 6-18 ore, a seconda della prova.
Il DNA modificato viene incubato in ghiaccio per 5 minuti, alla soluzione vengono poi aggiunti 750 µl di Binding buffer, posto in una colonna e centrifugato a 5.000 x g per 30 secondi, questo viene ripetuto due volte. Vengono aggiunti 750 µl di Wash
buffer e centrifugato per 1 minuto. Vengono aggiunti 50 µl di soluzione di
desolfonazione e incubato per 10 minuti poi centrifugata per 30 secondi. La membrana viene lavata due volte con 750 µl di Wash buffer e centrifugata per 30 secondi. Infine viene effettuata una centrifugata a vuoto per eliminare i residui di tampone e la membrana viene eluita con 30 µl di Eluition buffer incubato per 1 minuto e recuperata tramite centrifugazione di 1 minuto.
2.4.1b TRATTAMENTO CON SODIO BISOLFITO IN ACCORDO CON IL PROTOCOLLO SIGMA
Il Kit Imprint™ DNA Modification Kit (SIGMA) è stato utilizzato seguendo la procedura One-step Modification descritta qui di seguito.
1.1 ml di DNA modification solution è stato aggiunto ad una fiala contenente la DNA
Modification Powder, la soluzione è stata agitata energicamente per due minuti o
comunque fino a chiarificazione, eventualmente aiutando la solubilizzazione con due minuti a 65 °C. Sono stati poi aggiunti 40 mL di Balance Solution. 110 µL della soluzione appena preparata sono stati uniti a 10 µL di H20 contenente da 250 ng a 1
119
µg di DNA, la provetta è stata quindi incubata per 6 minuti a 99 °C e poi immediatamente posta a 65 °C per 90 o 180 minuti.
La composizione dei reagenti non è reso noto dalla casa produttrice il Kit.
Dopo la modificazione del DNA è necessaria la desolfonazione e purificazione del campione che richiede prima di tutto la preparazione di mini-colonne. Per ogni campione una mini-colonna è stata posta su una provetta da 2 ml e sono stati aggiunti 300 µL della Capture Solution lasciati incubare per 1 minuto.
Sulla mini-colonna così preparata è stata posta la soluzione di DNA al termine della incubazione e centrifugata a 12.000 x g per 20 secondi. Sono quindi stati aggiunti 200 µL della soluzione preparata aggiungendo 8.2 ml di etanolo assoluto a una provetta di Cleaning Solution e centrifugata per 20 secondi a 12.000 x g . A questo punto sono stati aggiunti 50 µl della soluzione Balance/Ethanol Wash Solution preparata unendo 10 mL of Balance Solution a 1.1 ml di 90% etanolo. A un’incubazione di 8 minuti a temperatura ambiente, è seguita una centrifugazione per 20 secondi a 12.000 x g. La colonna è poi stata lavata con 200 µL di etanolo 90% centrifugando per 40 secondi a 12.000 x g. Infine il DNA è stato eluito dalla colonna con 10 µL of Elution Solution incubando per 1 minuto and poi centrifugando per 20 secondi a 12 000xg. Il DNA modificato è ora pronto per essere utilizzato nelle PCR, ma può anche essere conservato a –20 °C per un massimo di 2 mesi.
120 2.4.1c TRATTAMENTO IN ACCORDO CON IL PROTOCOLLO SENZA KIT
E’ stata effettuata una prova con l’uso diretto del sodio bisolfito senza utilizzare kit.
A 250 ng DNA in un volume di 18 µl vengono aggiunti 7 µl di NaOH 3M ed incubati per 15 minuti a 42 °C e successivamente 5 minuti a 100 °C.
Alla soluzione di DNA denaturato vengono aggiunti 208 µl di soluzione di sodio bisolfito 2,8 M e 12 µl di idrochinone 185 mM e portata a pH 5 con circa 3 µl NaOH 10 M per un volume totale di 220 µl. La desolfonazione e purificazione del DNA trattato è effettuata secondo la procedura del Kit Chemicon.
121
1 1 µg DNA
foglie Chemicon - - 10’ a 37 °C - 18 ore a 50 °C
2 1 µg DNA
foglie Chemicon - - 10’ a 37 °C - 16 ore a 55 °C
3 1 µg DNA
foglie Chemicon
Digestione con SspI, trattamento con
Proteinasi K
10’ a 100 °C 10’ a 37 °C - 6 ore con ciclo:100 °C per 30’’,
55 °C per 20’
4 250 ng DNA
foglie Chemicon
Digestione con SspI, trattamento con
Proteinasi K
15’ a 80 °C 15’ a 37 °C 10 mM
Formate beads di agarosio 6 ore con ciclo: 100 °C per 30’’,
55 °C per 20’
5 250 ng DNA
foglie Chemicon
Digestione con SspI, trattamento con
Proteinasi K
5’ a 100 °C 15’ a 50 °C - 1 ora a 95 °C
6 250 ng DNA
foglie Sigma
Digestione con SspI, trattamento con
Proteinasi K
6’ a 99 °C 15’ a 50 °C - 90’ a 65 °C
7 250 ng DNA
foglie Sigma
Digestione con SspI, trattamento con
Proteinasi K
6’ a 99 °C 15’ a 50 °C - 180’ a 65 °C
8 250 ng DNA
foglie Nessuno
Digestione con SspI, trattamento con
Proteinasi K
5’ a 100 °C 15’ a 42 °C 10 mM 6 ore con ciclo: 95 °C per 30”,
55 °C per 15’ 9 500 ng DNA embrioni 1 µg DNA foglie Chemicon
Digestione con SspI, trattamento con
Proteinasi K
10’ a 100 °C 15’ a 37 °C -
2’ a 100 °C, 25’ a 55 °C, 2’ a 100 °C, poi per 6 ore con ciclo: 100 °C 1’30”,
55 °C per 30‘. Infine “overnight” a 55 °C
122
2.4.2 Amplificazioni dei campioni trattati con sodio
bisolfito
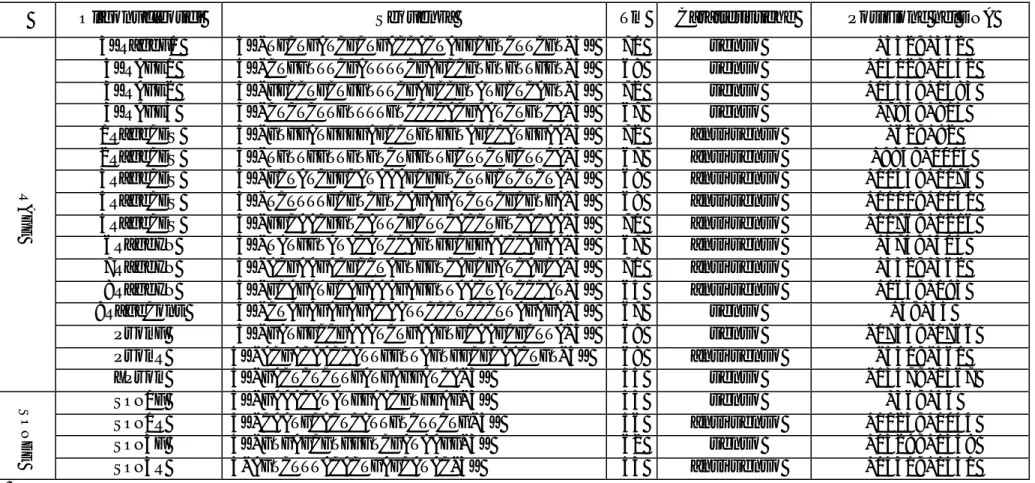
Sono state condotte due amplificazione successive utilizzando come stampo nella prima PCR il DNA derivato dai trattamenti con sodio bisolfito descritti sopra e nella seconda PCR la miscela della prima. Una serie di primer, mostrata nella tabella 2.2 è stata utilizzata in combinazioni diverse al fine di individuare la più idonea all’amplificazione delle Is1 e Is2.
I PCR:
Concentrazione finale Template: DNA trattato 5 µl
Buffer 1X MgCl2 1,25 mM
dNTPs 0,20 mM P1 specifico 0,5 µM P2 specifico 0,5 µM Taq Polimerasi 5 U\µl H2O q.b. fino a 50 µl
94° 94° 42° 72° 72° 2’ 30’’ 30’’ 1’ 30’’ 7’ 35 cicli
123 II PCR Concentrazione finale Template: I PCR 1 µl Buffer (10X) 1X MgCl2 1,25 mM dNTPs 0,20 mM P3 specifico 0,4 µM P4 specifico 0,4 µM Taq Polimerasi 5 U\µl H2O q.b. fino a 50 µl
I prodotti di amplificazione delle dimensioni attese sono stati purificati su gel, clonati e sequenziati.
2.4.2a SOFTWARE UTILIZZATI
Programma Descrizione Indirizzo internet:
BiQAnalyzer http://biq-analyzer.bioinf.mpi-sb.mpg.de
CyMATE
Analisi sequenze convertite da bisolfito
http://www.gmi.oeaw.ac.at/CyMATE CpGfinder Isole CpG http://www.softberry.com/
MatInspector Cis-acting sequence http://www.genomatix.de/ ClustaW Fasta Blast Allineamenti http://www.ebi.ac.uk/ 94° 94° 42° 72° 72° 2’ 30’’ 30’’ 30’’ 7’ 35 cicli
124
2.4.3 Clonaggio
I prodotti di amplificazione sono clonati secondo il protocollo del “TOPO-TA Cloning Kit” (Invitrogen). La procedura qui descritta è utilizzata ogni volta sia stato necessario effettuare un clonaggio
2.4.3a INSERZIONE DELL’AMPLIFICATO NEL DNA PLASMIDICO In una provetta da 0.5 ml è stata preparata la miscela di legame:
4 µl Prodotto di PCR 1 µl TOPO Vector 1 µl Salt Solution H2O q.b fino a 6 µl
La miscela è stata incubata a 23 °C, per 30 minuti e messa in seguito in ghiaccio.
2.4.3b TRASFORMAZIONE DELLE CELLULE
Sono stati aggiunti 2 µl della miscela di legame in una provetta contenente una coltura di cellule di “One Shot Chemically Competent E. coli”, rese chimicamente competenti ad accogliere il DNA plasmidico. Per consentire la trasformazione, in altre parole l’inserimento del plasmide nelle cellule batteriche, è necessario uno “shock” termico che è eseguito come descritto: la provetta è stata incubata in ghiaccio per 30 minuti, quindi rapidamente trasferita a 42 °C per 30 secondi e subito dopo di nuovo in ghiaccio per 2 minuti. Successivamente sono stati aggiunti 250 µl di SOC
125 (brodo di coltura) e di seguito è stata effettuata un’incubazione di un’ora a 37 °C in agitatore orizzontale. Quindi 100 µl della coltura di cellule trasformate sono stesi accuratamente su piastre di substrato solido di coltura (preriscaldate a 37 °C). Queste sono state incubate a 37 °C o.n., per permettere la crescita batterica. La selezione delle colonie viene fatta col metodo della distinzione blu-bianco: nel plasmide è infatti presente l’operone Lac-Z che codifica per il polipeptide beta-galattosidasi; quest’enzima è in grado di scindere la molecola X-gal (5-bromo-4-cloro-3-indolil-beta-Dgalattopiranoside) in 5-bromo-4-cloroindolil e 5,5’-dibromo-4,4’-dicloroindigo che è di colore blu. Poiché durante il clonaggio, il frammento s’inserisce all’interno dell’operone, viene interrotta la sequenza per la β-galattosidasi che non è più sintetizzata. Le cellule che contengono l’inserto non saranno in grado di scindere lo X-gal presente nel substrato, quindi saranno di colore bianco; mentre quelle prive dell’inserto metabolizzeranno lo X-gal colorandosi di blu. Le colonie bianche sono state sospese singolarmente in provette con LB e ampicillina, e fatte crescere o.n. a 37 °C. I cloni si conservano in glicerolo al 20% a -80 °C.
2.4.3c SELEZIONE DEI CLONI
Nella fase di amplificazione possono crearsi artefatti, in quantità non visualizzabili su gel di agarosio, che sono però clonati contemporaneamente agli amplificati specifici. Tali artefatti, che possono essere della stessa dimensione degli amplificati specifici, presentano, ad entrambe le estremità, lo stesso oligonucleotide (P1/P1 e P2/P2). E’ necessario quindi individuare le colonie bianche effettivamente contenenti l’inserto d’interesse. A tale proposito è stata effettuata una PCR da colonie, in cui è utilizzata direttamente un’aliquota della sospensione batterica. Devono essere impiegati non più di 2 µl di sospensione batterica, in volumi di 20 µl totali di miscela di amplificazione. Le condizioni di PCR sono le stesse già descritte, è stato però
126 prolungato a 10 minuti il tempo iniziale di denaturazione, prima della fase ciclica, per ottenere lisi delle cellule batteriche e liberazione del DNA plasmidico, che deve essere impiegato come stampo. Sono stati considerati positivi i cloni i cui amplificati sono ottenibili solo con la miscela di incubazione contenente entrambi gli oligonucleotidi, e che, visualizzati su gel di agarosio, risultano essere delle dimensioni attese. SOLUZIONI UTILIZZATE: LURIA BERTANI Bactotriptone 10g Yeast extract 5g NaCl 5 g H2O 1 l AMPICILLINA
10 mg/ml solubilizzata in alcool etilico al 70%.
SUBSTRATO SOLIDO PER PIASTRE Bactotriptone 8 g Yeast extract 5 g NaCl 5 g Bacto-Agar 15 g H2O 1 l IPTG 100 mM solubilizzato in acqua X-GAL 40 mg\ml solubilizzato in dimetilformammide
127 Il DNA plasmidico dei cloni selezionati è stato estratto in accordo con il protocollo “Wizard Plus SV Minipreps DNA Purification System”. Circa 20 µl di crescita dei cloni conservati a -80 °C sono stati sospesi in 6 ml di substato LB, contenente ampicillina, e incubati a 37 °C o.n. Al termine dell’incubazione la crescita batterica è stata centrifugata a 10000 rpm per 5 minuti, in centrifuga da tavolo. Il surnatante è stato scartato, le cellule impacchettate sono state nuovamente sospese in 250 µl di tampone di sospensione contenente RNasi. Quindi sono stati aggiunti 250 µl di tampone di lisi e la miscela incubata a temperatura ambiente per 5 minuti. Successivamente sono stati aggiunti 10 µl di Proteasi Alcalina ed incubato per 5 minuti ed infine aggiunti 350 µl di soluzione neutralizzante. Al fine di eliminare la fase precipitata, è stata effettuata una centrifugazione a 14000 rpm per 10 minuti; il surnatante, contenente il DNA plasmidico, è stato recuperato e messo in una provetta munita di filtro. Quindi, è stata condotta una centrifugazione a 14000 rpm per 1 minuto e la soluzione che attraversa il filtro è stata scartata. Il filtro è stato lavato due volte, rispettivamente, con 750 µl e 250 µl di tampone di lavaggio, centrifugando a 14000 rpm per 1 minuto e successivamente per 2 minuti. Un’ulteriore centrifugazione nelle stesse condizioni è stata eseguita per eliminare i residui del tampone di lavaggio. Il DNA, presente nel filtro, è stato recuperato aggiungendo 50 µl di H2O
deionizzata e centrifugando a 14000 rpm per 1 minuto. 2.4.3e SEQUENZIAMENTO DEL DNA
I plasmidi contenenti gli inserti sono stati sequenziati in entrambe le direzioni (usando gli oligonucleotidi M13R e M13F) con un sequenziatore automatico da parte della ditta MWG (Ebersberg, Germany). Sono sufficienti 1,5 µg circa di DNA che deve essere inviato in provette da 1,5 ml.
128
Oligonucleotidi Sequenza Tm Caratteristiche Posizione nel DNA
CNG2F 5'-CTTGTGATATATGCATCATCCAAGGTCCTA-3’ 64 senso -360;-390
CG1R 5'-CCAATCAATAGTTAGTTGGCAGA-3’ 57 antisenso -275;-298
CG1F 5'-TAGCTAGTGGAGATGGATATGAT-3' 57 senso -665;-689
CNG1R 5'-GGACCTTGGATGATGCATATATCACAAGAA-3’ 64 antisenso -362;-392
Is1F 5'-ATGTAATGGTAAAGTTATAGT-3’ 48 senso -1060;-1081
Is1R 5'-CTATATTTCTTAATCCAATCAATA-3’ 51 antisenso -285;-261
Is1Fnest 5'-ATGTTTAAGATATATGTAGATATA-3’ 49 senso -960;-936
Is1Rnest 5'-AATATCTAAATTATTTCATAATAAT-3’ 47 antisenso -415;-390
Is2F 5'-ATTATTATGAAATAATTTAGATATT-3’ 47 senso -415;-390
Is2R 5'-TTCCAACACCAAACATAATAACTAT-3’ 55 antisenso 413;438
Is2Fnest 5'-TATTGATTGGATTAAGAAATATAGAA-3’ 52 senso -285;-311
Is2Rnest 5'-TATCTATATTATATATATACACACAC-3’ 52 antisenso +264;+290
Pippo 5'-CCCATAAAATTAAAAAAACATTAT-3’ 49 antisenso +144;+168
Nero 5'-TCTTTAAAACCTTRAATAATA-3’ 45 antisenso -356;-377
Blu 5'-GTTGATAYATGAAATTTTAT-3' 44 senso -834;-854
Rosso 5'-TYYAGATTTATTGAGTTTTT-3' 45 senso -713;-734
Viola 5'-ACATATTTCCTTRATTTATA-3' 44 antisenso -461;-481
Giallo 5'-AAATTATGATTAAATYTGTT-3’ 42 senso -292;-312
Arancio 5'-GTGGTTAAATCAAATGTATT-3’ 47 senso -155;-175
METILAZIONE
Indaco 5'-TATTATATATATACACACAC-3’ 45 antisenso +264;+284
CHI-SR 5’-GTAGATGGAGAGTGTCAG-3’ 54 antisenso +1671;+1689
CHI-SF 5’-TATGCTCAGTGTAAAGAC-3’ 50 senso +1532;+1550
CHI-TcDNA 5’-CTAGAGAGAGACAATTCC-3’ 51 senso +5;+13
CHI-REL 5’-CAATGCACTCATTGTCTTC-3’ 54 antisenso +1025;+1044
ANCHOR 5’-GACCACGCGTCTCGATGTCGAC-3’ 72 - -
ACT3 5’-TYTCTGGWGGWGCAACCACC-3’ 68 antisenso -
RT
-PCR
129
Oligonucleotidi Sequenza Tm Caratteristiche Posizione nel DNA
3’RageF1 5’-TGCTGATCGCTGACCACTAGGCGTCTTCGT-3’ 71 senso +332;+362
3’RAGE1 5’-CTGGTTTCGATTTTCGAGCCGTGTGTTGGT-3’ 68 senso +1512;+1542
3’RAGE2 5’-GGCCTGCTGGTTTCGAGCCGTATGCTCAGT-3’ 72 senso +1553;+1583
3’RAGE3 5’-CTCTCTTGTTTTGTCCCCACGAATCTGTCA-3’ 67 senso +785;+815
1RageCDS 5’-GTGGATGGGGAGCCTGTGGTAGCCATGGAA-3’ 72 antisenso +62;+92
2RageCDS 5’-TGTTGGTTGTGTCTGGTTGCTTCTGCTTCA-3’ 67 antisenso +984;+1014
3RageCDS 5’-GCTATCGGCATAAAGCGGTCTTGCTCTCTA-3’ 68 antisenso +1045;+1075
4RageCDS 5’-TCTTTTGCGTCGTCAGAGATCTTCGCGTGA-3’ 68 antisenso +1111;+1141
5RageCDS 5’-GGCAACGGTCATTCGCTTCACCTGTCACAA-3’ 70 antisenso +1176;+1206
6RageIN 5’-TATGGTATACATCCAGTGGCGGAACCAGAA-3’ 67 antisenso +374;+404
7RageIN 5’-ACGAAGACGCCTAGTGGTCAGCGATCAGCA-3’ 71 antisenso +332;+362
8RageIN 5’-GCAGATGCAGAAAGAGGTTAACTATCCCAT-3’ 65 antisenso +163;+193
9RageCont 5’-CTAGAGAGAGACAATTCCCTCCCTTAGAGA-3’ 67 senso +5;+35
PromF 5’-GATGGCCGAAATCTGAAGTGCAAGCGCTTA-3’ 68 senso -1736;-1766
PromR 5’-ACGACAACCATTGGTTAGTGGCGCAACTGT-3’ 68 antisenso -530;-560
RAGE
XProm 5’-GACTCTCTTGATGAGGATCA-3’ 55 senso -1347;-1367
SON1F 5’-GAAACATATGGAACGTGGAG-3’ 55 senso +36;+56
SON1R 5’-CAATGCACTCATTGTCTTCTG-3’ 56 antisenso +1023;+1044
SON3F 5’-GTGAGCGTGGGTCGATAAGG-3’ 62 senso +1329;+1349
SONDE
SON3R 5-AGTCTTTACACTGAGCATAC-3’ 53 antisenso +1531;+1551