1
INTRODUZIONE
LE TALASSEMIE
Le talassemie rappresentano un gruppo eterogeneo di disordini ereditari caratterizzati dalla diminuzione o assoluta mancanza di sintesi di una o più catene globiniche, α o β, che si traducono in più o meno gravi anomalie nella sintesi dell’emoglobina e successiva minore sopravvivenza dei globuli rossi (1, 2). Le talassemie derivano da alterazioni a carico dei geni globinici, in particolar modo da delezioni più o meno estese o da mutazioni puntiformi di varia natura i cui effetti si possono manifestare nei processi di trascrizione e traduzione, portando alla totale assenza o riduzione della sintesi proteica, ad uno squilibrio nella velocità di sintesi oppure alla produzione di catene altamente instabili (3). La condizione di talassemia non è associabile quindi ad un unico difetto genetico, ma è piuttosto un gruppo di alterazioni che producono effetti clinici simili.
1. Emoglobina ed ematopoiesi
Il trasporto di ossigeno dai polmoni ai tessuti viene effettuato dall’emoglobina, una molecola proteica complessa e altamente specializzata contenuta nei globuli rossi. Ogni globulo rosso ne contiene almeno 300 milioni. La porzione proteica dell’emoglobina è costituita da due coppie di catene polipeptidiche ognuna delle quali si associa ad un anello protoporfinico definito eme che lega reversibilmente il ferro. Mentre quest’ultimo rimane lo stesso indipendentemente dall’età, le catene polipeptidiche si associano tra loro in modo variabile; esse vengono generalmente suddivise in due gruppi: il cluster α-globinico (comprendente le catene ζ e α) e il cluster β-globinico (comprendente le catene ε, γ, β e δ).
2
Come mostrato nella tabella seguente, nei globuli rossi dell’embrione, del feto e dell’adulto vi sono quindi, a seconda dell’età, 6 diversi tipi di emoglobina: 4 per l’embrione e per il feto e 2 per l’adulto.
Tipo di Hb Composizione Hb Gower 1 ζ2ε2 Hb Gower 2 α2ε2 Hb Portland ζ2γ2 Hb fetale α2γ2 Hb adulta A α2β2 Hb adulta A2 α2δ2
Tabella 1. Tipi di emoglobina.
Le catene globiniche presentano una struttura estremamente complessa che assicura loro la possibilità di legare velocemente l’ossigeno a livello polmonare e rilasciarlo gradualmente a livello periferico. Le globine si differenziano tra loro sia per il numero che per il tipo di aminoacidi che le compongono; le catene α sono infatti costituite da 141 residui aminoacidici, le catene non α da 146 residui (4). Ogni catena globinica viene prodotta sotto il controllo di un gene strutturale che ne regola l’espressione. Come mostra la figura seguente, nell’uomo i geni globinici sono raggruppati in clusters ed ogni cluster è costituito sia da geni strutturali, presenti in singola copia, attivi dal punto di vista trascrizionale, sia da pseudogeni o copie di geni che con l’evoluzione hanno perso le loro capacità funzionali. Inoltre vi sono anche delle regioni di regolazione, costituite da residui nucleotidici altamente conservati (4).
Figura 1. Struttura dei geni globinici. Figura tratta da “Diagnosis of
3
Tutti i geni globinici, a dimostrazione della comune origine, presentano una struttura simile (4, 5):
- ogni gene presenta tre regioni codificanti, gli esoni, e due regioni intercalari, gli introni, di lunghezza diversa per ogni gene globinico;
- alle estremità 5’ e 3’ di tutti i geni globinici ci sono delle sequenze, UTR, che vengono trascritte ma non tradotte;
- all’estremità 5’ ogni gene globinico presenta una regione promoter fondamentale per la corretta trascrizione del gene stesso.
Le due coppie di geni codificanti per la catena α sono localizzate sul cromosoma 16 mentre i geni codificanti le restanti catene polipeptidiche sono strettamente connesi tra loro e localizzati sul cromosoma 11.
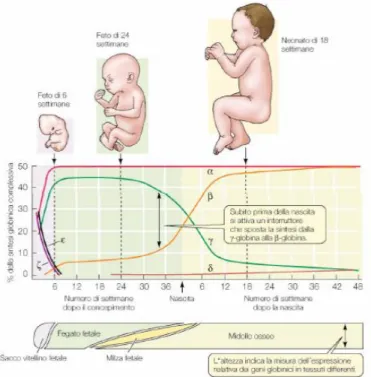
La struttura dell’emoglobina umana si modifica durante lo sviluppo (Fig. 2): durante la vita intrauterina infatti i geni codificanti le globine vengono attivati e disattivati in sequenza attraverso processi di metilazione e demetilazione secondo un fenomeno definito switching emoglobinico (5, 6, 7, 8, 9): dal concepimento, attraverso le tre fasi, embrionale, fetale e post-natale, fino all’età adulta vengono sintetizzati sei tipi diversi di emoglobina.
Figura 2. Espressione tempo e tessuto-specifica delle diverse catene
globiniche umane. Figura tratta da “Biologia L’informazione e l’eredità. Savada, Orians, Heller; Zanichelli 2005”.
4
Come mostra la tabella seguente, inoltre, il processo emopoietico si localizza in distretti e organi diversi a seconda dell’epoca di sviluppo considerata (10).
Fase di sviluppo
Sede emopoiesi Hb principalmente sintetizzate
Periodo embrionale
Sacco vitellino Hb Gower 1 Hb Gower 2 Hb Portland
Le emoglobine sintetizzate durante il periodo embrionale presentano maggiore affinità per l’ossigeno.
8-10° settimana –
6° mese di gestazione
Fegato e milza Hb fetale
In questa fase si verifica il primo switching emoglobinico: iniziano infatti ad esser prodotte le catene globiniche γ e l’emoglobina fetale rappresenta in questo periodo circa il 90% dell’emoglobina totale. Rispetto all’emoglobina adulta quella fetale presenta una maggiore affinità per l’ossigeno permettendo un efficace trasferimento dell’ossigeno dal sangue materno a quello fetale attraverso la placenta.
6-7° mese di gestazione –
epoca postnatale
Midollo: inizialmente ossa lunghe e piatte,
successivamente vertebre, sterno, coste
e creste iliache
HbA HbA2
In questa fase si verifica il 2° switching (il vero switching emoglobinico): compare infatti l’HbA che nel nato a termine raggiunge il 30% circa del totale. L’altra emoglobina tipica dell’età adulta, la HbA2, raggiunge i livelli tipici della vita post-natale, 2-3,4%, solo dopo l’anno di vita. Tabella 2. Variazione tempo e tessuto-specifica del processo emopoietico.
A partire dal 6° mese di vita quindi, in condizioni normali, i globuli rossi contengono il 98% di HbA, tracce di HbF e il 2% di HbA2. Durante la vita adulta una piccola percentuale di HbF continua infatti ad essere espressa ed i suoi livelli possono aumentare anche di dieci volte in conseguenza a vari fattori quali età, sesso, peculiarità genomiche come mutazioni puntiformi a carico del cluster β o in geni correlati (11). L’evento più interessante, e quindi più studiato, in questo processo di sintesi globinica è rappresentato dallo switching dell’emoglobina, in particolare il secondo, attraverso cui si verifica la soppressione del gene γ-globinico, accompagnata dall’aumento complementare dell’espressione del gene β-globinico (12). Si sa ormai che tale processo presenta infatti un’implicazione terapeutica sia nella talassemia che nella drepanocitosi. Il gene γ può sostituire dal punto di vista funzionale il gene β alterato (13): sono stati eseguiti a questo proposito vari studi che prevedono la riattivazione del gene γ. Il meccanismo alla base dello switching non è ancora noto anche se l’ipotesi più accreditata prevede l’esistenza di specifici fattori di trascrizione
5
in grado di influenzare l’espressione stadio-specifica dei geni per le globine (14). L’influenza dell’espressione del gene γ-globinico nell’outcome dei pazienti talassemici viene dimostrata dal notevole miglioramento clinico indotto dalla coeredità di un disordine definito HPFH (Hereditary Persistent of Fetal Hemoglobin), caratterizzato da un’anomala espressione dei geni γ-globinici con percentuali aumentate di HbF (dal 2,5 al 20% del totale).
2. Patogenesi
La sintesi dell’emoglobina è un processo complesso e finemente regolato in modo che il numero delle catene α sia perfettamente pari a quello delle catene β. Quando queste condizioni non si verificano il risultato è un difetto dell’emoglobina. Le talassemie rappresentano un gruppo di anemie emolitiche causate infatti dalla mancanza o deficit di sintesi di una o più catene globiniche. La ridotta o assente sintesi di una globina porta, insieme alla sintesi normale delle altre globine non colpite dal difetto genetico, alla formazione di globuli rossi contenenti al loro interno aggregati delle catene in eccesso con conseguente instabilità. L’anemia conseguente dipende sia dall’eritropoiesi inefficace, caratterizzata dalla morte prematura degli eritrociti intramidollari, che dall’emolisi con conseguente accorciamento della vita media eritrocitaria.
3. Classificazione
Le talassemie derivano da alterazioni a carico dei geni globinici, in particolar modo da delezioni più o meno estese o da mutazioni puntiformi di varia natura. La condizione di talassemia non è associabile quindi ad un unico difetto genetico, ma è piuttosto un gruppo di alterazioni genetiche diverse che producono effetti clinici simili (2).
E’ quindi più opportuno distinguere tra classificazione genetica e clinica della patologia stessa (Tab. 3).
6
Classificazione Sottotipo Criteri di classificazione
Genetica α-talassemia β-talassemia δβ-talassemia
δ-talassemia εγδβ-talassemia
Classificazione basata sul tipo di catena globinica sintetizzata in modo inefficace.
Clinica talassemia minor talassemia intermedia
talassemia major
Classificazione basata sul quadro
ematologico e gravità delle manifestazioni cliniche indipendentemente dal genotipo. Tabella 3. Classificazione genetica e clinica delle talassemie.
4. Epidemiologia
Le varie forme di talassemia presentano incidenze estremamente variabili a seconda del paese considerato (approssimativamente 4,4/10.000 nati vivi). I primi casi descritti risalgono al 1925 in pazienti di origine mediterranea. Successivamente è stata riscontrata una diffusione piuttosto elevata di questa patologia in Africa, Medio Oriente, India, Asia sud-orientale (2). L’incidenza maggiore è presente in Grecia (5-10%), Cipro (10-15%), Libano, Israele, Bulgaria, Romania (7%) (4). L’Italia è uno tra i paesi più colpiti: vi si contano almeno 3 milioni di β-talassemici, la maggioranza dei quali sono eterozigoti, cioè “portatori sani”. In particolare la distribuzione di α- e β-talassemia è pari al 5-10% in Sicilia e nel Salento, del 15% in Calabria e in Sardegna e arriva a percentuali superiori, pari al 25%, nelle regioni meridionali della Sardegna. Fin dagli inizi degli studi sulla talassemia alcuni ricercatori notarono che la sua area di presenza coincideva con quella della malaria: infatti si è potuto constatare come in alcune aree geografiche dove si ha un’alta predisposizione alla malaria, i soggetti talassemici risultavano avvantaggiati poiché più resistenti all’attacco da parte del Plasmodium falciparum, l’agente eziologico della malaria (3, 15) ed anche se non è conosciuto l’esatto meccanismo, è stato ipotizzato che la ragione di tale resistenza sia da ricercare nell’alterazione del ciclo intra-eritrocitario del parassita, attraverso l’inibizione dell’invasione delle emazie o l’interferenza con la lisi cellulare (16). Si tratta quindi di un esempio di polimorfismo bilanciato con selezione dell’allele talassemico, cioè patologico, rispetto a quello normale.
7
5. Prevenzione
Negli ultimi anni il numero delle nascite di nuovi pazienti talassemici è andato riducendosi, grazie ad un programma di prevenzione e diagnosi prenatale attuato nelle regioni a più alto rischio. Come per tutte le malattie croniche e mal curabili, anche per la talassemia la prevenzione è infatti di importanza fondamentale nella gestione complessiva della malattia. Oggi tale prevenzione, anche se costosa e difficile, è possibile attraverso l’individuazione di tutte le coppie a rischio: attraverso uno screening esteso, un’accurata diagnosi e consulenza genetica delle coppie e la diagnosi prenatale è infatti possibile ridurre l’incidenza della malattia.
8
α–TALASSEMIA
L’ α-talassemia, frequente soprattutto in Asia, in alcune regioni del bacino del Mediterraneo e in Africa, è causata da delezioni, di varia entità, dei geni α-globinici; possedendo ciascun individuo quattro geni α-globinici (2), due per ciascun cromosoma 16, si distinguono una vasta gamma di quadri clinici eterogenei in base al numero di geni interessati dall’alterazione genetica (Tab. 4).
Sottotipo Genotipo Quadro clinico Hb principalmente sintetizzate
α-talassemia major Mutazione di tutti i geni α
Grave idrope fetale
con conseguente
morte intrauterina
(23-38 settimane) o sopravvivenza limitata
a poche ore del
neonato.
- Hb Bart (HbB), formata da quattro catene γ incapace di trasportare ossigeno ai tessuti periferici.
- Hb Portland, costituita da due catene ζ e due γ. α-talassemia intermedia Mutazione di 3 geni α L’aspettativa di vita di questi pazienti risulta generalmente normale; il quadro clinico è caratterizzato da anemia lieve-moderata, microcitosi, ipocromia, splenomegalia. Il
quadro anemico può tuttavia peggiorare in corso di episodi infettivi richiedendo un supporto trasfusionale. - HbH, costituita da quattro catene β. I tetrameri β, insolubili, provocano la morte midollare degli eritroblasti e la formazione di precipitati emoglobinici circolanti, definiti corpi di Henz.
α-talassemia minor Mutazione di 2 geni α Condizione generalmente benigna e silente associata a microcitosi ma non ad anemia. Le mutazioni producono in questo caso solo una riduzione nell’espressione dei geni α con conseguente minore quantità di α globine. α-talassemia silente Mutazione di 1 gene α Condizione benigna e silente non associata
ad alterazioni
ematologiche.
La mutazione non determina conseguenze sul fenotipo.
9
β-TALASSEMIA
La β-talassemia rappresenta la forma più comune tra le sindromi talassemiche. Essa è caratterizzata da una riduzione quantitativa o dalla totale assenza di sintesi delle catene β, che risultano strutturalmente normali. Il risultato di questo fenomeno è rispettivamente la diminuzione e la mancanza di molecole HbA funzionali nel periodo postnatale (1).
1. Classificazione
Esistono due forme principali di talassemia β:
- β0: caratterizzata dalla mancanza completa della sintesi della catena β (evenienza molto rara);
- β+: associata a diminuzione della sintesi della catena β (sintesi residua pari al 10% circa).
Dato che ogni individuo eredita un solo gene per la catena β da ogni genitore queste due forme si classificano a loro volta in omozigoti ed eterozigoti. La forma omozigote si associa a quadri clinici eterogenei che vanno dalla dipendenza trasfusionale (talassemia major) ad una condizione di anemia di lieve o media gravità (talassemia intermedia).
La talassemia major o morbo di Cooley, è la forma più grave di anemia emolitica congenita. Tale forma, definita β0,è caratterizzata in genere dalla totale assenza di HbA in quanto causata dalla trasmissione di due alleli del tipo β0.Le manifestazioni cliniche compaiono generalmente al 4-6° mese di vita, quando fisiologicamente si verifica il passaggio dalla prevalente produzione di catene γ a quella di catene β e δ: questa forma si presenta con pallore ingravescente, subittero ed epato-splenomegalia; si associano spesso manifestazioni sistemiche come anoressia, scadimento delle condizioni generali e febbricola. Nella fase conclamata il paziente non trattato presenta i segni dell’anemia con emolisi intramidollare e periferica. L’esame cardiologico può evidenziare cardiomegalia secondaria allo stato ipercinetico. Quando la sintesi delle catene β non è tale da soddisfare le richieste fisiologiche di produzione di emoglobina vengono messi in atto dei meccanismi di compenso che in poco tempo
10
risultano tuttavia insufficienti: iperplasia del midollo rosso ed emopoiesi extramidollare. Inoltre gli eritrociti che si vengono a formare a livello midollare risultano alterati dalla presenza di aggregati di catene α e risultano poco emoglobinizzati per cui, estremamente instabili, non riescono a sopravvivere nello stesso midollo se non per pochi giorni in un processo definito eritropoiesi inefficacie. L’iperplasia midollare spiega le modificazioni ossee responsabili anche della tipica facies “microcitemica” caratterizzata da cranio rotondo e ingrossato, bozze frontali prominenti, naso con radice piuttosto infossata e ali larghe, zigomi pronunciati, prominenza della mandibola. L’esame radiologico del cranio mostra allargamento della diploe con possibile scomparsa del tavolato esterno e alterazione delle trabecole della spongiosa. La neoformazione di trabecole disposte radialmente conferisce il tipico aspetto del “cranio a spazzola”. L’attivazione di focolai di eritropoiesi extra-midollare conduce allo sviluppo di epato-splenomegalia a cui concorrono anche l’emosiderosi dei pazienti trasfusi e l’aumento della funzione emocateretica della milza. La presenza in circolo di eritrociti immaturi, ancora nucleati, dipende per lo più dai focolai di eritropoiesi extra-midollare dove non esistono meccanismi di controllo per il passaggio in circolo degli eritrociti. Gli esami ematochimici mostrano un quadro di anemia microcitica ipocromica, incremento del numero dei reticolociti e iperbilirubinemia indiretta, incremento di ferritina e sideremia secondario all’emolisi e all’eritropoiesi inefficace. Lo striscio del sangue periferico evidenzia alterazioni morfologiche dei globuli rossi. Nel caso di un trattamento trasfusionale adeguato il bambino appare pressoché normale in quanto la soppressione dell’iperplasia midollare indotta dalla trasfusioni previene le modificazioni scheletriche e la mancanza in circolo di globuli rossi alterati evita lo sviluppo di splenomegalia. Tuttavia, il progressivo sovraccarico di ferro introdotto con le trasfusioni determina, a partire dai 10 anni circa, una serie di complicanze di tipo cardiaco, epatico ed endocrinologico che rendono necessaria una terapia ferrochelante.
Il termine talassemia intermedia non si riferisce al genotipo, che può essere omozigote o eterozigote per alterazioni β lievi oppure omozigote per alterazioni β gravi associate a difetti dei geni α- o γ-globinici, ma al quadro clinico che risulta di gravità intermedia tra la forma major e la forma minor di talassemia (1). Questi individui sono infatti caratterizzati spesso da anomalie di entrambi i geni β, ma una o
11
entrambe le mutazioni sono lievi. L’esordio clinico è più tardivo rispetto alla forma major, lo sviluppo psico-fisico è normale, le deformazioni ossee sono scarse o assenti, il grado di anemia è variabile mentre si ha costantemente epato-splenomegalia con siderosi epatica e frequente colelitiasi. Il quadro ematologico è qualitativamente analogo a quello della forma major, tuttavia l’anemia è meno spiccata anche se dalla terza decade di vita possono rendersi necessarie le trasfusioni. La sopravvivenza di questi pazienti è variabile essendo in alcuni lunga, anche in assenza di supporto trasfusionale. Anche i pazienti affetti da talassemia intermedia possono tuttavia sviluppare un quadro di emosiderosi conseguenza dell’incrementato assorbimento intestinale di ferro indotto dall’aumentata eritropoiesi inefficace (1). E’ quindi importante il monitoraggio del sovraccarico di ferro che, in caso di valori di ferritinemia elevati, deve essere eseguito anche attraverso l’utilizzo delle tecniche di secondo livello (T2*RM, SQUID) (17). I più importanti meccanismi genetici in grado di determinare un quadro di talassemia intermedia sono tre: la presenza in omozigosi (o in eterozigosi con una mutazione di maggiore gravità) di mutazioni β-talassemiche lievi o silenti con residua produzione di catene β, la coereditarietà di mutazioni α-talassemiche con conseguente minore produzione di catene α e quindi riduzione dello sbilanciamento esistente tra le catene globiniche, la coereditarietà di determinanti genetici responsabili di un’aumentata sintesi di catene γ. Attraverso questi meccanismi diminuisce il rapporto esistente tra catene α e catene non- α (β o γ) con conseguente minore precipitazione delle catene globiniche α e quindi minore instabilità eritrocita ria (1).
La forma eterozigote della β-talassemia (definita anche talassemia minor o minima o stato di portatore asintomatico di β-talassemia o trait talassemico), al contrario, si presenta con fenotipi molto più lievi: questi individui presentano in genere un basso livello di contenuto emoglobinico (MCH), un diminuito volume cellulare (MCV), lievi alterazioni morfologiche dei loro globuli rossi allo striscio periferico, un aumentato livello di HbA2 associato ad un variabile incremento di HbF, un diminuito rapporto di sintesi delle catene β/α che solo occasionalmente si associa ad anemia. In condizioni normali il trait talassemico non si associa a importanti conseguenze cliniche anche se il riconoscimento di questa forma è essenziale ai fini della profilassi eugenetica della talassemia.
12
2. Patogenesi
La β-talassemia è causata da oltre 200 mutazioni diverse (1); diversamente dalla α-talassemia non vi è, se non in rari casi, ampia delezione del gene globinico: nella maggior parte dei casi si tratta di delezioni di uno o più esoni o di mutazioni puntiformi di una o poche basi, che interferiscono con la funzionalità del gene stesso (18). I meccanismi per lo più in gioco sono rappresentati da mutazioni puntiformi di tipo non senso con arresto della trascrizione del gene o comportanti inserzione di una singola base con conseguente lettura sfalsata del gene, mutazioni di sequenze introniche, con conseguente alterata processazione intranucleare del RNA messaggero o mutazioni della regione promoter che determinano riduzione della sintesi globinica (1). Le mutazioni patogenetiche della β-talassemia vengono classificate in funzione della regione colpita (Tab. 5).
Tipo di mutazione Meccanismo patogenetico
Mutazioni del promotore Ridotta efficacia di trascrizione: le catene globiniche vengono prodotte ma in quantità inferiore (β+talassemia).
Mutazioni del sito di taglio e della poliadenilazione del pre-mRNA
Sintesi di mRNA più lunghi e quindi più instabili del normale con conseguente β+talassemia. Mutazioni non senso oppure di
frameshift: difetti di traduzione dell’RNA
- A livello del codone di inizio: β0talassemia. - A livello del codone di termine: β+talassemia. Delezioni Alterazioni geniche rare nell’ambito della
β-talassemia: la patologia conseguente risulta in questo caso tanto più grave quanto più estesa è la regione coinvolta.
Tabella 5. Classificazione delle mutazioni patogenetiche della β-talassemia.
3. Diagnosi
Da alcuni anni la talassemia major dell’adulto si presenta con quadri clinici più lievi grazie alla diagnosi precoce: l’identificazione di queste forme fin dalla prima infanzia è fondamentale al fine di programmare un efficace trattamento. Ai fini diagnostici l’esame di primo livello è quello emocromo-citometrico che definisce il livello di emoglobina, il numero dei globuli rossi, il loro volume e la loro morfologia. Per la diagnosi di talassemia major e intermedia si ricorre quindi alla quantificazione delle
13
frazioni dell’emoglobina contenute nei globuli rossi che permettono di definire la percentuale di HbA2 e HbF. Per poter eseguire la diagnosi in epoca neonatale lo studio delle frazioni dell’emoglobina non è sufficiente in quanto nei neonati è fisiologico ottenere frazioni diverse di HbA2 e HbF. Si utilizzano pertanto altre procedure come la biosintesi in vitro delle globine e l’analisi del DNA. La condizione di eterozigosi non comporta, come già accennato, alcuna conseguenza né prima né dopo la nascita per cui non costituisce di per sé obiettivo di indagine e rappresenta generalmente solo un possibile esito di esame condotto per ricercare ed escludere lo stato di omozigosi. La diagnosi di eterozigosi è invece importante in epoca adulta al fine di individuare eventuali coppie “a rischio”.
4. Complicanze sistemiche
Negli ultimi decenni la corretta applicazione del supporto trasfusionale e della terapia ferrochelante ha determinato un miglioramento in termini di morbilità e mortalità dei pazienti affetti da β-talassemia. L’aumento di sopravvivenza di questi pazienti ha però fatto emergere complicanze a lungo termine e patologie secondarie: epatopatie, cardiopatie ed endocrinopatie, causate principalmente dalla emosiderosi, sono alcune delle più frequenti complicanze osservabili. Nelle tabelle seguenti vengono elencate le principali complicanze sistemiche della β-talassemia.
Epatopatia
Un quadro di epatopatia cronica di varia gravità si riscontra nel 90% dei pazienti affetti da talassemia major dopo i 15 anni di età (19).
Patogenesi - Sovraccarico di ferro - Infezioni croniche virali
- Accumulo di endotossine o altri metalli - Alterazioni immunologiche
- Predisposizione genetica
- Carenza di vitamine e di sostanze antiossidanti
Caratteristiche Anatomia patologica: fibrosi portale, formazione di ponti che delimitano pseudolobuli e quindi, negli anni, cirrosi. Nei restanti pazienti il quadro è quello di un’epatite cronica attiva o cronica persistente.
14
Cardiopatia
La cardiopatia insorge molto lentamente e in modo progressivo, diventando spesso evidente clinicamente solo quando la funzione del ventricolo è ormai compromessa o quando compaiono aritmie sintomatiche. Dopo la comparsa di manifestazioni cliniche, tuttavia, l’evoluzione può essere rapida, portando all’insorgenza di scompenso cardiaco refrattario (20, 21, 22, 23).
Patogenesi - Sovraccarico di ferro - Anemia cronica
- Infezioni (pericarditi, miocarditi) - Deficit di GH
Caratteristiche Principale causa di morbilità e mortalità.
L’anemia cronica induce una cardiopatia con interessamento delle sezioni destre e importante componente dilatativa, insufficienza tricuspidale e ipertensione polmonare. L’emocromatosi determina, invece, interessamento delle sezioni di sinistra con riduzione della compliance ventricolare, grave insufficienza mitralica e riduzione della cinesi parietale globale.
Tabella 7. Complicanze sistemiche: cardiopatia.
Endocrinopatie
In teoria tutte le ghiandole endocrine possono essere coinvolte dal danno conducendo a quadri clinici di ipogonadismo (50%), ipotiroidismo (10%) e ipoparatiroidismo (9%), che rimangono le complicanze endocrine più frequenti in questi pazienti, ma anche di compromissione della crescita, diabete, ipocorticosurrenalismo, disturbi del metabolismo del calcio, osteopenia ed osteoporosi (1, 24, 25, 26, 27).
Patogenesi - Sovraccarico di ferro: esisterebbe una correlazione tra elevati livelli di ferritina (superiori a 1000 ng/mL) e danno endocrino. Inoltre dagli studi eseguiti utilizzando la risonanza magnetica encefalica emerge la presenza di alterazioni strutturali ipofisarie riconducibili al deposito marziale, che, aumentando la produzione di radicali liberi e la perossidazione lipidica, induce alterazioni cellulari a livello delle membrane mitocondriali, lisosomiali e citoplasmatiche.
Altri fattori patogenetici: - Anemia cronica
- Epatopatia (alterazione della sintesi e del metabolismo ormonale)
Ipotiroidismo I dati sulla funzione tiroidea risultano discordanti; tuttavia gli studi più recenti mostrano una condizione di eutiroidismo in circa l’80% dei pazienti, di ipotiroidismo subclinico (caratterizzato cioè da incremento isolato di TSH, con normali valori di FT3 e FT4) nel 4% e di ipotiroidismo franco nel 12% (28).
15
Endocrinopatie
Deficit staturale Comune è il riscontro di deficit staturale nell’ambito dei pazienti affetti da talassemia major: la patogenesi rimane tuttavia complessa e non completamente chiarita. In letteratura vari autori dimostrano che anche la terapia ferrochelante con Desferrioxamina (DFO) possa contribuire al deficit di crescita: il farmaco sarebbe infatti in grado di inibire la sintesi di DNA, la proliferazione dei fibroblasti e la formazione del collagene; può inoltre causare deficit di zinco che a sua volta comporta riduzione dei valori di fosfatasi alcalina, un enzima zinco-dipendente. Tutto ciò conduce a displasia ossea e riduzione della velocità di crescita (29). Inoltre la crescita staturale viene ulteriormente compromessa da altre complicanze associate quali l’iposurrenalismo, l’ipotiroidismo e l’ipogonadismo (30). Per quanto riguarda invece la secrezione di GH i dati presenti in letteratura sono discordanti forse perché basati su gruppi ristretti di pazienti trattati con protocolli trasfusionali e di terapia ferro-chelante diversi. Un ruolo importante sembra attribuibile anche alla somatomedina; infatti la secrezione di IGF-1 e della sua binding protein IGFBP-3 risulta spesso ridotta come conseguenza del danno epatico cronico (31).
Ipogonadismo Interessa circa il 50-100% dei pazienti affetti da talassemia major (24). Questo può essere riconducibile, in parte, al danno testicolare o ovarico indotto dal sovraccarico marziale. Tuttavia, nella maggioranza dei casi, la funzione gonadica è compromessa secondariamente ad un’alterazione dell’asse ipotalamo-ipofisario conseguente a sua volta al sovraccarico di ferro a livello dell’ipofisi. Le cellule adenoipofisarie e in particolare quelle gonadotrope sono infatti molto suscettibili allo stress ossidativo indotto dai radicali liberi prodotti come conseguenza del sovraccarico marziale. Il quadro clinico conseguente più frequente è rappresentato dal ritardato o assente sviluppo puberale; in una percentuale variabile lo sviluppo puberale spontaneo è più o meno completo ma spesso, nel corso degli anni, la funzione sessuale e riproduttiva tendono a peggiorare anche in questi pazienti (32).
16
Endocrinopatie
Diabete Lo sviluppo del diabete è stato per lo più correlato all’effetto tossico del ferro sulle cellule β. In realtà sembra esistere anche un processo autoimmune attivato dal sovraccarico di ferro. Infatti, quest’ultimo indurrebbe un processo infiammatorio cronico aspecifico con conseguente fibrosi: in particolare il danno della membrana delle cellule β agirebbe come fattore scatenante l’autoimmunità (33). Altri fattori patogenetici ipotizzati sono rappresentati dal danno epatico con conseguente alterazione della sensibilità all’insulina, la predisposizione genetica individuale, lo stato puberale, la compliance alla ferrochelazione. Lo sviluppo di diabete nei pazienti affetti da talassemia major è graduale: sia ha una prima fase caratterizzata da insulino-resistenza con iperinsulinismo compensatorio e normale tolleranza glucidica e una seconda fase con ridotta tolleranza glucidica, progressivo esaurimento delle cellule β fino all’esordio clinico di diabete (34). Fondamentale è in questo caso la diagnosi precoce in quanto un’intensificazione della terapia ferro-chelante può consentire una riduzione del dosaggio insulinico così come è in grado di prevenire l’insorgenza del diabete, se iniziata precocemente (35).
Metabolismo P-Ca
- Ipocalcemia. Le cause più comuni sono rappresentate da: ipoparatiroidismo, deficit di vitamina D, danno epatico. - Ipoparatiroidismo. Ha una prevalenza del 10-11% nell’ambito
dei pazienti talassemici sopra i 16 anni (36): esso sembra dipendere in parte dal sovraccarico marziale delle paratiroidi e in parte conseguenza del riassorbimento osseo secondario all’espansione midollare. Si manifesta clinicamente con ipocalcemia, iperfosfatemia, normali o ridotti livelli di fosfatasi alcalina, normali livelli di vitamina D (35). La terapia è sostitutiva e si basa sulla somministrazione di calcio e metaboliti attivi del colecalciferolo.
Iposurrenalismo La ghiandola surrenalica viene interessata dal sovraccarico marziale e può, se alterata nella sua funzionalità endocrina, influenzare lo sviluppo puberale.
17
Patologia ossea
Già nelle prime descrizioni del quadro clinico della talassemia major, fatte da Cooley nel 1925, venivano descritte alterazioni ossee che rappresentano la conseguenza di espansione midollare, osteoporosi e eritropoiesi extramidollare (37).
Patogenesi - Anemia cronica - Ipogonadismo - Diabete
- Ipoparatiroidismo - Ipotiroidismo
- Terapia ferro-chelante
Caratteristiche - L’espansione midollare può raggiungere, nei pazienti non trasfusi o ipotrasfusi, valori pari a 15-30 volte i valori normali provocando distorsione e fragilità ossea. Le ossa craniche, già in ambito pediatrico, sono allargate con la comparsa di bozze frontali e posteriori, ipertrofia mascellare con retrazione del labbro superiore, prominenza dell’arcata dentaria superiore e conseguente malocclusione. Morfologicamente le ossa craniche presentano assottigliamento dei tavolati esterno e interno, dilatazione della diploe con assottigliamento delle trabecole ossee e dilatazione degli spazi midollari. Si associa inoltre una reazione del periostio che provoca una neoformazione di trabecole ossee perpendicolari alla calotta cranica con conseguente comparsa del caratteristico cranio a spazzola. Anche le ossa lunghe sono coinvolte da queste alterazioni: qui, tuttavia, prevalgono i processi osteolitici con conseguente assottigliamento della corticale mentre sono modesti i segni di neoformazione ossea (38). La colonna vertebrale di questi pazienti può presentare deformità, scoliosi, cifosi, collassi vertebrali con conseguenti danni midollari. Tutte queste alterazioni si osservano generalmente nei pazienti non trasfusi o ipotrasfusi dove lo stimolo da parte dell’anemia è elevato: i nuovi protocolli trasfusionali consentono di prevenire questi quadri patologici. I regimi attuati oggi, mantenendo valori di emoglobina pre-trasfusionale pari a 9-10 g/dL, sopprimono l’espansione midollare e limitano così le lesioni ossee conseguenti.
- L’ipogonadismo ipogonadotropo gioca un ruolo determinante nello sviluppo di osteopenia-osteoporosi (39, 40). La terapia ormonale sostitutiva porta infatti ad un incremento dei parametri di densità minerale ossea (41). Inoltre i pazienti con un ritardato o assente sviluppo puberale presentano un quadro di osteoporosi più severa rispetto a quelli che hanno presentato un normale sviluppo puberale.
- La DFO può presentare effetti tossici a carico dello scheletro (42): vari sono gli autori in letteratura che descrivono quadri di osteoporosi in pazienti adeguatamente chelati in cui le alterazioni ossee non sono riconducibili né all’anemia cronica né al sovraccarico marziale; questi dati suggerirebbero come la ferro-chelazione di per sé possa contribuire al danno osseo in pazienti anche pre-puberi che non presentano ancora alcuna complicanza endocrina (ipogonadismo, ipoparatiroidismo, ipotiroidismo, diabete) (43).
18
5. Strategie terapeutiche convenzionali
La β-talassemia major non trattata risulta costantemente fatale. Le strategie terapeutiche a disposizione hanno contribuito a determinare il prolungamento e il miglioramento della qualità di vita dei pazienti con quasi totale scomparsa delle manifestazioni un tempo tipiche della malattia, anche se l’unico trattamento definitivo è rappresentato dal trapianto di midollo osseo. Negli anni ’60 la terapia trasfusionale è diventata il gold standard per il trattamento dei pazienti talassemici consentendo di mantenere livelli di emoglobina adeguati: questo ha permesso di ottenere un notevole miglioramento della qualità di vita a breve termine, ma non di migliorare la sopravvivenza: i pazienti, a causa delle complicanze secondarie al sovraccarico di ferro, avevano un’aspettativa di vita ridotta (12-24 anni) (44). La terapia convenzionale attuale, basata sull’impiego di emotrasfusioni periodiche e un’adeguata terapia ferro-chelante, ha portato ad un notevole prolungamento della sopravvivenza di questi pazienti che oggi possono raggiungere età superiori ai 45 anni. Il più utilizzato protocollo terapeutico consiste in:
- mantenimento dei livelli emoglobinici entro il range di normalità, mediante un adeguato regime trasfusionale allo scopo di eliminare lo stimolo sul midollo osseo;
- eventuale splenectomia in età pediatrica per evitare la distruzione eritrocitaria e l’aggravamento dell’anemia;
- prevenzione e rimozione dell’eccesso di ferro di origine trasfusionale attraverso l’utilizzo di opportuni agenti chelanti.
5.a. Regime trasfusionale
Le emotrasfusioni rappresentano, attualmente, il primo approccio terapeutico alla β-talassemia; gli scopi della terapia trasfusionale sono vari. Le trasfusioni consentono principalmente di mantenere un apporto di emoglobina necessaria al corretto trasporto periferico di ossigeno, indispensabile al mantenimento della crescita e delle funzioni dei vari organi e tessuti. La terapia trasfusionale permette inoltre di
19
intervenire sulle principali manifestazioni e complicanze della β-talassemia major: attraverso la soppressione dell’eritropoiesi midollare viene infatti arrestata l’espansione del midollo osseo e la comparsa di alterazioni scheletriche cranio-facciali. Viene inoltre inibita l’eritropoiesi extra-midollare e l’assorbimento gastrointestinale di ferro. La riduzione dell’eritropoiesi inefficace porta infine ad un minore rilascio in circolo di eritroblasti alterati e di precipitati di catene α-globiniche con conseguente minore ipersplenismo e splenomegalia (45). Inizialmente, allo scopo di ottenere questi obiettivi, si utilizzavano regimi di ipertrasfusione con gravi conseguenze associate al sovraccarico marziale. Per tale motivo, nel corso degli anni, l’approccio terapeutico si è modificato: attualmente si segue un protocollo trasfusionale che prevede il mantenimento di un livello di emoglobina pre-trasfusionale compreso tra 9 e 10 g/dL (46); tale regime trasfusionale sembra infatti associarsi ad un adeguato equilibrio tra inibizione midollare e accumulo di ferro. L’emoglobina post-trasfusionale non dovrebbe invece superare i 15 g/dL. E’ indicato l’inizio del supporto trasfusionale in presenza di grave sintomatologia che generalmente si manifesta quando il livello di emoglobina risulta al di sotto di 7 g/dL; la maggior parte dei pazienti affetti da talassemia major riceve la prima trasfusione entro la fine del primo anno di vita. Tuttavia, a dimostrazione dell’esistenza di diverse forme di β-talassemia major, alcuni bambini non ricevono supporto ematico fino all’età di 4-5 anni. I pazienti sottoposti a terapia trasfusionale incorrono inevitabilmente in alcune complicanze. Presentano una maggiore facilità a contrarre infezioni: epatite B e C, infezione da CMV; tuttavia, grazie alle vaccinazioni, le infezioni da HBV sono ormai rare, mentre rimangono frequenti le altre. Il sovraccarico di ferro rappresenta un’altra conseguenza inevitabile del regime trasfusionale: esso provoca danni cellulari a causa probabilmente della formazione delle specie reattive dell’ossigeno (ROS) che alterano ossidativamente le proteine cellulari e i lipidi. L’emosiderosi conseguente rappresenta la principale causa di morbilità e mortalità in questi pazienti.
20
5.b. Terapia ferro-chelante
Il trattamento trasfusionale raccomandato per i pazienti talassemici prevede l’esecuzione di trasfusioni di emazie concentrate ogni 2-3 settimane, con frequenza variabile da paziente a paziente. Lo schema suggerito determina in genere un consumo di sangue pari a 100-200 mL/Kg/anno che equivale ad un accumulo di 0,3-0,6 mg/Kg di ferro al giorno (47). E’ proprio dal catabolismo del sangue trasfuso che dipende il grave sovraccarico marziale e le numerose complicanze ad esso legate: l’organismo umano non possiede infatti mezzi efficaci che impediscano l’accumulo del ferro in eccesso o ne garantiscono la sua eliminazione. La quota in eccesso rimane in parte in circolo andando a costituire il pool di ferro libero plasmatico noto anche come NTBI (Non-Transferrin Bound Iron) e in parte si accumula nei tessuti sotto forma di deposito (ferritina ed emosiderina) e ferro labile intracellulare (LIP) (48). Tale accumulo tissutale si verifica in modo lento e graduale in tutti gli organi, principalmente fegato, cuore e ghiandole endocrine (ipofisi, tiroide, paratiroide, pancreas), che presentano una maggiore concentrazione di recettori per la transferrina, ma con velocità molto diverse a seconda dell’organo coinvolto (più lento nel cuore rispetto al fegato) (49). La tossicità del ferro non legato a proteine di deposito o di trasporto è legata al suo ruolo come catalizzatore nelle reazioni di ossidoriduzione con conseguente formazione dei radicali liberi (ROS). Questi ultimi possono innescare reazioni libere a catena reagendo con qualsiasi molecola biologica posta nelle vicinanze tra cui DNA e RNA causando mutazioni, rottura della doppia elica e cancerogenesi, e membrane biologiche (mitocondri, lisosomi e membrana sarcoplasmatica) dove inducono un processo autocatalitico di perossidazione lipidica. I radicali secondari che vengono prodotti possono inoltre diffondere dal sito di origine determinando danni a distanza. L’organismo umano mette in atto una serie di meccanismi anti-ossidanti che non sono tuttavia in grado di annullare il danno ossidativo. La terapia ferro-chelante, associata al regime trasfusionale, è quindi l’unica opzione terapeutica in grado di limitare i danni indotti dal sovraccarico marziale. L’introduzione dei chelanti del ferro, avvenuta intorno agli anni ’70, ha modificato l’evoluzione naturale della malattia e prolungato la sopravvivenza dei pazienti (50). Queste molecole, grazie alla loro elevata affinità per il ferro ionizzato (Fe3+), sono in grado di indurne l’eliminazione consentendo il
21
raggiungimento di concentrazioni più sicure. Tuttavia questo processo avviene lentamente in quanto solo una piccola percentuale del ferro corporeo è disponibile alla chelazione: pertanto, se un paziente è già in una condizione di sovraccarico marziale, anche instaurando un trattamento chelante intensivo possono essere necessari mesi o anni per ridurre il ferro corporeo a livelli di sicurezza. Aumentando il dosaggio farmacologico si rischierebbe infatti di aumentarne gli effetti tossici venendo sequestrato anche il ferro necessario per il metabolismo tissutale. Il chelante ideale del ferro dovrebbe possedere alcune delle caratteristiche elencate nella tabella seguente.
Alta affinità e specificità per il ferro Bassa velocità di metabolizzazione
Elevata capacità di penetrazione tissutale e cellulare Non ridistribuzione del ferro
Ridotta tossicità Basso costo Somministrazione orale
22
Nelle tabelle seguenti vengono elencate le caratteristiche principali dei tre farmaci chelanti utilizzati oggi nella pratica clinica.
Desferrioxamina (DFO, Desferal®)
E’ il primo farmaco ferrochelante introdotto nell’utilizzo clinico e utilizzato per più di 40 anni. La DFO è un sideroforo naturale, prodotto dallo Steptomices pilosus, che chela il
ferro mascherandone i sei siti di coordinazione e inibendo la formazione dei ROS. Modalità di
somministrazione
Dato il suo elevato peso molecolare, che non ne contente l’assorbimento per os, la sua somministrazione avviene esclusivamente per via parenterale (s.c. o e.v.). La breve emivita plasmatica, circa 20 minuti, rende inoltre necessaria un’infusione prolungata, di almeno 12 ore, per 5-7 giorni la settimana (51) alla dose di 20-40 mg/Kg/die nei bambini e di 40-60 mg/Kg/die negli adulti.
Meccanismo d’azione
A causa dell’alto peso molecolare e della sua spiccata idrofilia questo farmaco non è in grado di penetrare nelle cellule per cui non agisce sottraendo il ferro né all’emoglobina né alle altre strutture proteiche cellulari di cui il ferro è costituente, ma chela il ferro libero o in transito tra transferrina e ferritina e soprattutto dal deposito epatico (52) determinandone l’escrezione attraverso urine (circa 70%) e feci (circa 30%): circa 0,15-0,5 mg di ferro escreto/Kg/die (53). La DFO, che rimuove efficacemente il ferro dal fegato, presenta effetti limitati sugli altri distretti, in particolare sul cuore (54, 55). Infatti il farmaco viene captato attivamente dagli epatociti consentendo la rimozione del ferro intraepatico in eccesso attraverso il sistema biliare (56) e quindi le feci. Il ferro escreto attraverso le urine invece deriva principalmente dal catabolismo degli eritrociti.
Tossicità L’utilizzo per via parenterale della DFO presenta molti limiti. Questi non sono semplicemente legati alla scarsa compliance conseguente la modalità di somministrazione, ma dipendono anche dagli effetti collaterali (ritardo di crescita, displasia scheletrica, reazioni cutanee locali, allergia, cataratta, ipoacusia neurosensoriale, deficit di zinco) che si possono manifestare soprattutto alle alte dosi. Questi svantaggi hanno indotto la necessità di nuove molecole, più economiche e maneggevoli nella pratica quotidiana.
23
Nel corso degli ultimi anni la formulazione di nuovi ferro-chelanti assumibili per os (Tab.14, Tab.15) ha permesso di migliorare notevolmente la compliance terapeutica.
Deferiprone (DFP, Ferriprox®)
La molecola del DFP (1,2 dimetil-3-idrossipiridina-4-uno, L1) è un farmaco chelante disponibile a partire dal 1999 nella pratica clinica. Il DFP è attualmente indicato nei pazienti affetti da talassemia major per cui la terapia con DFO è controindicata o
determina grave tossicità. Modalità di
somministrazione
Viene impiegato per via orale generalmente alla dose di 75 mg/Kg/die. Tuttavia, data la breve emivita (circa 160 minuti), questo farmaco deve essere assunto almeno 3 volte al giorno (1).
Meccanismo d’azione
Il farmaco è in grado di entrare nelle cellule (55) e legare il ferro favorendo la sua escrezione attraverso le urine. Inoltre ha un’azione specifica a livello cardiaco dove rimuove il ferro in eccesso prevenendo la cardiopatia (57): il peggioramento di una cardiopatia preesistente o l’esordio di complicanze cardio-vascolari è stato osservato infatti nel 4% dei pazienti trattati con DFP rispetto al 20% di quelli in terapia con DFO (58, 59). Probabilmente la migliore efficacia a livello cardiaco di questo farmaco dipende da varie caratteristiche della molecola tra cui il suo basso peso molecolare e la sua lipofilia che ne facilitano l’ingresso nelle cellule miocardiche (1). Tuttavia il deferiprone, se assunto abitualmente, è meno efficace rispetto alla DFO nella prevenzione dell’accumulo di ferro nel fegato: infatti l’escrezione biliare del ferro indotta dal DFP è trascurabile a causa della sua rapida inattivazione per glicuronoconiugazione a livello epatico (60).
Tossicità Il principale effetto collaterale del deferiprone è rappresentato dalla agranulocitosi (0,2-0,3 casi su 100 pazienti/anno) che richiede la sospensione della terapia. E’ quindi importante il monitoraggio dell’emocromo associato al controllo della funzionalità epatica, per il possibile rischio di ipertransaminasemia. Altri possibili effetti collaterali sono rappresentati dai sintomi gastrointestinali (nausea, vomito, dolore addominale), le artropatie, generalmente di lieve-modesta entità e transitorie, e il deficit di zinco (1).
Tabella 14. Farmaci chelanti: principali caratteristiche del deferiprone.
Data la diversa efficacia dei due farmaci, DFO e DFP, a livello epatico e cardiaco, sono stati condotti numerosi studi volti a stabilire l’utilità di una terapia “combinata” (45). Quest’ultima associa generalmente il deferiprone, assunto quotidianamente per os, alla desferrioxamina, somministrata tramite infusione da 2 a 7 giorni a settimana in funzione della gravità del sovraccarico di ferro (1). In questo caso la somministrazione orale di DFP consente di chelare il ferro depositato a livello cardiaco e di trasferirlo in circolo, mentre la successiva somministrazione parenterale di DFO permette di legare il ferro entrato in circolo e di eliminarlo attraverso feci e urine.
24
Deferasirox (DFX, ICL670, Exjade®)
Anche questo è un farmaco chelante orale; registrato negli Stati Uniti nel 2006 e in Europa nel 2007.
Modalità di somministrazione
Il farmaco viene somministrato per bocca ad un dosaggio di 20-30 mg/Kg una volta al giorno (emivita pari a 8-16 ore).
Meccanismo d’azione
Questa molecola si è dimostrata efficace nel ridurre il sovraccarico di ferro, soprattutto epatico, in maniera sovrapponibile alla DFO, presentando tuttavia un profilo di sicurezza maggiore e un’efficacia dose-dipendente (61, 62). La sua eliminazione avviene principalmente tramite le feci (1).
Tossicità I principali effetti collaterali associati al suo utilizzo sono a carico del sistema gastrointestinale, nausea, vomito, diarrea, dolore addominale (15%), e della cute, rash cutaneo (11%). Questi effetti sono generalmente lievi e raramente richiedono la sospensione del farmaco. Eventi avversi più rari come aumento reversibile delle transaminasi e aumento lieve e non progressivo della creatinina vengono generalmente gestiti attraverso la riduzione del dosaggio o la sospensione della terapia.
Tabella 15. Farmaci chelanti: principali caratteristiche del deferasirox.
La terapia chelante ha l’obiettivo di ridurre il sovraccarico di ferro già presente e ritardare la sua comparsa nei pazienti regolarmente trasfusi. Essa viene generalmente intrapresa dopo il secondo anno di vita e comunque dopo un certo numero di trasfusioni (generalmente 10, ferritinemia sierica > 1000 ng/mL): se iniziata precocemente può migliorare il profilo auxologico, lo sviluppo puberale e prevenire le complicanze sistemiche dell’emosiderosi (52). La sua efficacia deve essere tuttavia strettamente monitorata in ogni paziente; purtroppo non esiste un unico parametro in grado di definire con precisione il grado di sovraccarico di ferro. La determinazione routinaria dei livelli di ferritina sierica è sicuramente utile, anche se questi possono presentare oscillazioni indipendentemente dal sovraccarico marziale: ad esempio in occasione di febbre, processi infettivi o infiammatori, deficit di vitamina C o anche per le stesse emolisi cronica ed eritropoiesi inefficace. La determinazione dei livelli di ferritina non consente inoltre di valutare precisamente il grado di sovraccarico epatico e cardiaco, importanti dal punto di vista prognostico (1). Nel soggetto normale la ferritina sierica si trova per l’80% circa in forma glicosilata; nell’individuo talassemico politrasfuso è presente in quantità maggiore e per la maggior parte nella forma non glicosilata. Un altro parametro utile per valutare il rischio di sovraccarico marziale è rappresentato dalla determinazione del carico trasfusionale: pazienti che hanno
25
ricevuto cumulativamente > 120 mL/Kg di emazie concentrate o > 12 trasfusioni in un anno sono a rischio di sviluppare sovraccarico marziale. Per tale motivo è fondamentale registrare il volume di sangue trasfuso mantenendo aggiornata la storia trasfusionale del paziente. La valutazione della concentrazione di ferro intraepatico mediante biopsia fornisce una valutazione più accurata dei depositi di ferro nell’organismo se eseguita su campioni di peso adeguato e in assenza di cirrosi (63, 64): recenti studi hanno tuttavia dimostrato che la distribuzione epatica di ferro può essere disomogenea; inoltre, sebbene considerata sicura, la biopsia rappresenta comunque una tecnica invasiva che può comportare dolore e non è completamente esente da rischi. Inoltre, per i rischi ad essa associati, la biopsia non può essere utilizzata nel follow-up (1). Una metodica efficace nella valutazione dell’emosiderosi è rappresentata dalla Biosuscettometria Magnetica tramite SQUID (Superconducting Quantum Interference Device), tecnica innocua, sensibile e specifica che misura direttamente le proprietà magnetiche di ferritina ed emosiderina. Valuta inoltre la concentrazione di ferro su una porzione di organo, centrale, circa 10.000 volte più grande della porzione esaminabile attraverso la biopsia (65). Tuttavia, data la scarsa disponibilità di questa indagine, dovuta ai costi elevati, essa non può essere utilizzata di routine nella valutazione dell’emosiderosi (66). Inoltre esiste una dissociazione tra la siderosi cardiaca e quella epatica per cui la previsione del sovraccarico marziale nel cuore a partire dal contenuto di ferro intraepatico tramite SQUID non risulta affidabile in tutti i pazienti. Fondamentale è stata l’elaborazione di una nuova tecnica di risonanza magnetica (T2*) in grado di quantificare, in vivo, il grado di sovraccarico di ferro intracardiaco: questa tecnica permette oggi di identificare precocemente i pazienti ancora asintomatici a rischio di complicanze cardiovascolari. La diagnosi pre-sintomatica di grave siderosi del miocardio è dunque essenziale per una prognosi migliore: permette di instaurare un regime chelante intensivo precoce nonostante la totale assenza di sintomi e segni ecocardiografici di disfunzione. Come già detto la terapia con desferrioxamina non è in grado di prevenire la cardiopatia anche nei pazienti che si chelano adeguatamente. L’introduzione nell’uso clinico del deferiprone sta fornendo risultati concreti sia nella terapia che nella prevenzione delle complicanze cardiologiche. La disponibilità di più molecole, specialmente se somministrabili per os, permetterebbe un approccio migliore alla terapia ferro-chelante e il loro uso
26
combinato porterebbe a numerosi vantaggi terapeutici. Su queste basi vari sono i trials clinici che hanno valutato i risultati della terapia combinata con DFO e DFP (67, 68, 69). La terapia combinata ha determinato una significativa riduzione della ferritina e/o del ferro epatico con un conseguente miglioramento anche della siderosi miocardica e quindi della funzionalità ventricolare. I due farmaci sembrerebbero avere inoltre un effetto sinergico (70): infatti la combinazione di un chelante debole che ha una migliore capacità di penetrare all’interno delle cellule, con uno più potente, che penetra poco nelle cellule ma ha una maggiore escrezione urinaria, può risultare in un effetto complessivo migliore attraverso il trasferimento del ferro da una molecola all’altra.
5.c. Splenectomia
Nell’ambito del paziente talassemico, la splenomegalia è riconducibile fondamentalmente a 3 cause principali: attivazione dell’emopoiesi extramidollare, sviluppo di congestione splenica secondaria a ipertensione portale e insufficienza cardiaca e incremento della funzione emocateretica (71). La splenectomia, accanto alle trasfusioni, rappresenta un altro cardine della terapia della talassemia major. Tutti i talassemici infatti sviluppano inevitabilmente splenomegalia che può diventare di entità tale da peggiorare significativamente la qualità di vita del paziente stesso. Tuttavia, per il possibile sviluppo di complicanze post-operatorie a medio e lungo termine, a differenza di un tempo, la splenectomia viene oggi indicata solo in presenza di specifiche condizioni (71): presenza di un fabbisogno ematico superiore di 1,5 volte a quello di un paziente già splenectomizzato, splenomegalia grave e tale da indurre compressione locale e dolore, presenza di marcata leucopenia e piastrinopenia conseguente all’ipersplenismo. La predisposizione a contrarre infezioni batteriche nei pazienti splenectomizzati può essere ridotta tramite la vaccinazione preventiva pre-operatoria contro i batteri capsulati, pneumococco e meningococco, e quindi la somministrazione di un’adeguata profilassi antibiotica dopo l’esecuzione dell’intervento (17).
27
5.d. Trapianto di midollo osseo
Al momento attuale non esiste comunque alcun trattamento definitivo per nessuna forma di talassemia ad eccezione del trapianto di midollo osseo che, in presenza di donatore compatibile, può portare a guarigione completa. I rischi per il paziente non sono tuttavia irrilevanti: questa opzione terapeutica comporta un’adeguata valutazione dei rischi correlati alla possibilità di rigetto e alla terapia chemioterapica cui il paziente si deve sottoporre.
6. Strategie terapeutiche alternative
Nel tentativo di identificare nuove terapie alternative sono stati esplorati vari approcci tra cui la terapia genica e l’induzione della emoglobina fetale.
6.a. Terapia genica
La mancanza totale o parziale della sintesi di catene globiniche β rende fondamentale l’incremento della produzione di qualsiasi catena globinica non α con l’obiettivo di ridurre lo sbilanciamento alla base della talassemia. Questo risultato può essere ottenuto tramite terapia genica, potenziando l’espressione dei geni globinici β. Gli studi di terapia genica della β-talassemia hanno come obiettivo principale quello di sviluppare nuovi sistemi sperimentali in grado di sostituire le funzioni del gene alterato della catena β con quelle di un gene “sano”, opportunamente inserito nelle cellule eritroidi del paziente. Il problema principale è rappresentato dallo sviluppo di un vettore adeguato che sia in grado di penetrare all’interno delle cellule trasportando il gene con lo scopo di inserirlo nel genoma della cellula ospite. Tali caratteristiche sono proprie del DNA di alcuni virus tra cui i retrovirus. Potenzialmente, tuttavia, ogni virus risulta dannoso per la salute umana per cui è importante rendere il vettore virale innocuo. Per questo il genoma retrovirale utilizzato in terapia genica è privo dei geni essenziali per la propagazione del virus. Altre molecole utilizzabili in terapia genica
28
sono rappresentate dai vettori lenti virali, ottenuti ad esempio dal virus HIV-1, e i ribozimi ossia molecole in grado di legare e degradare specifici prodotti genici. Si è riusciti ad esempio a realizzare ribozimi in grado di sopprimere gli mRNA delle catene α-globiniche con conseguente minore precipitazione delle stesse e minori danni ai precursori eritroidi della β-talassemia.
6.b. Induzione di HbF
Un altro approccio terapeutico molto studiato nell’ambito dei pazienti talassemici è rappresentato dall’induzione dell’emoglobina fetale. Nell’ambito della β-talassemia, infatti, la mancanza di catene β viene in parte compensata da un’aumentata sintesi di catene γ con conseguente incremento della emoglobina fetale: la maggiore espressione di catene globiniche γ, riducendo lo sbilanciamento esistente tra catene α e non- α, dovrebbe indurre un miglioramento dell’anemia (72). Valori elevati di HbF si associano infatti a fenotipi più lievi della patologia e quindi ad una prognosi migliore. Vari sono quindi gli studi volti a identificare molecole in grado di riattivare l’espressione delle catene γ in questi pazienti. Purtroppo molti degli agenti farmacologici testati a questo scopo (es. idrossiurea) determinano una soppressione dell’eritropoiesi e quindi, pur aumentando i livelli di HbF, non presentano effetti significativi sulla risoluzione dell’anemia. Inoltre l’aumentata espressione delle catene γ, potendosi associare ad un concomitante incremento nell’espressione delle catene α, non risulta sempre associato ad un miglioramento clinico in quando non riduce lo sbilanciamento esistente tra le catene globiniche (72).
Normalmente, in epoca adulta, la produzione di HbF è ridotta all’1-2% del totale in quanto viene limitata ad una sottopopolazione eritrocitaria definita F-cell (73). Un numero significativo di mutazioni, variabili in entità e posizione, sono associate ad un disordine definito HPFH (Hereditary Persistent of Fetal Hemoglobin) caratterizzato da un’anomala espressione dei geni γ-globinici. Questo fenomeno determina un incremento nel livello di HbF che può raggiungere valori medi che vanno da un 2,5% ad un 20-30%. Di per sé la produzione di HbF nei pazienti talassemici, come è già stato accennato, è maggiore rispetto alla popolazione generale. Probabilmente l’aumentata
29
sintesi delle catene γ è il risultato di vari fattori tra cui l’eritropoiesi inefficace. L’effetto è inoltre amplificato da una sopravvivenza selettiva dei precursori eritroidi che sintetizzano più catene γ. Pertanto gli individui β-talassemici, eterozigoti o omozigoti, hanno un incremento variabile nei loro livelli di HbF con conseguente miglioramento del quadro clinico dovuto alla riattivazione dei geni γ e in cui l’aumentata produzione di HbF riesce, almeno in parte, a supplire alla carenza di HbA (74). I soggetti talassemici che presentano in coeredità un fenotipo HPFH presentano un ulteriore miglioramento dell’outcome clinico. Le alterazioni che determinano un’elevata persistenza di HbF non sono ancora completamente note ma le attuali conoscenze permettono di suddividerle in due categorie principali: fenotipo HPFH deletion, caratterizzato dalla presenza di delezioni interessanti il cluster genico β-globinico, e il fenotipo HPFH non deletion, in cui è presente l’intero cluster β. Quest’ultima condizione sarebbe infatti riconducibile a mutazioni puntiformi a carico della regione promotrice del gene γ-globinico con conseguente sintesi post-fetale persistente. La regione promotrice del gene codificante per le γ-globine rappresenta pertanto un potenziale target per farmaci in grado di aumentare i livelli di HbF e contrastare quindi il fenotipo talassemico.
Uno degli obiettivi nella terapia della β-talassemia è quello di aumentare la sintesi delle catene γ per compensare il deficit di quelle β. Varie osservazioni sperimentali dimostrano come i geni globinici non siano repressi in modo irreversibile nelle cellule differenziate per cui la loro espressione può essere riprogrammata in presenza di specifici fattori di regolazione. La riattivazione del gene γ-globinico nelle cellule eritroidi può essere realizzata sui vari livelli che ne determinano il silenziamento. Per raggiungere tale scopo sono state testate varie molecole, chimiche e naturali, ma nessuna è stata ad oggi autorizzata per essere utilizzata in questi pazienti come terapia convenzionale: la maggior parte sono infatti poco specifiche, di efficacia limitata e potenzialmente carcinogeniche (74). Le basi genetiche del fenomeno dello switching emoglobinico sono state studiate per decenni: i meccanismi molecolari sottostanti sono stati solo in parte chiariti. La variabilità di risposta individuale agli agenti farmacologici induttori di HbF può essere in parte spiegata sulla base del diverso background genetico (75).
30
Gli agenti farmacologici in grado di indurre l’espressione dell’emoglobina fetale vengono generalmente classificati in funzione della loro struttura chimica e del meccanismo d’azione sfruttato.
- Agenti ipometilanti. Il DNA, nella cromatina, è metilato in punti specifici correlati strettamente all’attività ed all’espressione genica. La prima dimostrazione che la sintesi di HbF potesse essere indotta farmacologicamente risale al 1982 quando è stato osservato che un farmaco citotossico, la 5-azacitidina, era in grado di stimolare la produzione di HbF nel topo tramite inibizione delle metil-transferasi. La metilazione del DNA rappresenta infatti uno dei meccanismi di repressione genica, catalizzato dalla famiglia enzimatica delle transferasi. Gli inibitori della metil-transferasi, 5-azacitidina e il suo analogo 5-aza-2’-desossicitidina (decitabine), agiscono sull’ipometilazione del DNA, un meccanismo che annulla la down-regulation di vari geni tra cui quelli che controllano la sintesi delle catene γ. Infatti, sia il gene per le γ-globine che quello per le β-globine risultano ipometilati durante la vita intrauterina, mentre in epoca adulta il gene γ è metilato. Questo fa ipotizzare che la metilazione sia uno dei meccanismi coinvolti nello switching dell’emoglobina e in particolare nella repressione del gene delle γ-globine durante la vita post-natale. La 5-azacitidina è stato il primo farmaco in grado di indurre l’HbF utilizzato nella terapia dei pazienti talassemici dove è risultato efficace nell’induzione di HbF, pur presentando purtroppo vari effetti collaterali: causa marcata soppressione dell’eritropoiesi ed è potenzialmente cancerogeno; per questi motivi il farmaco è stato abbandonato nel trattamento della talassemia (75, 76).
- Idrossiurea (HU). Nell’individuo adulto è presente, come precedentemente accennato, una piccola percentuale di cellule eritroidi, definite F, contenenti una quota di HbF superiore e pari allo 0,7% circa. Sembra che esista una correlazione diretta tra l’attività proliferativa midollare e la produzione di cellule F per cui agenti citotossici, come l’idrossiurea, in grado di indurre la rigenerazione eritroide, sarebbero in grado di indurre la formazione di cellule F aumentando quindi la concentrazione di HbF. Questo farmaco, noto per il suo utilizzo nei disordini mielo-proliferativi (leucemie acute e disordini
31
mielodisplastici), agisce come inibitore del ribonucleotide riduttasi bloccando la sintesi del DNA e impedendo la formazione di desossiribonucleotidi a partire dai precursori ribonucleotidici. Inoltre agisce anche come donatore di NO che, attivando l’enzima guanililciclasi, tramite l’interazione con specifici fattori di trascrizione, aumenta l’espressione del gene γ-globinico. L’idrossiurea, attraverso meccanismi tuttora poco chiari, è in grado di indurre l’emoglobina fetale con risultati incoraggianti, seppur non omogenei; la notevole variabilità di risposta associata ai potenziali effetti di mielosoppressione e citotossicità, non permettono tuttavia il suo utilizzo come terapia convenzionale nei pazienti talassemici. Viene invece largamente utilizzato nell’ambito dei pazienti affetti da drepanocitosi dove è in grado di ridurre l’incidenza delle crisi vaso-occlusive e migliorare il profilo ematologico migliorando la prognosi e la qualità di vita dei pazienti (77).
- Inibitori dell’istone-deacitilasi. L’acido butirrico, un acido grasso con quattro atomi di carbonio, ed altri composti simili sono in grado di indurre l’emoglobina fetale probabilmente attraverso l’inibizione dell’enzima istone deacitilasi. L’espressione genica è infatti conseguenza della struttura cromatinica indotta dall’acetilazione e deacetilazione degli istoni che rispettivamente inducono e inibiscono la trascrizione genica (75). L’enzima istone-deacitilasi (HDAC) inibisce la trascrizione impedendo il legame degli specifici fattori di trascrizione: la sua inattivazione si associa quindi ad una maggiore espressione genica. Questo meccanismo è coinvolto anche nell’espressione genica delle catene γ e quindi nel fenomeno dello switching emoglobinico. Anche l’acido butirrico, come l’idrossiurea, è associato ad una risposta clinica estremamente variabile che sembrerebbe dipendere dal background genetico del paziente (78, 79).
- Eritropoietina. La somministrazione di Epo in pazienti affetti da β-talassemia conduce ad un incremento della sintesi di HbF (80). Oltre a stimolare la proliferazione eritroide, l’Epo possiede un effetto antiapoptotico e prolunga la sopravvivenza delle cellule eritroidi. La percentuale di HbF sintetizzata nelle cellule trattate con Epo dipende dalla durata della terapia e dalla dose