Università degli Studi del Piemonte Orientale “Amedeo
Avogadro”
Dipartimento di Scienze del Farmaco
Dottorato di Ricerca in Biotecnologie Farmaceutiche ed Alimentari
XXVII ciclo a.a. 2012-2015
ROLE OF p50 NF-
B IN CANCER RELATED
INFLAMMATION
Alessandro Ippolito
Supervised by Prof. Antonio Sica
Milioni di persone hanno visto la caduta della mela, ma Newton è stato
colui che ha chiesto “perchè”
Contents
Chapter 1
1Introduction
-Inflammation and cancer 3
-The transcription factor NF-B 11
-Macrophages 21
-Colorectal cancer 42
-Bibliography 51
Chapter 2
67Outline of the thesis 69
Chapter 3
73The p50 NF-κB subunit promotes intestinal tumor development and progression by shaping gut associated inflammation
Chapter 4
119Tumors promote p50-driven
transcriptional reprogramming to divert Interferon-γ-mediated myeloid
cell functions towards
immunosuppression
Chapter 5
157 DiscussionChapter 6
171 List of pubblications1
3
1.1 Inflammation and cancer
The idea of a relationship between inflammation and cancer dates back 1863, when Rudolf Virchow observed leukocyte infiltration in neoplastic tissues and hypothesized that the origin of cancer was at sites of chronic inflammation. Yet, it was only during the last decade that numerous studies undoubtedly demonstratedthe critical role of inflammation in tumorigenesis, and thatsome of the underlying molecular mechanisms have been elucidated [1]. Nowadays several lines of evidences [2-4] (Box 1) – based on a range of findings from epidemiological studies of patients to molecular studies of genetically modified mice – have led to a general acceptance that inflammation and cancer are linked. In pathological conditions, as
after tissue injury, the
inflammatory process is the first response of the body designed to “heal” the afflicted tissue. This involves activation and direct
migration of leukocytes
(neutrophils, monocytes and eosinophils) from the venous system to sites of damage. This cellular migration is controlled by a family of chemotactic cytokines, named chemokines, which possess a relatively high degree of specificity for specific leukocyte populations chemoattraction [5, 6], recruits downstream effector cells and dictates the natural evolution of the inflammatory response.
Hence, inflammation is a fundamental process both for physiological conditions and to protect the body against different exogenous and/or endogenous treats and it is strictly controlled and self-limiting: the disregulation of this mechanism can become an health-treatening event. In fact, in chronically
4
inflamed tissues, a subversion of cell death and/or repair programmes might occur, resulting in uncontrolled proliferation of cells that carries DNA mutations and predisposes tissues for cancer development.
Indeed, it is estimated that 20% of all cancers is associated with chronic infection and inflammation [7] and that underlying infections and inflammatory responses are linked to 15-20% of all deaths from cancer worldwide [8].
In addiction, a “smouldering” inflammation is present also in tumors not causally related to an obvious inflammatory process. Recent evidence have indeed demonstrated that different classes of oncogenes (e.g. RET [9], RAS and MYC) and tumor-suppressor genes (e.g.VHL, TGFβ and PTEN) regulate the expression of inflammation-related programs [10, 11].
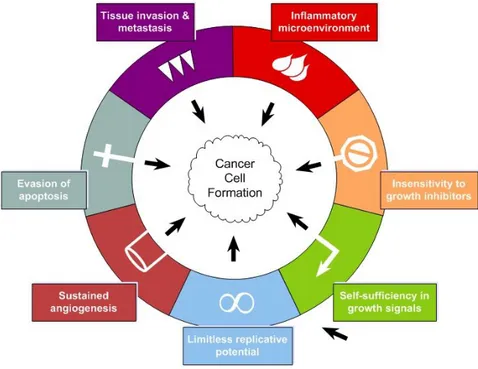
As well as clinical correlations, also molecular evidences show that infiltrating leukocyte can be involved in carcinogenesis and/or tumor invasion and metastasis, [12-15] indicating inflammation as the “Seventh hallmark of cancer”(Fig.1)[16-18].
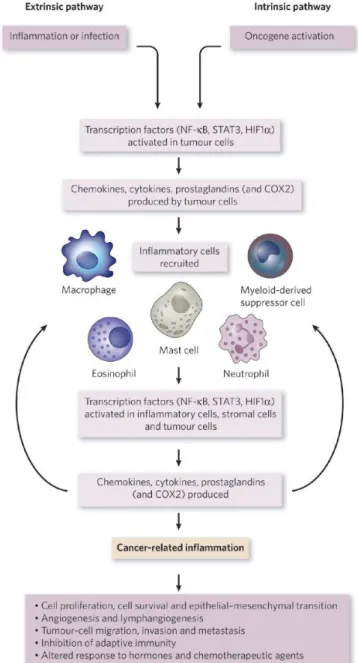
In some types of cancer, inflammatory conditions are present before a malignant change occurs (extrinsic pathway). Conversely, in other types of cancer, an oncogenic change induces an inflammatory microenvironment that promotes the development of tumours (intrinsic pathway) (Fig. 2).
The extrinsic pathway starts with unresolved and prolonged inflammatory conditions that produce activation of transcription factors and DNA mutations with consequent alteration of the physiological cellular processes like proliferation, survival and apoptosis.
On the other side, in the intrinsic pathway an early genetic event is necessary and sufficient for the development of tumor and directly promotes the build-up of an inflammatory microenvironment.
The activation of transcription factors, mainly nuclear factor-κB (NF-κB), signal transducer and activator of transcription 3 (STAT3) and hypoxia-inducible factor 1α (HIF1α), in tumour cells are the points in which the two
5
pathways converge and, as a consequence, link DNA mutations with the production of inflammatory mediators. These, in turn, recruit and activate various leukocytes, most notably cells of the myelomonocytic lineage, and activate the same key transcription factors in inflammatory cells, stromal cells and tumor cells, resulting in more inflammatory mediators being produced and a cancer-related inflammatory microenvironment being generated in a sort of positive feedback loop. This uncontrolled and non self-limiting cancer-related inflammation has many tumour-promoting effects including induction of genomic instability, alteration in epigenetic events and subsequent inappropriate gene expression, enhanced proliferation and resistance to apoptosis of initiated cells, induction of tumour angiogenesis and tissue remodelling with consequent promotion of tumour cells invasion and metastasis [19].
Figure 1. Inflammation as the seventh hallmark of cancer
In 2000, Hanahan and Weinberg proposed a model to define the six properties that a tumour acquires. These are unlimited replicative potential, ability to develop blood vessels (angiogenesis), evasion of programmed cell death (apoptosis), self-sufficiency in growth signals, insensitivity to inhibitors of growth, and tissue invasion and metastasis. Next studies indicate that this model should be revised to include cancer-related inflammation as an additional hallmark. Adapted from [17] and [18].
6
Figure 2. Pathways that connect inflammation and cancer.
Cancer and inflammation are connected by two pathways: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is activated by genetic events that cause neoplasia. These events include the activation of various types of oncogene by mutation, chromosomal rearrangement or amplification, and the inactivation of tumour-suppressor genes. Cells that are transformed in this manner produce inflammatory mediators, thereby
generating an inflammatory
microenvironment in tumours for which there is no underlying inflammatory condition (for example, breast tumours). By contrast, in the extrinsic pathway, inflammatory or infectious conditions augment the risk of developing cancer at certain anatomical sites (for example, the colon, prostate and pancreas). The two pathways converge, resulting in the activation of transcription factors, mainly nuclear factor- B (NF- B), signal transducer and activator of transcription 3 (STAT3) and hypoxia-inducible factor 1 (HIF1 ), in tumour cells. These transcription factors coordinate the production of inflammatory mediators, including cytokines and chemokines, as well as the production of cyclooxygenase 2 (COX2) (which, in turn, results in the production of prostaglandins). These factors recruit and activate various leukocytes, most notably cells of the myelomonocytic lineage. The cytokines activate the same key transcription factors in inflammatory cells, stromal cells and tumour cells, resulting in more inflammatory mediators being produced and a cancer-related inflammatory
microenvironment being generated.
Smouldering cancer-related
inflammation has many
tumour-promoting effects. From [19].
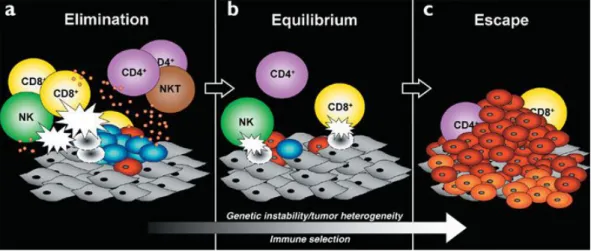
However, despite these evidences, genetic studies of mouse models have demonstrated that the inflammatory response supported by innate immune cells is crucial for the activation of an adaptive immune response capable of eliminating nascent tumors [20].
It is generally accepted that immune cells continuously recognize and destroy nascent tumor cells but, due to the genetic instability that characterized
7
neoplastic cells, the arising of new variants able to evade the immune surveillance results in tumor establishment and progression (immunoediting process; Fig.3) [21]. Only when tumor cells reach to escape the immune cells surveillance, the cancer-related inflammation develops. This is mostly dependent on cytokines and chemokines produced by tumor cells that subvert the anti-tumor activity of inflammatory cells toward a tumor-promoting condition. In this regard, several studies have emphasized that the “smouldering” inflammation associated with tumors is mainly oriented to tune the adaptive immune response.
Figure 3. Immunoediting process
Cancer immunoediting encompasses three processes: (a) Elimination corresponds to immunosurveillance. (b) Equilibrium represents the process by which the immune system iteratively selects and/or promotes the generation of tumor cell variants with increasing capacities to survive immune attack. (c) Escape is the process wherein the immunologically sculpted tumor expands in an uncontrolled manner in the immunocompetent host. In a) and b), developing tumor cells (blue), tumor cell variants (red) and underlying stroma and nontransformed cells (gray) are shown; in c, additional tumor variants (orange) that have formed as a result of the equilibrium process are shown. Different lymphocyte populations are as marked. The small orange circles represent cytokines and the white flashes represent cytotoxic activity of lymphocytes against tumor cells. From [21].
Myeloid cells in cancer
Myeloid cells are the most abundant haematopoietic cells in the human body and have diverse functions. All myeloid cells arise from multipotent haematopoietic stem cells (HSCs) that develop into mature myeloid cells through sequential steps of differentiation. The three groups of terminally differentiated myeloid cells — macrophages, dendritic cells (DCs) and
8
granulocytes — are essential for the normal functions of the innate and adaptive immune systems [22].
In addition to their physiopatological roles, infiltrating myeloid cells are an abundant component of solid tumours. Thanks to molecular interactions with tumour and stroma, cells of the myeloid lineage recruited at tumour site change their transcriptional program toward a pro-tumoural phenotype that supports tumour growth inducing immunosuppression, angiogenesis and tissue remodeling. It is well recognized that tumor-derived factors (TDFs), such as cytokines, chemokines and inflammatory messengers like prostaglandins, act in paracrine or systemic fashion to ‘reprogram’ non-cancerous host cells to promote tumour progression.
Myelopoiesis is a tightly regulated process of cellular development occurring in the bone marrow. Consequently, chronic exposure of the bone marrow microenvironment to non-physiologic levels of ordinarily tightly regulated myelopoietic-like growth factors corrupts the normal process of myeloid cell development and differentiation. This phenomenon drives the increase of circulating myeloid cells in tumour-bearing hosts, originally termed “emergency myelopoiesis”, and it is associated with a partial blockade of myeloid cell differentiation and a consequent accumulation of highly immunosuppressive, immature myeloid cells (iMCs) [17, 22]. Indeed, many TDFs are myelopoietic factors making the myeloid compartment a major target of this ‘tumour reconditioning’ [23]. For example, cytokines such as granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-(G-CSF), stem cell factor (SCF; also known as KIT ligand), vascular endothelial growth factor (VEGF) and IL-3 are described to promote myelopoiesis and to contribute, in part, to a blockade of myeloid cell maturation. Myeloid deficiencies can occur at developmental and/or functional levels in essentially all myeloid lineages. To distinguish “normal” myeloid cells from their dysfunctional counterparts, the latter
9
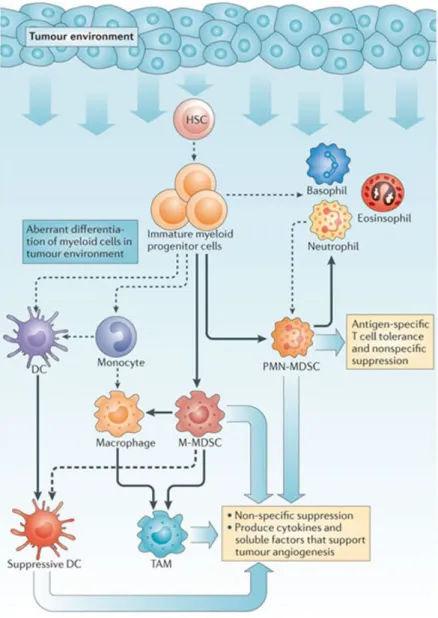
populations have been variously renamed myeloid-derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs), tumour-associated neutrophils (TANs), immature DCs or tolerogenic DCs (Fig.4).
Figure 4. Myeloid cells in cancer.
Factors produced in the tumour microenvironment by tumour cells and stromal cells promote the aberrant differentiation of myeloid lineage cells. The dotted lines show the
normal pathways of myeloid cell differentiation from immature myeloid precursor cells to dendritic cells, macrophages and
granulocytes. The solid bold lines indicate the
aberrant pathways of
myeloid cell
differentiation that occur in cancer, in which the tumour environment can promote the development
of various immunosuppressive populations, including monocytic MDSCs, polymorphonuclear MDSCs, suppressive DCs and TAMs and TANs. From [22].
These classifications are largely based on assays (in vitro and/or in vivo) that measure how these cells affect immune activation or tumour growth and on surface marker expression. Among these, for example, we have recently identified subsets of MDSCs and TAMs based on the expression of
retinoic-10
acid-related orphan receptor (RORC1/RORγ) in human and mouse tumor bearers. In these disfunctyonal myeloid cells, RORC1 orchestrates myelopoiesis by suppressing negative (Socs3 and Bcl3) and promoting positive (C/EBPβ) regulators of the key transcriptional mediators of myeloid progenitor commitment and differentiation to the monocytic/macrophage lineage (IRF8 and PU.1). RORC1 supported tumor-promoting innate immunity by protecting MDSCs from apoptosis, mediating TAM differentiation and M2 polarization, and limiting tumor infiltration by mature neutrophils [24].
11
1.2 The Transcription Factor NF-kB
The NF-B transcription factor family is considered the central mediator of the inflammatory process and a key participant in innate and adaptive immune responses; moreover during the last years it has been proved to play a crucial role in cancer development [25].
NF-κB is an inducible transcription factor that regulates immediate and long-lived cellular responses to environmental changes. NF-κB is evolutionarily conserved and plays a critical role in many biological systems, above all the immune system, where it acts as the major orchestrator of the transcriptional responses to many different stimuli. The engament of several immune receptors such as B and T cell receptor (BCR, TCR), Toll Like Receptors (TLRs), Tumor Necrosis Factor Receptor (TNFR) or CD40 triggers NF-kB activation which in turn results in the expression of cytokines, growth factors and effector enzymes [26-28]. NF-κB also regulates the expression of genes outside the immune system, playing a crucial role even in embryo, mammary gland, skin, bone and nervous system development and physiology [29-35]. At present, more than 150 genes under control of NF-B have been identified, as a demonstration of its vast spectrum of biological functions [36]. Indeed, it is very well known that NF-B disregulaton is linked to various pathological situations.
Misregulation of NF-κB activity, such as constitutive activation, could be associated with pathological conditions such as rheumatoid arthritis, asthma, intestinal bowel diseases (IBDs), multiple sclerosis and cancer [37-41].
Given this great variety of biological roles, a better understanding of NF-κB pathways could provide the basis for the development of therapeutic strategies with a relevant impact on human diseases.
12
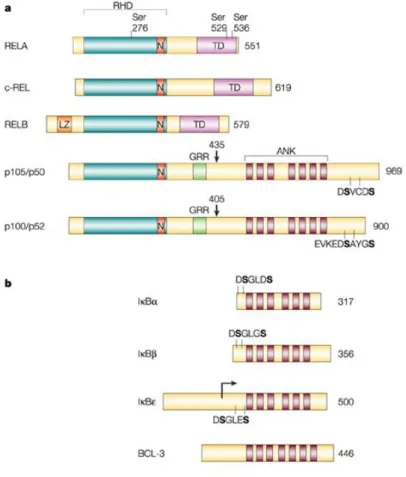
NF-κB family
NF-κB family is composed of five members: RELA (p65), RELB, cREL, NF-κB1 (p105-p50) and NF-κB2 (p100-p52). All these proteins possess a conserved 300-amino acid REL homology domain (RHD) that is located toward the N-terminus of the proteins and is responsible for dimerization, binding to inhibitors of nuclear factor κB (IκBs) and binding to DNA. Instead, the carboxy-terminal non-homologous transactivation domain (TAD), which strongly activates the transcription of targeted genes, is present only in cREL, RELB and RELA. p105 and p100 after proteolytic degradation generate p50 and p52 [28] which lack the transactivation domain. Therefore if they form homodimers they still bind the DNA consensus sites, but they don’t activate transcription [16] (Fig.4).
Each member of NF-κB family except for RELB can form homodimers as well as heterodimers with one another but the main activated form of NF-κB is the heterodimer composed by p65 and p50 or p52.
Mice lacking cREL, NF-κB1 (p105-p50) or NF-κB2 (p100-p52) subunits display a normal development except some defects in lymphocytes activation, whereas p65 or RELB knockout mice are embryonic lethal by liver degeneration and died postnatally from multiorgan inflammation, respectively. Mice lacking more than one subunit, for instance p50-/-p52-/- or p50-/-RelB-/- display more severe phenotypes demonstrating redundancy between NF-κB members.
NF-κB regulation: IκB proteins
Inhibitor of nuclear factor κB (IκB) family comprises four members: IκBα, IκBβ, IκBε and BCL-3 (Fig.6). These proteins share ankyrin repeats which mediates protein-protein interactions [16].
IκBα, IκBβ and IκBε exhert their regulatory function binding NF-κB proteins and masking their Nuclear Localization Sequence (NLS). So, the complexes IκBs-NF-κB cannot translocate into the nucleus and are retained in the
13
cytoplasm in inactive forms. Unlike the other members of IκB family, BCL-3 binds specifically to p50 and p52 homodimers and induce the transcription of NF-κB regulated genes [16, 42].In addition,IκB proteins act only masking p65 NLS, but p50 NLS remains accessible [43-46].
Figure 6: Mammalian NF-κB- and IκB-family members.
A. NF-κB family comprises five members: RELA (p65), cREL, RELB, p105/p50 (NF-κB1) and p100/p52
(NF-κB2). LZ: leucine-zipper motif. B. IκBs family contains IκBα, IκBβ, IκBε and BCL-3, and it is identified by the presence of many ankyrin (ANK) repeats. The amino-acid sequences of the sites of induced phosphorylation of IκBα, IκBβ, IκBε for their degradation are shown (DSGLDS, DSGLGS and DSGLES, respectively). Proteolytic processing of p105 and p100 at residues 435 and 405 (as indicated by arrows), respectively, generates the p50 and p52 NF-κB proteins. The glycine-rich region (GRR) and the carboxy-terminal sites of inducible phosphorylation (in the DSVCDS and EVKEDSAYGS sequences for p105 and p100, respectively) are required for processing. Phosphorylation of RELA at Ser276, Ser529 and Ser536 is important for its transactivation activity. The size of each human protein is shown on the right (number of amino acids). From [47].
14
NF-κB Activation
κB activation is mainly dependent on IκBs degradation thus leaving NF-κB complexes free to translocate into the nucleus, bind promoter and enhancer regions containing κB sites with the consensus sequence GGGRNNYYCC (N=any base, R=purine, Y=pyrimidine) and activate gene transcription. In addition to IκBs, also post-translational modification, like acetylation or phosphorylation, can modulate NF-κB activation. For example, IL-1/TNFα-induced phosphorylation of p65 Ser276 after IκBα degradation is essential for the efficient binding of p65 to the transcription co-activator CREB-binding protein (CBP) [4, 48]. Similarly, phosphorylation of p65 Ser529 enhances its transcriptional activity [4, 49] and the loss of p65 phosphorylation influences both its DNA binding and transactivation activities.
Triggering of many different receptors can induce NF-κB activation that is initiated upon phosphorylation of IκBs by IκB Kinases (IKK). IKK is a complex made by kinase subunits IKKα and IKKβ and the regulatory subunit IKKγ or NEMO (NF-κB Essencial Modifier) [50, 51].
Upon phosphorylation by IKKs, IκB proteins are recognized and ubiquitinated by ubiquitin ligases [52, 53] leading to NF-κB activation.
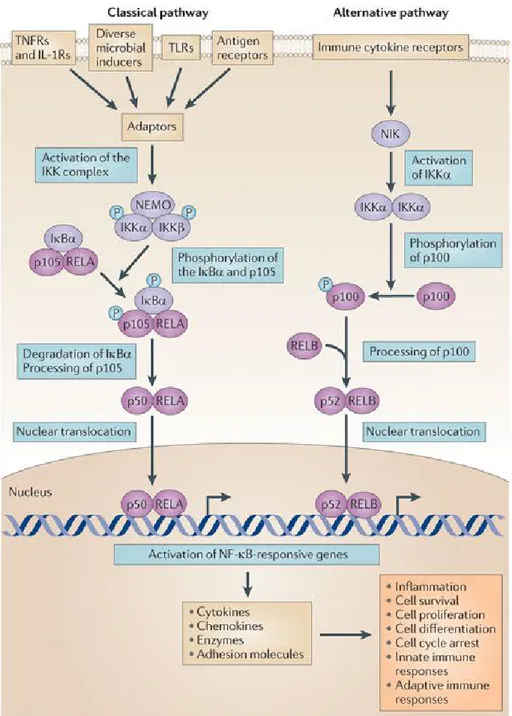
NF-κB could be activated through two different pathways: classical and alternative (Fig.7).
Classical pathway
The common or classical NF-κB signalling pathway is particularly active in innate immunity [54, 55] and is activated predominantly by the subunit IKKβ in a NEMO dependent manner. The released NF-κB dimers, that in this pathway are predominantly p65-p50 heterodimers, go to the nucleus and activate gene transcription [51]. Many different pattern recognition receptors (PRRs) has evolved to recognize microbial invaders and are able to activate NF-κB classical pathway; among these, there are TLRs, members of the CATERPILLAR/NOD
15
family of cytoplasmic receptors, scavenger receptors and the complement system.
TLRs are evolutionarily conserved PRRs that recognize molecules characteristic of various classes of microbes [56]. The function of TLRs as arbitrators of self/non-self discrimination highlights their central role in innate immunity as well as in the initiation of the adaptive immune response.
Signalling through TLRs leads to activation of canonical IKKs complexes, degradation of IκBs and activation of RELA and cREL containing NF-κB dimers. TLR signalling to NF-κB is divided into two pathways: those that are MyD88 (myeloid differentiation primary response gene 88)-dependent and those that are MyD88-independent.
The beginning of an inflammatory response is strictly dependent from NF-κB classical pathway. Signals coming from the environment lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation –
rubor, calor, dolor and tumor (redness, heat, pain and swelling, respectively).
Alternative pathway
The alternative pathway of NF-κB activation (Fig.6) is particularly active in cells of the adaptive immunity, such as B and T lymphocytes. This pathway is independent of IKKβ and NEMO [57, 58], but it is dependent of IKKα homodimers, which selectively phosphorylate p100 associated with RELB. Therefore, the consequence is the release of active RELB-p52 heterodimers [59, 60].
Activation of NF-κB downstream B cell receptor (BCR) and T cell receptor (TCR) is a critical step for mounting adaptive immune responses allowing antigen specific maturation and proliferation of lymphocytes into effector cells [61].
16
Figure 7: Classical and alternative NF-κB pathway.
Protein levels and activity of signalling molecules can be regulated through post-translational modifications such as phosphorylation, ubiquitylation and acetylation. The activation of nuclear factor-κB (NF-κB) ultimately results in the transcription of genes that encode pro-inflammatory factors and factors that influence cell proliferation. IκBα, NF-κB inhibitor-α (also known as NF-κBIα); IKK, IκB kinase; IL-1R, interleukin-1 receptor; NEMO, NF-κB essential modulator (also known as IKKγ); NIK, NF-κB-inducing kinase (also known as MAP3K14); TLR, Toll-like receptor; TNFR, TNF receptor. From [62].
17
NF-κB and cancer
NF-B exerts a great variety of biological roles; this means that disregulations of NF-B pathways can have broad deleterious consequences.
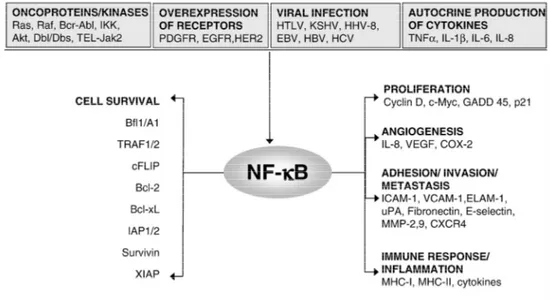
For its function in activating the transcription of genes important for cell proliferation (e.g. cyclin D1, Myc) survival (cIAPs, A1/BFL1, BCL-2, c-FLIP) adhesion, and angiogenesis (e.g. IL-8, VEGF) [4], NF-B is considered a potential molecular bridge between inflammation and cancer [4].
In fact, as a master regulator of inflammation, NF-κB triggers the transcription of several proinflammatory mediators such as IL-1β, TNFα, IL-6 and IL-8. These factors are able to induce higher NF-κB activation, thus providing a positive feedback loop at the site of inflammation which creates an environment in which DNA damage, cell proliferation, transformation and survival and consequently cancer initiation, growth and progression are facilitated [36]. In addition, NF-κB is involved not only in tumor development at early stages, but also in the migration, invasion and metastasis of malignant cells. The invasive capacity of malignant cells can increase in the presence of inflammatory cytokines such as TNFα, IL-1β and IL6 [2]. In particular TNFα is a potent stimulator of epithelial-mesenchimal transition by breast cancer cells [63] for its ability of activate NF-κB signaling. NF-κB was also found to promote metastatization in a genetic mouse model of prostate cancer, in which inactivation of IKKα was found to reduce metastatic spread [64].
In many cancers, NF-κB is constitutively active, even if the exact mechanism that sustain this activation is not fully understood and several mechanisms have been proposed (Fig.8), such as IL-1β and TNFα production, shorter IκBα half life or IκBα mutations [65-69].
For these reasons, NF-κB represents an ideal therapeutic target for the development of new anti-tumor strategies. Proteasome inhibitors as well as IKK inhibitors block NF-κB activation. Similarly, inhibitors of histone acetylation
18
can inhibit NF-κB as well as stimulation of histone deacetylase like HDAC3[70] [70].
Another mechanism to block NF-κB activation is the transfer of mutated genes that encode for NF-κB inhibitory proteins, most commonly IκB genes. IκB are mutated at the site of phosphorylation or ubiquitination, therefore they cannot be degraded.
In many studies, the super repressor of NF-κB was delivered into intestinal epithelial cells through adenoviral vectors and inhibition of NF-κB was very successful. These studies are very important because they provide a possible in vivo approach for the treatment of intestinal malignances [71].
Figure 8: Constitutive NF-B activation in tumours
p50/NF-κB1
The role of p50 and its precursor p105 in cell physiology and function is very complex. Although originally considered a repressor of transcription, p50 could also be a transcriptional activator: the balance between pro- and anti-inflammatory activity of p50 depends on cell type and environmental conditions.
19
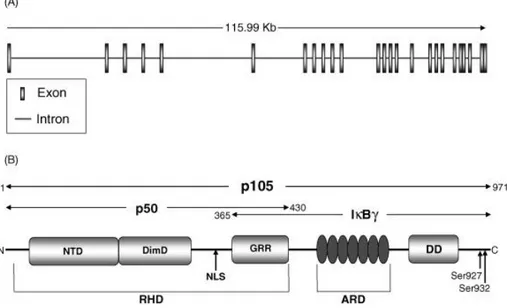
The NF-κB1 gene encodes two functional proteins: p50 and p105. It is thought that a third protein, IκBγ, could be generated by alternative splicing (Fig.9)[72].
p105 is the precursor of p50 which is the active form of the protein and could form dimers with itself and with other NF-κB subunits.
Since p50 homodimers mainly act as repressors of transcription, and given the importance of NF-κB during inflammation, it is likely that they act also as repressors of inflammation. Indeed, it has been demonstrated that in LPS-tolerant macrophages increased expression of the p50 subunit of NF-κB directly results in the downregulation of LPS-induced TNFα production, whereas in p50 -/- macrophages long-term pre-treatment with LPS was unable to induce
tolerance. In line, our group has demonstrated that TAMs express a tolerant pro-tumoral phenotype that is controlled by massive nuclear accumulation of p50 NF-B [73].
Figure 9: NF-κB1 gene and protein structure.
A. NF-κB1 gene is 115.6 kb long and encodes a 3452 bp-long transcript containing 25 exons. B. Protein structure of
p105, p50 and IκBγ. p105 is a 971 amino acids peptide containing a Rel homology domain (RHD), seven ankyrin repeats in the ankyrin repeat domain (ARD) and the death domain (DD). The RHD includes the N-terminal domain (NTD), dimerisation domain (DimD), nuclear localisation sequence (NLS) and glycine-rich region (GRR). p105 is phosphorylated at serine residues 927 and 932, a signal for poly-ubiquitination and subsequent degradation by the 26S proteasome releasing the active subunit p50. p50 spans amino acids 1–430 which encompass the RHD, in this form the
20
NLS is exposed and promotes p50 nuclear translocation. IκBγ spans amino acids 365–969 and contains the GRR, ARD and DD. From [74].
Even if p50 homodimers lack the transactivation domain, they may activate gene expression by recruiting trascritional co-activators. In particular, Cao et al. demonstrate that in LPS (or other TLRs agonists) stimulated macrophages, p50 homodimers form complexes with the transcriptional co-activator CREB binding protein. This complex binds to the IL-10 promoter and stimulates transcription of this anti-inflammatory gene [75]. In fact, p50-/- mice display reduced IL-10 production and increased TNFα and IL-12 production [76]. Accordingly, we have shown that LPS stimulated p50-/- TAMs recover an IL-12highTNFαhighIL-10low phenotype and that this correlates, in vivo, with tumor growth inhibition [73]. Further, a detailed analysis of the role of p50 NF-κB homodimer in macrophage functions revealed that its nuclear accumulation, both in TAMs and LPS-tolerant macrophages, not only mediates a status of unresponsiveness (tolerance) toward pro-inflammatory signals, but actually plays a role of key regulator of M2-driven inflammatory responses [77]. Accordingly, p50-deficient mice show exacerbated M1-driven inflammation and defective capacity to mount allergy and helminth-driven M2-polarized inflammatory reactions [77]. Hence p50 NF-κB regulates the orientation of macrophage polarization, playing a crucial role in the control of M1- vs M2-driven inflammation.
For all these reasons, p50 could represent a good therapeutic target for human diseases; in one way, p50 could be inhibited to enhance M1 pro-inflammatory response in situation when it is strongly required (e.g. tumours), whereas in the other way, its activity could be augmented to promote exhintion of exacerbates inflammatory conditions (e.g. IBDs, rheumatoid arthritis, multiple sclerosis).
21
1.3 Macrophages
A century ago Metchnikoff received the Nobel Prize for the discovery of macrophages and innate immunity. After their first discovery, macrophages acquired many different functions both immunological and non-immunological; they have roles in almost every aspect of an organism’s biology from developmet and homeostasis to repair through immunity. Resident macrophages regulate tissue homeostasis by acting as sentinels and responding to changes in physiology as well as challenges from outside. Unfortunately, in many cases, these homeostatic and reparative functions can be subverted by continuous insults, resulting in diseases such as chronic inflammation, autoimmune diseases, obesity and cancer (Fig.10).
Macrophages play an indispensable role in the immune system with decisive functions in both innate and acquired immunity. In innate immunity, resident macrophages provide immediate defence against foreign pathogens and coordinate leukocyte infiltration [78]. Macrophages contribute to the balance between antigen availability and clearance through phagocytosis and subsequent degradation of senescent or apoptotic cells, microbes and possibly neoplastic cells. Their role is essential for triggering, instructing and terminating the adaptive immune response. Macrophages collaborate with T and B cells, through both cell-to-cell interactions and fluid-phase mediated mechanisms, based on the release of cytokines, chemokines, enzymes and reactive radicals.
Macrophage origins and differentiation
In mammalian, macrophages are found in all tissues and display great anatomical and functional diversity. The density of macrophages changes in many tissues during development [79].
22
Macrophages are differentiated cells of mononuclear phagocytic lineage [80] that are characterized by a specific phenotype and by the expression of particular markers, none of which are entirely restricted to the lineage [81]. In mice, macrophages express CD11b, F4/80, CD68, colony-stimulating factor-1 receptor (CSF-1R; CD115) and do not express Gr1. In humans they are described as CD68+CD33+CD14+ cells.
Figure 10: Macrophages in Development, Homeostasis and Disease
Macrophages play many developmental roles shaping the architecture of tissues ranging from the brain to bone to the mammary gland. Once development is over macrophages modulate homeostasis and normal physiology through regulating diverse activities including metabolism and neural connectivity and by detecting damage. These trophic and regulatory roles however, are often subverted by continuous insult and macrophages contribute to many diseases that are often associated with aging. EAE: Experimental Autoimmune Encephalomyelitis; IBD: Inflammatory Bowel Disease. From [82].
23
The term “macrophage” comes from the Greek, emphasising the ability of these cells to phagocyte. Lots of definition were given to these cells during time; nowadays macrophages are classified basing on ontogeny and phagocytosis by the term mononuclear phagocytic system (MPS) [83]. It includes bone-marrow derived precursor cells, monocytes in peripheral blood and mature macrophages in tissues. In the MPS schema, adult macrophages are end cells of the mononuclear phagocytic lineage and tissue macrophages derive from circulating monocytes that originate in BM.
More recently the classification of the MPS has been refined because several studies pointed out that macrophages have several origins during ontogeny and each of these different lineages persist into adulthood displaying great diversity [84]. In addition, some dendritic cell subsets can differentiate from monocytes and macrophages [85].
The origin of mononuclear phagocytic cells in ebryo is complex: in mice, the first population of macrophages is observed at embryonic day (E)7.5 and is of maternal origin [86]. Embryonic macrophages derive from the primitive ectoderm of the yolk sac and do not go through a monocytic stage but differentiate directly from mesenchymal progenitor cells [87]. By E10.5 to E11 liver represents the main site of hematopoiesis followed then by a second wave of progenitor cells from the aorta-gonads-mesonephros region of embryo [88, 89]. After birth, the bony structures are formed and, from that moment, the bone marrow become the source of circulating monocytes (resident Ly6C- and inflammatory Ly6C+ in mice) [90]; at this stage, the MPS is established [83].
Nowadays, this first model of the MPS formation has been expanded. In fact, in a few tissue, such kidney and lung, macrophages were shown to have a chimeric origin (Hematopoietic Stem Cells [HSC]- and yolk sac-derived) as well as Langerans cells (LC) of the skin, which have a mixed origin from yolk sac and fetal liver [91, 92]. So it is possible to identify at least three lineages of
24
macrophages in mouse during different stages of development and persisting in the adult (Fig.11).
Figure 11. Macrophage Lineages Re-Defined in Mice
The mononuclear phagocytic system in adults derives from at least three sources. The first is the yolk sac (YS) that results in progenitors that populate all tissues and their progeny persist throughout life as F4/80 bright resident macrophages. These lineages are largely regulated by CSF1R. The second from the fetal liver is less well defined but seems to contribute to adult LCs perhaps through a progenitor derived from the YS. The third lineage derives from the bone marrow (BM) to give circulating monocytes and their progeny F4/80low macrophages and DCs. In this case the Ly6c+ monocytes give rise to classical DCs under the regulation of FLT3 and these are continuously replenished. Other macrophages that are F4/80low also emanate from Ly6c+ monocytes and in some cases such as kidney and lung, co-exist with those derived from YS to give chimeric organs. The exact role of the patrolling Ly6c+ macrophages remains unclear, as is the contribution of fetal liver to adult tissue macrophages. From [82].
Yolk sac macrophages first appear at E9.0 in both mouse and rat, and develop without passing through a monocytic intermediate stage [93]. They are the primary source of microglia, the resident macrophages of the central nervous system [91], and also give rise to a minor fraction of Langherans cells (LCs) [92]. The major fraction of adult LCs derives from fetal monocytes generated in the fetal liver from E12.5 and recruited into fetal skin around E14.5 [92]. Fetal monocytes also contribute to populations of adult macrophages in lung alveoli [94, 95] and in the heart [96]. Using fate-mapping to distinguish cells arising from primitive versus definitive hematopoiesis initially suggested that adult
25
macrophage populations in lung, dermis, and spleen arise predominantly from definitive hematopoiesis with negligible contribution from yolk sac-derived macrophages [91]. However, a new approach exploiting the differential dependence of macrophages on the transcription factor c-Myb has since indicated that c-Myb-independent yolk sac-derived macrophages may be the sole origin of macrophages in the lung, liver, and pancreas, as well as of microglia and LCs[97]. In this scenario it seems that the expression of c-Myb between early and late stage is different and many reports indicate that primitive hematopoiesis in yolk sac can occur in the absence of c-Myb [98] because the earliest yolk sac progenitors, which give rise to microglia, do not express c-Myb [99], whereas progenitors from definitive hematopoiesis both express and depend upon c-Myb [100-102]. Because fetal monocytes are absent in c-Myb-deficient embryos [97, 103] and c-Myb expression is upregulated during fetal monopoiesis, it is likely that the change in the progenitors fate between the yolk sac and the fetal liver is orchestrated by c-Myb. Microglia represents an exception because it have a unique origin, arising from yolk sac macrophages that maintain themselves by proliferating in situ throughout adulthood, and not from fetal monocytes [91, 99].
In addition, recent studies have shown that even in absence of hematopoietic stem cells, yolk sac progenitors were capable of giving rise to the major tissue resident population of macrophages in skin, spleen, pancreas, liver, brain and lung [97].
Hence, the idea that macrophages derive from circulating monocytes has been questioned. In fact, complete loss of CD16+ monocytes in humans appears
to be of little consequence [104] and in this scenario the function of monocytes needs to be re-defined. It is possible that patrolling monocytes (Ly6C-) act to maintain vessel integrity and to detect pathogens while inflammatory monocytes (Ly6C+) are recruited only to site of infection or injury or to tissues that have continuous cyclical recruitment of macrophages such as the uterus.
26
The development of macrophages from monocytes is regulated by several growth factors. Early studies indicate that, in mice and rats at least, the most important of these was the colony stimulating factor 1 receptor (CSF1R) which not only drove the differentiation of macrophages from progenitors, but also controlled their proliferation and viability in vitro. More recent studies, basing on the ablation of Csf1r, demonstated a severe depletion of macrophages in many, but not all, tissues [105]. These new data opened a different scenario and following experiments confirmed that CSF1R is expressed both on yolk sac macrophages and fetal monocytes, but only the development of the former is dependent on CSF1R [91, 92]. In fact, for example, Hoeffel and Ginoux discovered that the major fraction of adult LCs is derived from fetal monocytes that are generated in the fetal liver independently of CSF-1R expression [91, 92].
Macrophages in development
Macrophages present important roles in tissue development as demonstrated by the cluster of abnormalities that characterize Csf1 null mice, which lack many macrophages populations. This mice survive to adulthood because of extra-medullary hematopoiesis in the spleen and liver [80].
As professional phagocytes, macrophages perform critical functions in the remodelling of tissues, both during development and in the adult animal. For istance, during erythropoiesis, maturing erythroblasts are surrounded by macrophages, which ingest the extruded erythrocyte nuclei. This function of macrophages is of critical importance because in its absence, erythropoiesis is blocked and lethality ensues [106]. Remodeling deficiencies in the absence of macrophages have been noted in many other tissues, suggesting a general requirement for macrophages in tissue remodeling and morphogenesis [80, 107].
27
Macrophages also regulate angiogenesis through a variety of mechanisms, among which there is the synthesis and release of Wnt7b that delivers a cell death signal to the vascular endothelial cells (VEC) and either its absence or the absence of macrophages drives vascular over-growth [108]. This is not restricted to the vascular arm of the circulatory system since macrophages also play roles in lymphangiogenesis during development [109].
Also brain development is influenced by a specific population of macrophages called microglia, whose presence is dependent on CSF1R signalling [110, 111] that promotes neuron viability [80] , modulates neuronal activity [112], prunes synapse during development [113] as well as expresses a range of neuronal growth and survival factors including NGF [80].
Macrophage phenotypes
Peculiarity of macrophages is their plasticity, that means the ability to finely modulate their programs in response to differentmicroenvironmental conditions [114].
The diversity of macrophages functions has led to various classification attempts. “Classical activated” M1 macrophages are characterized by high levels of inducible nitric oxide syntase (iNOS) and the production of reactive oxygen intermeadiateds (ROI). They are potent effector cells and strongly antigen presenters involved in T helper 1 (Th1)-cell-mediated immune resolution of infection [115]. Signals that led to M1 polarization (LPS, IFN) trigger the activation of NFκB- and STAT1- pathways with subsequent transcription of NFκB- and STAT1- dependent proinflammatory cytokines (e.g. 12 and IL-23). On the contrary the “alternative activated” M2 macrophages, which respond to Th2-type cytokines, such as IL-4 and IL-13, are involved in fibrosis, scavenge debris, tissue remodelling and repair, angiogenesis and humoral immunity [116] and are able to tune inflammatory response. They show strong activation of
28
arginase pathway with generation of ornithine and polyamines [12] (Fig.12). M2-polarizing signals generally inhibit the expression of M1 cytokines and chemokines. These inhibitory effects principally relay on STAT-3 dependent mechanisms [117] and the direct inhibition of NF-κB [47].
Figure 12: M1 and M2 macrophages.
In the presence of interferon (IFN)–, lipopolysaccharide and other microbial products, monocytes differentiate into M1 macrophages. In the presence of macrophage colony-stimulating factor (CSF-1), interleukin (IL)-4, IL-13, IL-10 and immunocomplexes in association with either IL-1R or TLR-ligands, monocytes differentiate into M2 macrophages. M1 and M2 subsets differ in term of phenotype and functions. M1 cells have microbial activity, immuno-stimulatory functions and tumor cytotoxicity. M2 cells have high scavenging ability, promote tissue repair and angiogenesis and favour tumor progression.
Signals from the microenvironment are able to drive macrophages towards M1 or M2 polarization; moreover macrophages are exposed to a multiplicity of opposite signals in vivo with different temporal pattern. Nevertheless there is considerable plasticity between distinct types: M1 and M2 polarization states are often referred to as the extremes of a continuum (Fig.13) [116].
When tissues are damaged following infection or injury, inflammatory monocytes (Ly6C+ in mice) are recruited from the circulation and differentiate into macrophages as they migrate into the affected tissues [90]. These recruited
29
macrophages exhibit a M1 pro-inflammatory phenotype in the early stages of a wound healing response.
Figure 13: M1 and M2 macrophages are the extremes of a continuum. M1 and M2 macrophages, the extremes of a continuum. Essential properties of polarized macrophage populations are shown. For M1 cells, molecules induced by IFN-γ and LPS are shown in green. For M2 cells, molecules induced by IL-4 and IL-13 are shown in yellow, those induced by IL-10 in red and those induced both by IL-4 and IL-13, and
IL-10, in blue.
Macrophages exposed to the classic activation signals, IFN-γ and LPS,
express opsonic receptors (e.g. FcγRIII/CD16), whereas type II macrophages are characterized by
abundant levels of non-opsonic receptors (e.g. the MR). M1 cells also have a higher ratio of reduced-to-oxidized
glutathione, with
opposite effects of IFN-γ and IL-4 on the
reductive status.
Components of the IL-1 system are differentially regulated in polarized macrophage populations. IL-4, IL-13 and glucocorticoid hormones induce expression of the IL-1 type II decoy receptor, whereas IFN-γ and LPS inhibit it. IFN-γ and LPS upregulate the signaling type I receptor, and IL-1R accessory protein (IL-1RacP). IL-4 and IL-13 induce IL-1ra production and inhibit IL-1. Therefore, pro- and anti-inflammatory components of the IL-1 system are coordinately regulated by signals that polarize macrophages in a type I or type II direction. IL-10 upregulates the CC chemokine receptors CCR1, CCR2 and CCR5. By contrast, CXCR2 and CXCR4 are partially downregulated under the same conditions. An increase in CCR2 expression is also observed in monocytes exposed to dexamethasone. IL-4 and IL-13 do not modify the expression of CC chemokine receptors but induce functional CXCL8 (IL-8) receptors in human monocytes. By contrast, monocytes exposed to LPS or IFN-γ downregulated CCR1, CCR2 and CCR530. and 73.. Similar to what was reported for DCs, exposure of monocytes to classical proinflammatory signals induces the expression of functional CCR7 and the effect is inhibited by IL-10. Abbreviations: DC, dendritic cell; IFN, interferon; IL, interleukin; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; MR, mannose receptor; ra, receptor agonist; ROI, reactive oxygen intermediates; TLR, toll-like receptor; TNF, tumor necrosis factor. From [116].
30
Although these inflammatory macrophages are initially beneficial because they facilitate the clearance of invading organisms, they also trigger substantial collateral tissue damage because of the toxic activity of reactive oxygen and nitrogen species [118]. Indeed, if the inflammatory macrophage response is not quickly controlled, it can become pathogenic and contribute to disease progression, as seen in many chronic inflammatory and autoimmune diseases [119, 120].
To counteract the tissue damaging potential of the inflammatory macrophage response, macrophages undergo apoptosis or switch into a M2 anti-inflammatory or suppressive phenotype that dampens the pro-anti-inflammatory response, while facilitating wound healing [121].
So, the pathophysiological adaptations to regulate over-exuberant inflammation serve as an important mechanism for host protection and one of the classic examples of such a protective mechanism is endotoxin tolerance [122-126].
Tolerance is a state of hyporesponsiveness acquired after prolonged exposure of macrophages to inflammatory agents including bacterial products such as LPS. Tolerant macrophages enter into a transient unresponsive state and are unable to respond to further challenges with endotoxin. This phenomenon has been observed both in vitro and in vivo in animal models as well as in humans [123, 124, 126-130]. Tolerance is caused by a profound “gene expression reprogramming” in macrophages [122, 126, 129] which develop poor inflammatory capacity coupled with upregulation of anti-inflammatory cytokines. Overall this characteristics contribute to protection against septic shock and increased phagocytosis allow efficient bacterial clearance (Fig.14).
Interestingly, many of the characteristic of endotoxin-tolerant monocytes/macrophages resemble that of the immunosuppressive M2 macrophages [77].
31
Figure 14: phenotype of endotoxin tolerance monocytes/macrophages.
A. Upon endotoxin re-challenge with gram-negative bacteria or LPS, monocytes/macrophages show a drastic
downregulation of inflammatory cytokines (e.g. TNFα, IL-6, IL-1β, IL-12) but an upregulation of anti-inflammatory cytokines like IL-10, TGFβ and IL-1RA as compared to non-tolerized cells challenged with the same stimuli. The tolerant monocytes/macrophages also show an impaired antigen presenting capacity correlated with decreased expression of HLA-DR and some co-stimulatory molecules. In contrast, these cells show upregulated expression of a number of scavenging/C-type lectin receptors like MARCO, CD64, CLEC4a 10 and 11 is linked to enhanced phagocytic capacity. Upregulation of negative regulators of TLR4 signaling like IRAK-M is also a characteristic of these cells. (+) denotes upregulation, while (-) denotes downregulation during ET. B. Schematic representation of the biphasic nature of sepsis wherein an initial overt inflammatory phase leads to a later phase of immunosuppression or “immunocompromise”. In parallel with these events, monocytes/macrophages also “switch” from an inflammatory phenotype to an endotoxin tolerant or refractory state. Monocytes respond to the systemic infection by triggering an inflammatory reaction characterized by overt levels of inflammatory cytokines and chemokines (e.g. TNFα and IL-6). However, as sepsis progresses, these monocytes become refractory to further endotoxin challenge whereby they fail to upregulate inflammatory cytokines. Instead, they start producing anti-inflammatory cytokines (TGFβ, IL-10) which promote immunosuppression. Under these conditions, there is a high risk of developing secondary infections, which may lead to mortality. From [131].
We previously demonstrated that, like M2 skewed macrophages, also tolerant macrophages display accumulation of p50 NF-κB subunit in the nucleus, and subsequent defective NF-κB activation (Fig.15) [132]. The importance of p50 for the acquisition of a tolerant phenotype is also demonstrated by the fact that
32
lack of p50 in murine macrophages prevents the development of tolerance. Macrophages lacking p50 do not dowregulate pro-inflammatory cytokines and are not able to upregulate anti-inflammatory factors even after prolonged exposure to LPS [77].
Figure 15: plasticity of NF-kB function in inflammation and endotoxin tolerance.
This figure represents how different combinations of NF-kB hetero- and homodimers can switch-on or -off the same genes under inflammatory or endotoxin tolerant conditions. During overt inflammation (as seen in the first phase of sepsis), the p65/p50 NF-kB heterodimer is responsible for the transcription of inflammatory genes like TNFA, IL12 etc. During endotoxin tolerance, there is an overexpression of p50/p50 NF-kB homodimers, which lack a transcription activating domain. This causes p50/p50 NF-kB homodimers to occupy the promoters of inflammatory genes and thereby prevent p65/p50 NF-kB heterodimer binding and gene transcription. Conversely, p50/p50 NF-kB homodimer triggers the transcription of genes like IL10, TGFB1 and COX2 [133]. RelB/p65 NF-kB heterodimers present in the endotoxin tolerant cells also prevent the transcription of inflammatory genes. Finally, the accumulation of IkBa and IkBe in endotoxin tolerant cells also prevents NF-kB activation. From [131]
Macrophages in cancer – Tumor Associated Macrophages
The major population of leukocyte infiltrating tumors is represented by macrophages.
33
M2-like phenotype which supports immune escape, tumor growth and malignancy exerting crucial tumor-promoting functions (e.g. induction of tumor cell proliferation and angiogenesis, incessant matrix turnover) [134, 135] and suppress the adaptive immune response [136, 137]. This activities ultimately have an important impact on disease progression [12]. Clinical studies have demonstrated a correlation between high frequency of TAM and the poor prognosis for many different human tumors including lymphoma, cervix, bladder, breast and lung cancers [138]. One of the most important characteristic of TAMs include their ability to directly affect tumor growth through promotion of tumor angiogenesis as well as the survival and metastasis of tumor cells [3, 116, 139-141].
TAMs are recruited to the tumor by a tumor-derived chemotactic factor, originally identified as CCL2, also known as MCP-1. Actually, other chemokines and molecules active on TAMs were detected in neoplastic tissues such as M-CSF, VEGF and angiopoiein-2 [139]. These factors have been showed to promote macrophages recruitment as well as macrophages survival and proliferation and their expression correlates with tumor growth.
Cytokines network at tumor site has a central role in TAMs recruiting and differentiation. Immunosuppressive cytokine IL-10 and transforming growth factor β (TGF β) as well as PGE2 produced by both cancer cells (ovary) and TAMs [116] contribute to a general suppression of anti-tumor activities.
Also in terms of cytotoxicity and expression of inflammatory cytokines, TAMs resemble the M2 macrophages: both are poor producers of nitic oxide (NO) [142, 143] and of ROIs; both are poor antigen presenting cells and not only they are unable to trigger Th1 polarized immune responses, but also they induce T regulatory cells (Treg) [116] and suppress T cell activation and proliferation [116, 144]. Moreover TAMs are unable to produce IL-12, even upon stimulation with IFN and LPS [145]. TAMs express high levels of both
34
scavenger receptor-A (SR-A) [146] and the mannose receptor (MR) together with other M2 markers like Arginase I, YM1, FIZZ1, MGL2.
Angiogenesis is an M2-associated function which represents a key event in tumor growth and progression. Lin and colleagues [147] demonstrated a slower rate of progression to malignancy and fewer pulmonary metastases in CSF-1 null mutant mice (that lack macrophage population) bearing spontaneous mammary carcinoma, than in CSF-1 wild type mice. These data are consistent with clinical findings that high number of TAMs often correlate with increase tumor vascularization. In several human cancer, TAMs accumulation has been associated with angiogenesis and with the production of angiogenic factors such as VEGF and platelet-derived endothelial cell growth factor [8]. Additionally, TAMs participate to the proangiogenic process by producing the angiogenic factor thymidine phosphorylase (TP), which promotes endothelial cell migration
in vitro and whose level of expression are associated with tumor
neovascularisation [148]. These pro-angiogenic TAMs are known as Tie-2 expressing monocytes (TEMs) because they are characterized by the expression of the angiopoietin receptor TIE2 [149, 150] and were found to constitute a small subpopulation of the total tumour infiltrating CD11b+ myeloid cells that could be distinguished from the majority of TAMs by their surface marker profile (Tie2+Sca-1+CD11b+), their preferential localization to areas of angiogenesis, and their marked pro-angiogenic activity [13, 14]. The selective elimination of these Tie2-expressing monocytes dramatically impaired angiogenesis in mouse tumours and induced substantial tumour regression.
TAMs also express molecules which affect dissolution of connective tissues. These include enzymes which regulate the digestion of the extracellular matrix, such as MMPs, plasmin, urokinase-type plasminogen activator (uPA) and the uPA receptor (Fig.16).
TAMs can also be potent immunosuppressors of the cytotoxic activity of CD8+ T cells in progressing tumors: a high stromal TAMs infiltration inversely
35
correlates with CD8+ T cell number [151]. This immunosuppression is mediated, at least in part, by nitrosylation of T cell receptors via ARG1, iNOS and peroxynitrite, inducing T cell apoptosis [22].
For these reasons TAMs have been described as “obligate partners for tumor-cell migration, invasion and metastasis” [141].
Figure 16: TAMs produce several factors that favour tumor growth and spreading.
NF-kB in TAMs
Activation of the transcriptional factor NF-κB is a necessary event promoting transcription of several proinflammatory genes. TAMs display a defective NF-κB activation in response to M1 polarizing signals LPS and TNF [145]. The defect in NF-κB was shown to be associated to the over expression of nuclear p50 NF-κB homodimers which inhibit the transcription of proinflammatory genes [73]. The defective NF-κB activity was seen in TAMs isolated from
36
advanced stages tumors and it is in apparent contrast with TAMs NF-κB dependent pro-tumorigenic functions observed in murine models of inflammation-associated liver and colorectal cancer [152, 153]. This discrepancy might reflect a dynamic change in the tumor microenvironment during the transition from early-neoplastic events to advanced tumor stages, which would result in progressive modulation of the NF-κB activity expressed by infiltrating inflammatory cells and progressive conversion of the TAMs from an M1 to an M2 macrophage phenotype (Fig.17).
Figure 17: tumor immunoediting and progression: macrophage polarization.
During tumor progression a gradual switching of macrophage polarization, M1 versus M2, is paralleled by the gradual inhibition of NF-kB activity. These events concur to establish permissive conditions for tumor growth and spread. From [21].
Although these experimental and clinical results, some evidence does not fit into this general pattern. For example, in certain tumors or subset of tumors, the presence of inflammatory cells is associated with better prognosis (for example, eosinophilis in colon tumors and TAMs in a subset of breast tumors and pancreatic tumors). In fact, appropriately activated macrophages can kill tumor cells although in most cases their tumor-promoting properties prevail [144]. The
37
importance of this balance is evident in psoriasis where a marked chronic inflammatory response is not associated with an increased risk of developing skin cancer [154]. This because psoriasis is a M1/Th1-mediated disease and consequently the inflammatory microenvironment that develops has antitumor features. For these reason not only the ablation, but also the repolarization of TAMs from a M2 to a M1 status is considered an interesting therapeutic strategy.
TAMs as therapeutic targets
It is now clear that myeloid cells infiltrating in the tumor represent an important player that can initiate and support tumor development. In addition to these pro-tumoral activities, TAMs can also modulate the efficacy of various form of anticancer therapy. Based on this, both the recruitment and activation of TAMs are are considered putative targets for therapeutic intervention.
The major strategy so far is based upon genetic experiments targeting genes specifically involved in pro-tumoral macrophages phenotype like CSF-1. In this case the approach is based on anti CSF-1 receptor neutralizing antibodies or small molecule inhibitors interfering with this pathway. For example, TAMs depletion by anti-CSF1 antibodies enhanced the efficacy of combination chemotherapy (cyclophosphamide, methotrexate, and 5-fluoro-uracil) in chemoresistant, human breast cancer xenografts grown in immunodeficient mice [151]. Similarly, TAMs depletion improved the efficacy of paclitaxel in mouse models of mammary tumors [155]. Small molecule inhibitors to CSF1R have also been shown to deplete some populations of TAMs and to dramatically enhance responses to chemotherapy. This effect is at least in part consequent to the removal of macrophage-mediated immunosuppression during the tumor recovery period [151, 156].
38
Furthermore, low-dose irradiation of tumors activates macrophages to orchestrate T cell immunotherapy [157] while the therapeutic efficacy against tumors of Trabectedin in mouse model derives from its ability to directly kill mononuclear phagocytes, including monocytes and macrophages [158].
The role of macrophages in modulating the antitumor efficacy of chemotherapy is very complex and poor understood also because it can be based on both direct and indirect effects. In this regard, innate immune cells like macrophages, are known to activate local antigen presenting cells and increase the immunogenicity of the tumour by inducing the release of danger signals from the tumours cells [159]. These signals can stimulate innate immune responses by operating as adjuvants. This event has been defined as Immunogenic Cell Death (ICD). ICD is an immunogenic type of death that is characterized by a well-known series of events that include: the pre-apoptotic exposure of calreticulin (CRT) and other endoplasmic reticulum proteins at the cell surface (heat-shock proteins, HSPs), the increasing extracellular release of adenosine triphosphate (ATP) during the blebbing phase of apoptosis, and the post-apoptotic release of the chromatin-binding protein high-mobility group B1 (HMGB1) [159]. These molecules act together to promote presentation of tumour antigens [160-162]. Once on the cell surface, CRT serves as an ‘‘eat-me’’ signal, stimulating the engulfment of dying tumour cells and their apoptotic debris by macrophages and immature dendritic cells [160, 163, 164]. Similarly, HSP90 has been demonstrated to be a crucial mediator of immunogenicity [163]. ATP molecules released by dying cells constitute a potent chemotactic signal for myeloid cells including monocytes/macrophages [164] and DC precursors [165]. Cancer cells respond to ICD inducers by secreting ATP, lysosomal exocytosis, and plasma membrane blebbing [164, 166]. Only a few chemotherapeutics are known to induce ICD and Doxorubicin is among them.
39
The contribution of TAMs to the modulation of tumor responses to chemotherapy can vary markedly among different cytotoxic agents and tumor models. For example, the antitumor activity of the taxane docetaxel involves the depletion of immunosuppressive (M2-like) TAMs and the concomitant activation or expansion of antitumoral (M1-like) monocytes. Indeed, in vivo T cell assays showed that docetaxel-treated monocytes/MDSCs are able to enhance tumor-specific cytotoxic T cell responses [167].
TAMs may also release “chemoprotective” factors. Shree and colleagues showed how lysosomal enzymes, cathepsins B and S, secreted by TAMs protected cancer cells from paclitaxel-induced cell death [168]. In addition, also the efficacy of Doxorubicin and Etoposide was seen to be reduced by these TAMs-secreted cathepsins. Furthemore, a recent study demonstrates how the release of these cathepsins from lysosomes by TAMs is induced after 5-fluoro-uracil and Gemcitabine treatment [169]. The chemoprotection produced by cathepsins is correlated to an increase in the production of IL-1β by TAMs, which, in turn, stimulates the secretion of IL-17 by CD4+ T cells, blunting the anticancer effects of chemotherapy.
TAMs may support tumor chemoprotection also by providing survival signals to tumor initiating/cancer stem cells (CSCs). For example, TAMs were found to protect lung and colon CSCs from Cisplatin by releasing milk fat globule-epidermal growth factor 8 protein (MFG-E8) which, in turn, activates STAT3 pathway [170]. In addition, TAMs depletion was demonstrated to improve T cell responses and the efficacy of chemotherapy in pancreatic cancer model, in part by decreasing the tumor-initiating capacity and STAT3 activation of CSCs [156].
Many studies demonstrate the contribution of TAMs to the cytotoxicity of therapeutic monoclonal antibodies (moAbs) [171]. In fact, TAMs express surface receptors for the Fc fragment of antibodies and enable them to engange
40
Trastuzumab, a moAb against the human epidermal growth factor receptor-2 (HER2), not only interrupts HER2 signaling in breast cancer cells, thereby slowing their proliferation rate, but also induces Fcγ receptor (FcγR)-mediated activation of macrophage cytotoxicity [172] and priming of antigen-specific CD8+ T cell responses [173]. Macrophages also enhance lymphoma elimination in mice in response to rituximab, a moAb against CD20, primarly through FcγR-dependent ADCP [174]. Furthemore, high number of TAMs correlates with better prognosis in rituximab-treated patients [175].
Tumor irradiation is widely used to treat many cancers types. Early studies correlated high TAM number in mouse with poor tumor responses to irradiation [176]. Recent data suggest that DNA damage induced by irradiation promotes the transcription of Csf1 via the v-abl Abelson murine leukemia viral oncogene homolog 1 (ABL1) kinase, which, in turn, recruits CSF1R-expressing myeloid cells (including TAMs) that enhance post-irradiation tumor regrowth. Indeed, a CSF1R inhibitor improved tumor response to radiotherapy in a prostate cancer model [177]. Antibody-mediated depletion of Cd11b+ myeloid cells in human head and neck tumors grown in immunodeficient mice also reduced tumor regrowth after therapy [178]. In a model of orthotopic human glioblastoma, local irradiation dramatically enhanced tumor infiltration by CD11b+ myeloid cells [179]. It has been proposed that TAMs activity in post-irradiated tumors is similar to that of M2-like macrophages driving tissue repair after injury [180]. TAMs drive reparative mechanisms in tumours after not only radiotherapy, but also treatment with vascular-targeting agents.
Docetaxel have a strong antitumor activity dued to the depletion of immunosuppressive (M2-like) TAMs and the concomitant activation or expansion of antitumoral (M1-like) monocytes in 4T1-Neu mammary tumour implants [167]. Moreover, Trabectedin, a DNA-damaging agent approved for soft tissue sarcomas, inhibited the growth of mouse fibrosarcomas primarily by depleting mononuclear phagocytes, including monocytes and TAMs [158].
![Figure 5. Role of RORC1 myelopoiesis associated with cancer. From [24]](https://thumb-eu.123doks.com/thumbv2/123dokorg/4805123.49528/16.773.163.610.370.723/figure-role-rorc-myelopoiesis-associated-cancer.webp)