4 . MATERIALI E METODI
4.1 Caratteristiche della Libreria Genomica
Le caratteristiche della Libreria Genomica utilizzata sono riportate nelle seguenti tabelle:
Library Name HA_HBa Common Name Sunflower
Genus Helianthus
Species annuus
Cultivar HA383
Library Type BAC
Vector pIndigoBac536
DNA Fragmentation HindIII Selectable Marker Chloramphenicol
Number of Clones 202752
Number of Plates 528
Number of Filters 11
Average Insert Size 125
Genome Size 3000
Genome Equivalents 8.3
Constructed by Chris Saski,Monica Munoz
Organization CUGI-Clemson University Genomics Institute (USA)
4.2 Preparazione sonda
Gli oligonucleotidi SONDA 1 F e SONDA 3 R sono stati utilizzati per l’amplificazione di una regione comprendente il CDS e il 3’UTR del gene
HaL1L per la preparazione di una sonda (448 bp) che comprende il
dominio conservato (Fig. 4.1). La sonda è stata costruita in modo tale da riconoscere i membri della famiglia delle HAP3 di tipo LEC da utilizzare nelle ibridazioni per lo screening della libreria.
Dal clone HaK-3, conservato a –80 °C, con ansa sterile è stata raccolta un’aliquota di cellule che è stata inoculata in una provetta da 1,5 ml contenente 1 ml di substrato LB ed ampicillina. La coltura è stata lasciata crescere su un agitatore per 14-16 ore a 37 °C. Una diluizione opportuna della crescita è stata utilizzata come stampo per le reazioni di PCR.
HaL1L H.annuus ----CTAGAGAGAGACAATTCCCTCCCTTAGAGAGAGAAACATATGGAACGTGGAGGAGG 56 DH0ALL10ZA09ZZM1 GGTTCTAGAGAGAGACAATTCCCTCCCTTAGAGAGAGAAACATATGGAACGTGGAGGAGG 60 QHE14C23.yg.ab1 --- DH0AG18ZF09RM1 --- TC15033 --- BQ911236 --- CD851495 ---
HaL1L H.annuus TTTCCATGGCTACCACAGGCTCCCCATCCACCCTACATCTGGGATGCAACAGCCGGATAT 116
DH0ALL10ZA09ZZM1 TTTCCATGGCTACCACAGGCTCCCCATCCACCCTACATCTGGGATGCAACAGCCGGATAT 120 QHE14C23.yg.ab1 --- DH0AG18ZF09RM1 --- TC15033 --- BQ911236 --- CD851495 ---
HaL1L H.annuus GAAGCAGAAGCAACCAGACACAACCAACACCACATCTACAGAAGACAATGAGTGCATTGT 176
DH0ALL10ZA09ZZM1 GAAGCAGAAGCAACCAGACACAACCAACACCACATCTACAGAAGACAATGAGTGCATTGT 180 QHE14C23.yg.ab1 --- DH0AG18ZF09RM1 --- TC15033 ---GTCGTCAAATGT 12 BQ911236 ---GTCGTCAAATGT 12 CD851495 ---T 1
HaL1L H.annuus TAGAGAGCAAGACCGCTTTATGCCGATAGCAAACGTGATCAGGGTCATGCGAAAGATCTT 236
DH0ALL10ZA09ZZM1 TAGAGAGCAAGACCGCTTTATGCCGATAGCAAACGTGATCAGGATCATGCGAAAGATCTT 240 QHE14C23.yg.ab1 --- DH0AG18ZF09RM1 ---ACTTCCCCAAATAAACCTTCAACGGCTCCAC---ATACTCTTC 40 TC15033 TCGAGAACAGGATCGGTTTTTGCCGATTGCGAATATCAGTCGGATTATGAAGAAAGGGTT 72 BQ911236 TCGAGAACAGGATCGGTTTTTGCCGATTGCGAATATCAGTCGGATTATGAAGAAAGGGTT 72 CD851495 TCGAGAACAGGATCGGTTTTTGCCGATTGCGAATATCAGTCGGATTATGAAGAAAGGGTT 61
HaL1L H.annuus GCCACCTCACGCGAAGATCTCTGACGACGCAAAAGAAACGATCCAAGAATGTGTTTCCGA 296
DH0ALL10ZA09ZZM1 GCCACCTCACGCGAAGATCTCTGACGACGCAAAAGAAACGATCCAAGAATGTGTTTCCGA 300 QHE14C23.yg.ab1 ---AAGATTGCCAAGGATGCCAAGGATACGGTGCAGGAATGCGTTTCTGA 47 DH0AG18ZF09RM1 AAACCCTAACGTCGTCATCGCCCACAGCA-AATCGTCACCGTT---GATCGT-CTTCCGC 95 TC15033 GCCGGCTAATGGCAAAATGTCAAAGGATGCAAAGGAGACTGTTCAGGAGTGTGTTTCTGA 132 BQ911236 GCCGGCTAATGGCAAAATGTCAAAGGATGCAAAGGAGACTGTTCAGGAGTGTGTTTCTGA 132 CD851495 TCCGGCTAATGGCAAAATGTCAAAGGATGCAAAGGAGACTGTTCAGGAGTGTGTTTCTGA 121 ** * * * * ** * ** * *** *
HaL1L H.annuus GTACATTAGTTTTGTGACAGGTGAAGCGAATGACCGTTGCCAACGCGAGCAGAGAAAGAC 356
DH0ALL10ZA09ZZM1 GTACATTAGTTTTGTGACAGGTGAAGCGAATGACCGTTGCCAACGCGAGCAGAGAAAGAC 360 QHE14C23.yg.ab1 GTTTATCAGTTTCATCACTAGCGAGGCTAGTGACAAATGCCAGAAAGAGAAAAGGAAGAC 107 DH0AG18ZF09RM1 TTCTCTC---TCTGACACTTGTCCGATGCCTCTCCGGTGATGAAGCTTATGAACTCCGAC 152 TC15033 GTTTATCAGCTTTGTCACTAGTGAGGCGAGTGATAAGTGTCAAAGGGAAAAGAGGAAAAC 192 BQ911236 GTTTATCAGCTTTGTCACTAGTGAGGCGAGTGATAAGTGTCAAAGGGAAAAGAGGAAAAC 192 CD851495 GTTTATCAGCTTTGTCACTAGTGAGGCGAGTGATAAGTGTCAAAGGGAAAAGAGGAAAAC 181 * * * ** * * ** * **
HaL1L H.annuus TATCACAGCTG-AAGATGTGTTGTGGG-CTATGAGCAAACTCGGGTTTGATGACTATATC 414 DH0ALL10ZA09ZZM1 TATCACAGCTG-AAGATGTGTTGTGGG-CTATGAGCAAACTCGGGTTTGATGACTATATC 418 QHE14C23.yg.ab1 GATCAACGGTG-ATGACTTGTTGTGGG-CGATGGCTACTTTAGGCTTCGAAGATTACATT 165 DH0AG18ZF09RM1 ACACACTCCTGGACCGTCTCCTTCGCCTCCTTCGATATCTTCGCGTTCGCCGGTAACGCC 212 TC15033 TATCAACGGTG-ATGATCTTCTATGGG-CAATGGCTACTTTAGGGTTTGAAGATTATATC 250 BQ911236 TATCAACGGTG-ATGATCTTCTATGGG-CAATGGCTACTTTAGGGTTTGAAGATTATATC 250 CD851495 TATCAACGGTG-ATGATCTTCTATGGG-CAATGGCTACTTTAGGGTTTGAAGATTATATC 239 ** ** * * * * * * * * * ** * * *
HaL1L H.annuus GAACCGTTGACTGTGTATCTTCATCGCTACAGGGAGTTTGATGGGGGTGAGCGTGGGTCG 474
DH0ALL10ZA09ZZM1 GAACCGTTGACTGTGTATCTTCATCGCTACAGGGAGTTTGATGGGGGTGAGCGTGGGTCG 478 QHE14C23.yg.ab1 GACCCACTCAAAGCTTACCTATCTAGGTACAGAGAGTTAGAGGGTGATAGTAAGGGGTCG 225 DH0AG18ZF09RM1 TTTTTCATTA-TCCGGCTCACGTTAGCTATCGGCAGG-AACCTGTCCTGCTCTCGAGCCG 270 TC15033 GAACCACTCAAGTTATACTTGACTAGATACAGAGAGATGGAGGGTGACACCAAGGGATCC 310 BQ911236 GAACCACTCAAGTTATACTTGACTAGATACAGAGAGATGGAGGGTGACACCAAGGGATCT 310 CD851495 GAACCACTCAAGTTATACTTGACTAGATACAGAGAGATGGAGGGTGACACCAAGGGATCC 299 * * * * ** * ** * * *
HaL1L H.annuus ATAAGGGGTG---AGCCGCTTGTCAAGAGG---GCTGCGGCTACTGCTGACCCTGGTCCG 528
DH0ALL10ZA09ZZM1 ATAAGGGGTG---AGCCGCTTGTCAAGAGG---GCTGCGGCTACTGCTGACCCTGGTCCG 532 QHE14C23.yg.ab1 GGTAGGGGTGGAGATGGATCTGGCAAGGGG---GAGCTGGGAGCATCATCTTGCTGTTCA 282 DH0AG18ZF09RM1 ATAAATCGTTC-TGTCCGCCGGACTCGTTGTCCGAATCCGCCATCGCTGATCGGAGAATT 329 TC15033 GCAAAAGGCCCACGAAGGACTGGCTAGGAG---AGACGGGGGGCAACCTGATCATAA-CG 366 BQ911236 GCAAAAGGCCCACTAATGACTGGCTAGGAG---AGACGGGGGGCAACCTGATCATAA-CG 366 CD851495 GCAAAAGGCC-ACGAAGGACTGGCTAGGAG---AGACGGGGGGCAACCTGATCATAA-CG 354 * * * * * * * *
HaL1L H.annuus TTTGGTATGGGTCCGTTTGTGCCTGGTTCTCACATGGGTCATCATAA-TGGGTTCTTTGG 587
DH0ALL10ZA09ZZM1 TTTGGTATGGGTCCGTTTGTGCCTGGTTTTCACATGGGTCATCATAA-TGGGTTCTTTGG 591 QHE14C23.yg.ab1 CTTATGCAAG--CCAAACACTCGTCGAACTTTCAAGCTTTGTGTTTACTGATTCCGTTGA 340 DH0AG18ZF09RM1 CAAGATCTGAA-GGATTAACGGCGGAGATTTGTGGGGACTGGTTTGGTTACAGCTTTTCA 388 TC15033 CACAT-CTTG--CTCATCAAGGTTCGTACTTGCAGGGTCTGGATTACGGGAGCTCACAGG 423 BQ911236 CACAT-CTTG--CTCATCAACGCTCCTACTTGCA--- 397 CD851495 CACAT-CTTG--CTCATCAAGGTTCGTACTTGCAGGGTCTGGATTACGGGAGCTCACAGG 411 *
HaL1L H.annuus TCCTGCTAGCATTGCTGGTTTCTTTAAGGACCCGTCGAGTGCGGCTGGCCAGTCTGGGCC 647
DH0ALL10ZA09ZZM1 TCCTGCTAGCATTGCTGGTTTCTTTAAGGACCCGTCGAGTGCGGCTGGCCAGTCTGGGCC 651 QHE14C23.yg.ab1 TTTCAATTCTGCGGTCAGCATAT--AATGTTTAAAC-ATGATTTATAACT--TCTATACC 395 DH0AG18ZF09RM1 TTTGGG--- 394 TC15033 GTCAA--CATGTGGTTCCAATGC--AAGGCAGAGATTAACATTAATGGGT--TCAAGATC 477 BQ911236 --- CD851495 GTCAA--CATGTGGTTCCAATGC--AAGGCAGAGATTAACATTAATGGGT--TCAAGATC 465
HaL1L H.annuus TGCTGGTTTCGAGCCGTA-TGCTCAGTGTAAAGACTAACTGGTTTCGATT----TTCGAG 702 DH0ALL10ZA09ZZM1 TGCTGGTTTCGAGCCGTA-TGCTCAGTGTAAAGACTAACTGGTTTCGATT----TTCGAG 706 QHE14C23.yg.ab1 GTATAGTTTTTATATGCAGTTTAAAATATATGTATTAA-ATATTTTGTTCATCCCCCCAA 454 DH0AG18ZF09RM1 --- TC15033 TGTTACGATTTAAAGCTAAAACGCAGCCGTCACATCAACAGGTCTTGTTTTAAACTCGAG 537 BQ911236 --- CD851495 TGTTACGATTTAAAGCTAAAACGCAGCCGTCACATCAACAGGTCTTGTTTTAAACTCGAG 525
HaL1L H.annuus CCGTGTGTTGGTTGGTTTGTTTTTTTTTCT-TGTTTGTTTGTC--TTCTTGGTCTTGG-A 758
DH0ALL10ZA09ZZM1 CCGTGTGTTGGTTGGGTTGTTTTTTTTTTTCTGTTTGGTNGNT--TCCTTGGCCCTGGGA 764 QHE14C23.yg.ab1 CATGTTTTTTCTTGGGGCATGAAAAAGTTTGTGTTTGTTTTCTGTTTGCTTCTCTTTTGT 514 DH0AG18ZF09RM1 --- TC15033 CATCTACT--- 545 BQ911236 --- CD851495 CATCTACT--- 533
HaL1L H.annuus GCAACAAATTACATTGGGTCAAACTTTGGCAAGG---CATGTAAAAACTGACACTCTC 813
DH0ALL10ZA09ZZM1 GCAACAAAATATATTGGGCCAAACTTTGGCCGGGGAGTGCNCAAAAAAAACAAAACTAAC 824 QHE14C23.yg.ab1 TAAGTTACTATCGTTGTATTGTATCTCTGCTTG--- DH0AG18ZF09RM1 --- TC15033 --- BQ911236 --- CD851495 ---
HaL1L H.annuus -CATCTAC--- 820
DH0ALL10ZA09ZZM1 ATATTTTCCTGCCGGCGGGCTTTGCCCGGTTGGCTCT 861 QHE14C23.yg.ab1 --- DH0AG18ZF09RM1 --- TC15033 --- BQ911236 --- CD851495 ---
Fig. 4.1: Allineamento di sequenza del gene HaL1L in H. annuus e delle EST scelte in banca dati per selezionare gli oligonucleotidi da utilizzare nello screening dei cloni della libreria genomica.
ATG codone d’inizio, TAA codone di stop di HaL1L. In giallo è evidenziato il domino. Primer utilizzati per lo screening della libreria genomica: in rosa TCF-TCR, in celeste AGIF-AGIR, in verde DHOF-DHOR, in blu QUEF-QUER.
In rosso i primer SON1F e SON3R utilizzati per la sonda.
- Marcatura della sonda
Il kit Ladderman TM Labeling Kit (Takara BIO INC., Japan) è stato utilizzato
per ottenere una sonda con elevata specificità richiesta per l’identificazione di specifiche sequenze di DNA in esperimenti di ibridazione. In una provetta si aggiungono i seguenti componenti:
DNA 1 µg Primer random 2 µl
H2O sterile o buffer TE x µl per un volume finale di 14 µl
Si riscalda a 95 °C per 3 minuti e poi subito in ghiaccio per altri 5 minuti. A questa miscela si aggiungono 2,5 µl di buffer 10X, 2,5 µl della miscela di dNTP, 5 µl di [α- 32P] dCTP (1.85 MBq, 50 µCi) e 1 µl di Bca DNA polimerasi
e il tutto viene incubato a 50 °C per 10 minuti. L’enzima è poi inattivato aggiungendo EDTA fino ad una concentrazione finale di 30 mM e la miscela è incubata a 95 °C per 3 minuti e quindi posta in ghiaccio.
- Pre-ibridazione e Ibridazione dei filtri
Per l’ibridazione dei filtri è stato utilizzato l’ULTRAhyb Ultrasensitive
Hybridization Buffer. Si effettua una pre-ibridazione a 42 °C per 30 minuti. La sonda marcata è diluita in un volume opportuno di 10 mM EDTA fino ad un volume finale di 50 µl e incubata a 90 °C per 10 minuti. La sonda è
quindi centrifugata brevemente, diluita in 0,5 ml di ULTRAhyb e trasferita nel tubo di pre-ibridazione. Non è necessario sostituire l’ULTRAhyb usato per la pre-ibridazione. Viene quindi eseguita una ibridazione overnight sempre a 42 °C.
- Lavaggi ed esposizione dei filtri
Il passaggio successivo prevede una serie di lavaggi dei filtri. Il primo lavaggio a bassa astringenza viene fatto con SSC 2X e SDS 0,1% per 10 minuti a temperatura ambiente. Questo primo lavaggio ha lo scopo di rimuovere il buffer in eccesso e la sonda non ibridata. Il secondo lavaggio ad alta astringenza viene fatto con SSC 0,5% o 0,1% e SDS 0,1%, 2 x 15 minuti a 37 °C. Tutti i lavaggi vengono fatti su agitatore.
Dopo i lavaggi i filtri sono avvolti nella pellicola evitando accuratamente che si asciughino e analizzati mediante Typhoon Variable Mode Image ( Perkin Elmer). Essi sono posizionati su opportuni schermi che sotto stimolazione indotta da laser emettono un segnale luminoso proporzionale alla quantità di radioattività. Tale segnale è raccolto e convertito in segnale elettrico che è digitalizzato e trasformato in immagine la quale può a sua volta essere analizzata mediante software forniti dalla casa produttrice l’apparecchio. Le informazioni ottenute dall’ibridazione hanno permesso l’identificazione di 20 cloni potenzialmente contenenti frammenti di DNA genomico relativo a HaL1L o altri componenti della famiglia (Fig.4.2).
a)
b)
Fig. 4.2: Filtro di ibridazione. Il filtro b) è lo stesso filtro a) a cui è stato sottratto il background. Nei cerchi sono evidenziati due segnali di ibridazione che corrispondono ad altrettanti cloni della libreria.
4.3 PCR per verifica dei cloni scelti della libreria
Lo screening della libreria genomica è stato effettuato mediante PCR utilizzando oligonucleotidi scelti basandosi sulla sequenza del gene
utilizzando i software Blast, Fasta e ClustaW (Tab. 4.1). Tra i 20 cloni analizzati mediante PCR è stato scelto il clone 135I01 che è stato poi sequenziato automaticamente dalla MWG (Ebersberg, Germany). Un volta ottenuta, la sequenza del BAC è stata analizzata con i seguenti programmi:
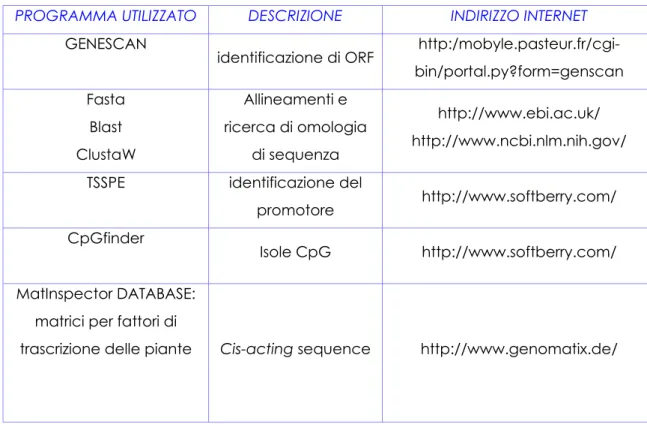
Tab. 4.2: Software utilizzati per analizzare la sequenza del BAC.
PROGRAMMA UTILIZZATO DESCRIZIONE INDIRIZZO INTERNET
GENESCAN
identificazione di ORF http:/mobyle.pasteur.fr/cgi-bin/portal.py?form=genscan Fasta Blast ClustaW Allineamenti e ricerca di omologia di sequenza http://www.ebi.ac.uk/ http://www.ncbi.nlm.nih.gov/ TSSPE identificazione del
promotore http://www.softberry.com/ CpGfinder
Isole CpG http://www.softberry.com/ MatInspector DATABASE:
matrici per fattori di
trascrizione delle piante Cis-acting sequence http://www.genomatix.de/
4.4 Estrazione dell’RNA
L’estrazione degli RNA è stata condotta, in accordo con il protocollo del “TriPure Isolation Reagent Kit” (Roche), sui diversi tessuti ed organi della linea pura HOR di girasole (Helianthus annuus L.). Le piante erano allevate in cella climatica a 23 ± 1 °C utilizzando un fotoperiodo 16/8 (luce/buio) ed una intensità luminosa di 200 µmol m-2 s-1. I campioni
utilizzati sono stati:
1. embrioni zigotici a diversi stadi di sviluppo (5, 10, 21, 28 giorni dalla data di impollinazione;
2. ovuli prelevati a 1-2 giorni dall’impollinazione;
3. ovari di fiori del disco prelevati a 1-2 giorni dall’impollinazione; 4. foglie cotiledonari di semi germinati da 10 giorni;
5. foglie giovani (2 cm di lunghezza);
6. foglie espanse di H. annuus (15 cm di lunghezza):
7. internodi di fusto (5°-8° nodo) di piante allo stadio di bottone fiorale
8. radici di piante allo stadio di bottone fiorale;
9. germogli vegetativi prelevati da piante di 21 giorni; 10. germogli fiorali prelevati da piante di 35-40 giorni;
I campioni prelevati sono stati immediatamente congelati in azoto liquido, quindi 300 mg di materiale sono stati polverizzati in mortaio mantenendoli sempre ghiacciati con azoto liquido. La polvere ottenuta è stata trasferita in una provetta da 2 ml ed incubata per 5 minuti a temperatura ambiente dopo l’aggiunta di 1 ml di Trizol. Successivamente, sono stati addizionati 200 µl di cloroformio e la provetta è stata agitata vigorosamente per 15 secondi, poi è stata lasciata a temperatura ambiente per 5 minuti ed infine centrifugata a 9000 rpm per 25 minuti in rotore Kroton A 8.24 a 4 °C. La fase superiore acquosa è stata recuperata e trasferita in un’altra provetta, dove sono stati aggiunti 500 µl d’isopropanolo ed il tutto è stato mescolato delicatamente. Per poter favorire la precipitazione del RNA la soluzione è stata incubata a temperatura ambiente per 10 minuti, quindi centrifugata a 9.000 rpm in rotore Kroton A 8.24 per 40 minuti a 4 °C. Il surnatante è stato scartato, mentre il precipitato formatosi è stato sottoposto a due lavaggi successivi per eliminare eventuali residui di cloroformio con 1 ml d’etanolo al 75%, centrifugando a 9.000 rpm in rotore Kroton A 8.24 per 20 minuti a 4 °C. Il surnatante è stato scartato e l’RNA precipitato è stato fatto asciugare all’aria per eliminare i residui d’etanolo. L’RNA precipitato è stato poi sciolto in acqua trattata con DEPC e conservato a –80 °C.
4.5 Corsa elettroforetica su gel di agarosio in condizioni
denaturanti
Il gel è stato preparato sciogliendo l’agarosio in H2O DEPC con aggiunta
di formaldeide e MOPS 10X preriscaldati a 65 °C. La miscela è stata versata sul supporto e lasciata polimerizzare. Una volta polimerizzato il gel è stato posto nel tampone di corsa (MOPS 1X) ed è stata effettuata una precorsa di 10 minuti a 60 mA. I campioni sono stati preparati aggiungendo 3 volumi di soluzione denaturante, incubandoli a 65 °C per 15 minuti. I campioni, così denaturati, sono stati tenuti in ghiaccio per 5 minuti e poi è stato aggiunto il tampone di caricamento in quantità pari a 1/5 del volume finale. Caricati i campioni, è stata effettuata una corsa per circa 2 ore a 50 mA , per poter verificare l’integrità dell’RNA estratto.
Soluzioni utilizzate :
GEL DI AGAROSIO 1% (70 ml): SOLUZIONE DENATURANTE:
Agarosio 0,7 g H2O DEPC 52,5 ml Formaldeide 37% 10,5 ml MOPS 10X 7 ml MOPS 10X 50 ml Formaldeide 37% 75 ml Formammide deionizzata 250 ml
MOPS 10X: TAMPONE DI CARICAMENTO:
Saccarosio 30%
Blu di bromofenolo q.b. 10mM Na2 EDTA
0,2 M MOPS pH 7.0
4.6 RT-PCR semiquantitativa
L’espressione del gene HaL1L è stata valutata mediante RT-PCR semiquantitativa, una tecnica che consente la co-amplificazione, nella stessa reazione di PCR, del cDNA di interesse e di un controllo interno (il trascritto di un gene costitutivamente espresso) utilizzando due coppie di oligonucleotidi specifici con temperatura di “annealing” analoghe. Questa tecnica è idonea ad evidenziare la presenza di trascritti rari o poco abbondanti come nel caso dei fattori di trascrizione. I frammenti da amplificare del controllo interno e del cDNA d’interesse devono essere compatibili tra loro, ovvero non devono formarsi prodotti ibridi dovuti al legame tra i due amplificati, e nella co-amplificazione, non devono essere prodotte bande aggiuntive rispetto a quelle attese. Affinché i dati ottenuti dalla RT-PCR semiquantitativa siano significativi, la reazione di PCR deve essere terminata quando i prodotti, sia del controllo interno che del cDNA di interesse, risultino evidenziabili e siano ancora all’interno della fase esponenziale.
4.7 Scelta degli oligonucleotidi per la β-actina
L’actina è una proteina espressa costitutivamente nelle piante e l’mRNA ad essa relativo costituisce lo standard interno con cui sono stati confrontati gli mRNA per l’HaL1L. Sulla base dell’allineamento di sequenze di specie vegetali vicine ad H. annuus disponibili in rete, sono stati scelti oligonucleotidi (ACT3 e ACT5) per l’amplificazione di un frammento del cDNA della β-actina della lunghezza di 243 bp distinguibile, ma paragonabile, al frammento, di 175 bp, ottenuto con i primer CHI-REL e CHI-TCDNA, per i cDNA di HaL1L (Tab. 4.1).
- Retro trascrizione
Per la retrotrascrizione degli RNA è stato seguito il protocollo del Super-script-Kit TM (Invitrogen).
La miscela per la retrotrascrizione è costituita da: • X µl RNA (1 µg)
• 1 µl oligonucleotidi per RT (500 µg/ml) • 1 µl dNTP (10 mM)
• H2O DEPC q.b. fino a 12 µl
La miscela è stata incubata per 5 minuti a 65 °C, poi è stata messa immediatamente in ghiaccio per altri 5 minuti. Successivamente, dopo aver brevemente centrifugato, alla miscela sono stati aggiunti, nell’ordine:
• 4 µl First Strand Buffer (5X) • 2 µl DTT (0,1M)
• 1 µl Inibitore delle Ribonucleasi
La miscela ottenuta è stata incubata a 42 °C per 2 minuti, quindi è stato aggiunto 1 µl di Superscript II RT (200 U/µl) ed il tutto è stato rimesso ad incubare per altri 50 minuti a 42 °C. Infine, la reazione è bloccata ponendo la provetta a 70 °C per 15 minuti.
-
Amplificazione del cDNA mediante PCR
La miscela di reazione della retrotrascrizione, contenente il cDNA sia per la β-actina che per l’HaL1L, è stata utilizzata come stampo nella reazione di amplificazione con il seguente protocollo:
Reagenti a concentrazione iniziale Concentrazione finale
MRT (Miscela di Reazione della
Retrotrascrizione) 0,5 µl MgCl2 (25mM) 4 µl Buffer 10X 1,5 µl dNTP (10mM) 0,2 µl Primer β-actina 0,1 µM Primer HaL1L 0,1 µM TaqPolimerasi (5U/µl) 0,15 µl H2O q.b. fino a 15 µl
L’amplificazione è stata condotta mediante PCR, utilizzando oligonucleotidi che non presentano regioni di complementarietà, in modo da evitare artefatti. In queste analisi è di fondamentale importanza conoscere il numero di cicli di PCR da usare in modo da ottenere amplificati che hanno raggiunto la saturazione. Ogni PCR, infatti, raggiunge il punto di saturazione quando l’aumento del prodotto di amplificazione non è più esponenziale, ovvero non è più proporzionale alla quantità di stampo iniziale poiché sono utilizzati come stampo anche i prodotti amplificati nei cicli precedenti. Per conoscere il punto di saturazione è stata condotta una PCR dalla quale, al termine di cicli successivi, sono state sottratte aliquote di campione e immediatamente trasferite in una adiacente macchina per PCR per subire la fase di estensione finale di 7 minuti a 72 °C. I prodotti di
amplificazione, ottenuti nell’intervallo compreso tra 22 e 32 cicli, sono stati analizzati su gel di Agarosio ed è stato deciso che il numero di cicli da utilizzare è compreso tra i 26 e i 30.
CICLI UTILIZZATI:
temperatura (°C) 95° 94° 53° 72° 72° tempo 3’ 30’’ 30’’ 25’’ 7’
numero cicli 26-30
- Corsa elettroforetica su gel di Agarosio
I prodotti amplificati mediante PCR sono stati sottoposti a corsa elettroforetica su gel di Agarosio. A seconda della banda attesa, corrispondente all’ampiezza dell’amplificato, è scelta la concentrazione del gel di Agarosio, variabile tra l’1% e il 2%. L’Agarosio è stato disciolto in un tampone Tris-acetato (TAE) 1X con 0,5 µg/ml di bromuro di etidio. Prima di effettuare la corsa elettroforetica ai campioni sono stati aggiunti 0,2 volumi di tampone di caricamento .
Soluzioni utilizzate :
TAMPONE DI CARICAMENTO TAMPONE TRIS-ACETATO (TAE) 10X
Saccarosio 60% p/v Orange G 0,1% p/v 25 mM Na2EDTA pH 8
Numero di cicli 22 24 26 28 30 32 M bp
Fig. 4.3: Visualizzazione su gel di Agarosio al 2% dei prodotti di amplificazione di HaL1L e β-actina ottenuti nell’intervallo compreso tra 22 e 32 cicli.
La figura 4.3 mostra che a 26 cicli la PCR è ancora nella fase esponenziale, mentre a 30 cicli il segnale di amplificazione della β-actina è a saturazione, quindi il numero dei cicli da utilizzare nella RT-PCR relativa deve ricadere in questo intervallo.
Le quantità relative di ogni prodotto di amplificazione sono state quantificate mediante lo “scanning” diretto del gel di Agarosio 2% con il densitometro UVP Image Store 5000 (Ultra Violet Product Ltd,
Tris-acetato 0,4 M
0,01 mM Na2EDTA pH 8
portato a pH 7 con acido acetico glaciale
Oligonucleotidi HaL1L:
TC-DNA/CHI-REL
HaL1L (175 bp) → β-actina (243 bp)→ -1353 - 872 - 603 - 310 - 234 - 194Cambridge, England) equipaggiato con l’UVP GelBase-GelBlot TM Windows Software. Per normalizzare i campioni relativamente alla quantità di RNA totale utilizzato e alla efficienza della sintesi di cDNA, le intensità delle bande di amplificazione del frammento di HaL1L sono state rapportate ai prodotti di amplificazione della β-actina. Un valore percentuale arbitrario (100%) è stato assegnato in ogni campione al livello della β-actina. È stata effettuata l’analisi della varianza e le medie sono state separate mediante il test di Tukey (con P = 0,05).
4.8 CLONAGGIO
I prodotti ottenuti dall’amplificazione sono stati clonati seguendo il protocollo del TOPO-TA Cloning Kit (Invitrogen). La procedura qui descritta è utilizzata ogni volta sia necessario effettuare un clonaggio.
- LIGATION - Inserzione dell’amplificato nel DNA plasmidico
In una provetta da 0,5 ml è stata preparata la miscela di legame : • 0,5 – 4 µl Prodotto di PCR
• 1 µl Salt Solution • 1µl TOPO vector • H2O q.b. fino a 6 µl
La miscela ottenuta è stata incubata a temperatura ambiente (intorno ai 22-23 °C), per un tempo variabile che va dai 30 secondi ai 30 minuti e trasferita successivamente in ghiaccio.
- Trasformazione delle cellule
Sono stati aggiunti 2 µl della miscela di legame in una provetta contenente una coltura di cellule di "One Shot Chemically Competent E.
coli”, rese chimicamente competenti ad accogliere il DNA plasmidico.
Per consentire la trasformazione, ovvero l’inserimento del plasmide nelle cellule batteriche, è necessario uno “shock” termico che è di seguito descritto: la provetta è stata incubata in ghiaccio per 30 minuti, quindi rapidamente trasferita a 42 °C per 30 secondi e subito dopo di nuovo in ghiaccio per altri 2 minuti. Successivamente sono stati aggiunti 250 µl di SOC (brodo di coltura) ed è stata fatta un’incubazione di un’ora a 37 °C in agitatore orizzontale. Quindi 100 µl della coltura di cellule trasformate sono stesi accuratamente su piastre di substrato solido di coltura (preriscaldate a 37 °C). Per permettere la crescita batterica le piastre sono state incubate a 37 °C o.n.. La selezione delle colonie viene fatta col metodo della distinzione blu-bianco: infatti, nel plasmide è presente l’operone Lac-Z che codifica per il polipeptide beta-galattosidasi; quest’enzima è in grado di scindere la molecola X-gal (5-bromo-4-cloro-3-indolil-beta-Dgalattopiranoside) in 5-bromo-4-cloroindolil e 5,5’-dibromo-4,4’-dicloroindingo che è di colore blu. Poiché durante il clonaggio, il frammento s’inserisce all’interno dell’operone, viene interrotta la sequenza per la beta-galattosidasi che non è più sintetizzata. Le cellule che contengono l’inserto non saranno in grado di scindere lo X-gal presente nel substrato e quindi saranno di colore bianco; mentre quelle prive dell’inserto metabolizzeranno lo X-gal colorandosi di blu. Le colonie bianche sono state sospese singolarmente in provette con LB (terreno liquido Luria Bertani) e ampicillina, e fatte crescere o.n. a 37 °C.
I cloni si conservano in glicerolo al 20% a – 80 °C.
- Selezione dei cloni
Durante l’amplificazione possono crearsi degli artefatti, clonati contemporaneamente agli amplificati specifici, non visualizzabili su gel di Agarosio. È necessario a questo punto individuare le colonie bianche contenenti effettivamente l’inserto d’interesse. Per tale scopo è
effettuata una PCR da colonie, in cui è utilizzata direttamente una aliquota della sospensione batterica come stampo. Nella miscela di amplificazione (volume finale 20 µl) devono essere impiegati non più di 2 µl di sospensione batterica. Le condizioni di PCR sono le stesse già descritte, è stato però prolungato a 10 minuti il tempo iniziale di denaturazione, prima della fase ciclica, per ottenere lisi delle cellule batteriche e liberazione del DNA plasmidico impiegato come stampo. Oltre ad utilizzare una miscela d’amplificazione contenente entrambi gli oligonucleotidi, sono state condotte separatamente anche amplificazioni con miscele contenenti singolarmente i due oligonucleotidi. Queste ultime servono ad evidenziare eventuali artefatti. Sono stati considerati positivi i cloni i cui amplificati sono ottenibili solo con la miscela di incubazione contenente entrambi gli oligonucleotidi, e che, visualizzati su gel di Agarosio, risultano essere delle dimensioni attese.
Soluzioni utilizzate
LB (LURIA BERTANI) liquido: Bactotriptone 10 g Yeast extract 5 g NaCl 5 g H2O 1 L Ampicillina:
10 mg/ml solubilizzata in alcool etilico al 70%. SUBSTRATO SOLIDO PER PIASTRE:
Bactotriptone 8 g Yeast extract 5 g NaCl 5 g Bacto-Agar 15 g H2O 1L IPTG: 100 mM solubilizzato in H2O
X-GAL:
40 mg/ml solubilizzato in dimetilformammide
4.9 Estrazione del DNA plasmidico
Il DNA plasmidico dei cloni selezionati è stato estratto in accordo con il protocollo “Wizard Plus SV Minipreps DNA Purification System”. Circa 20 µl di crescita dei cloni conservati a - 80 °C sono stati sospesi in 6 ml di substrato LB, contenente ampicillina, e incubati a 37 °C o.n.. Al termine dell’incubazione la crescita batterica è stata centrifugata a 10000 rpm per 5 minuti, in centrifuga da tavolo. Il surnatante è stato scartato e le cellule impacchettate sono state nuovamente sospese in 250 µl di tampone di sospensione contenente RNasi. Quindi sono stati aggiunti 250 µl di tampone di lisi e la miscela è stata incubata a temperatura ambiente per 5 minuti. Successivamente sono stati aggiunti 10 µl di Proteasi Alcalina e la miscela incubata per non più di 5 minuti. Successivamente sono stati aggiunti 350 µl di soluzione neutralizzante ed al fine di eliminare la fase precipitata, è stata effettuata una centrifugazione a 14000 rpm per 10 minuti; il surnatante, contenente il DNA plasmidico, è stato recuperato e messo in una nuova provetta munita di filtro. E’ stata condotta una centrifugazione a 14000 rpm per 1 minuto e la soluzione che attraversa il filtro è stata scartata. Il filtro è stato poi lavato due volte, la prima volta con 750 µl di tampone di lavaggio centrifugando a 14000 rpm per 1 minuto e successivamente con 250 µl di tampone di lavaggio centrifugando sempre a 14000 rpm per 2 minuti. Un’ulteriore centrifugazione nelle stesse condizioni è stata eseguita “a secco” per eliminare i residui del tampone di lavaggio. Il DNA, presente nel filtro, è stato recuperato aggiungendo 50 µl di H2O deionizzata e
4.10 Sequenziamento del DNA plasmidico
I plasmidi contenenti gli inserti sono stati sequenziati in entrambe le direzioni (usando gli oligonucleotidi M13R e M13F) con un sequenziatore automatico da parte della ditta MWG (Ebersberg, Germany). Il DNA da sequenziare deve essere inviato in provette da 1,5 ml e ne sono sufficienti 1,5 µg.
4.11 Isolamento gene WUSCHEL di H. annuus
Con le tecniche 5’3’ RACE e RAGE è stato possibile isolare l’intera sequenza del gene WUSCHEL di girasole Per questo scopo sono state allineate sequenze nucleotidiche di specie vegetali diverse (Fig. 4.4a; Fig. 4.4b) che non risultano molto conservate tranne che nella regione del dominio. Difficoltà sono state incontrate per individuare oligonucleotidi da utilizzare nelle reazioni di amplificazione poiché anche il motivo amminoacidico TLPLFP condiviso solo con il peptide WOX2 è poco conservato a livello dei residui nucleotidici.
A. thaliana MEPPQHQHHHHQADQESG---NNNNNKSGSGGYTCRQTSTRWTPTTE 44 L. sativa MET---QQQEQDLG---NKNNTS---YLCRQSSTRWTPTSD 32 L. esculentum ME---HQHN---IEDGG-KNSNNSFLCRQSSSRWTPTSD 32 P. hybrida METAQHQQNNQQHYLHQHLSIGQGTNIEDGS---NKNNSSN----FMCRQNSTRWTPTTD 53 P. trichocarpa MEP--HQQQPN---EDNN-GGAKGNFLCRQTSTRWNPTTD 34 A. majus MEP---QQQQQQQGNEQQDSQ---GIGKINN-GSGGSSFLCRQSSTRWTPTTD 46 ** * ***.*:**.**:: A. thaliana QIKILKELYYNNAIRSPTADQIQKITARLRQFGKIEGKNVFYWFQNHKARERQKKRFNGT 104 L. sativa QIRILKELYYNNGIRSPTADQIQRIAARLRHYGKIEGKNVFYWFQNHKARERQKKRFTPV 92 L. esculentum QIRILKDLYYNNGVRSPTAEQIQRISAKLRQYGKIEGKNVFYWFQNHKARERQKKRLIAA 92 P. hybrida QIRILKDLYYNNGVRSPTAEQIQRISAKLRQYGKIEGKNVFYWFQNHKARERQKKRLIAA 113 P. trichocarpa QIRILKELYYIKGVRSPNGAEIQQISARLRKYGKIEGKNVFYWFQNHKARERQKKRLTNE 94 A. majus QIRILKDLYYNNGVRSPTAEQIQRISAKLRQYGKIEGKNVFYWFQNHKARERQKKRFTAD 106 **:***:*** :.:***.. :**:*:*:**::************************: A. thaliana NMTTPSSSPN---SVMMAANDHYHPLLH--HHHGVPMQRPANSVNVKLNQDHHLYHHN 157 L. sativa PVTPPPPPPPPPPPPSTAILLPSPFSDHRH--HHH--HMHVNSHHPPQFYSQQHKLYTTH 148 L. esculentum ASATDNNNIS---SMQMIPHLWRSPDD---HHKYNTATTNPGVQCPSPSSHGVLPVV 143 P. hybrida ATTDNTNLPM---QMQFQRGVWRSSADDPIHHKY----TNPGVHCPSASSHGVLAVG 163 P. trichocarpa VPMQ---QRTAWK-PED-YYSYKY----SNRFSSASSSANTGVVTVG 132 A. majus HHHHMN---VPTIHNHHYKPPPV---YNKFSN-MNSGSFPSSSNGSPGFLTTP 152 : . . A. thaliana KPYPSFNNGNLNHASSGTECGVVNASNGYMSSHVYGSMEQDCSMNYNN---VGG-GWANM 213 L. sativa QISPSEGG---SSS---TGFMPVGYGYGSVAMEKSFK-ECSISQGESRVVGG-TSQNF 198 L. esculentum ---QTGNYGYGTLAM-EKSFRECSISPPGGS---YHQNL 175 P. hybrida ---QNGNHGYGALAM-EKSFRDCSISPGSSMSHH--HHQNF 198 P. trichocarpa ---QTDSHGYGSVTMQEKNSWDCSAPAGGSNGAGSGSMSNI 170 A. majus GS---HVGNYGYGSVAM-EKSFRECTISSTTDANVGGSMSQNI 191 . ** : : . :*: *:
A. thaliana DHHYSSAPYNF----FDRAKPLFGLEGHQEEEECGGDA---YLEHRR 253 L. sativa GSWVGSDSYS---YEKIKPEY--EAPEEDDDRGGE---LSAQME 234 L. esculentum TW-VGVDPYNNMSTTSPATYPFLEKSNNKHYE-ETLD--EEQEEEN---YQRGNSALE 226 P. hybrida AW-AGVDPYS---STTTYPFLEKT--KHFENETLEADEEQQEEDQENYYYQRTTSAIE 250 P. trichocarpa NYGSGVDINS---HSSSYAVFGQE---QEAAAKIE 199 A. majus AW-IGINNEY---HNPYTFIDTRKYMNGYDQTLEIEEEAE---ENYTAEIE 235 . . . A. thaliana TLPLFPMHGEDHINGGSGAIWKY---GQSEVRPCASLELRLN--- 292 L. sativa TLPLFPVNGGATMGSSHHDIIN--- 256 L. esculentum TLSLFPMHEENIISNFCIKHHE---SSGG----WYHSDNNNLAA---LELTLNSFP-- 272 P. hybrida TLPLFPMHEENISSFCNLKHQE---SSGGFYTEWYRADDNLAAARASLELSLNSFIGN 305 P. trichocarpa TLPLFPMLGEDISSSFNINNINPDFYYSSGCG----YGDYGNDTSSRTSLDLSLYSYNGQ 255 A. majus TLPLFPMHADIKQDTADY---FNGRLENGCPRASLELTLNSWFGN 277 **.***: . A. thaliana ---- L. sativa ---- L. esculentum ---- P. hybrida SS-- 307 P. trichocarpa PQDY 259 A. majus SKYN 281
Fig. 4.4a: Allineamento di sequenza aminoacidica del gene WUSCHEL (WUS) in diverse specie vegetali per la scelta di oligonucleotidi.
In giallo è stato evidenziato il motivo TLPLFLP, specifico per il peptide WUS e conservato nelle specie vegetali prese in esame(riquadro blu).
A. thaliana TGGTATCGACGTACGCTTCCTCTCTTCCCTATGCACGGTGAAGATCACATCAA L. sativa GCCCAGATGGAGACTCTTCCTCTCTTTCCAGTCAACGGTGGCGCCACCATGGG L. esculentum TCTGCTTTAGAAACTCTGTCACTTTTCCCCATGCATGAAGAGAACATCATCTC P. hybrida TCTGCAATAGAAACACTTCCACTTTTTCCAATGCATGAAGAGAACATTTCCAG P. trichocarpa CCAAAGATTGAGACTCTTCCTCTATTTCCAATGCTTGGTGAGGACATCAGTAG A. majus GCTGAGATTGAAACTCTTCCCTTATTTCCTATGCACGCAGATATAAAACAAGA
Fig. 4.4b: Allineamento delle sequenze nucleotidiche per geni WUSCHEL di specie diverse. In giallo la regione del motivo aminoacidico TLPLFLP. Codici di accesso: AJ012310 Arabidopsis thaliana, AM234747 Populus trichocarpa, AF481951 Petunia
hybrida, AJ538329 Lycopersicon esculentum (Solanum lycopersicum L.), AY162209 Antirrhinum majus, CLSX6179 Lactuca sativa.
4.12 5’ RAGE
La tecnica della 5’ RAGE è stata utilizzata nel Genome Walking per ottenere la sequenza genica dell’estremità 5’ del gene WUSCHEL. A questo scopo è stato utilizzato il kit “GenomeWalkerTM Universal Kit” (Clontech).
- Digestione
Il DNA genomico viene digerito con 4 diversi enzimi di restrizioni in dotazione del Kit, ovvero Dra I, EcoR V, Pvu II e Stu I. Con la digestione è stato possibile costruire 4 librerie genomiche.
Per ogni reazione sono state utilizzate differenti provette da 1,5 ml contenenti:
25 µl di DNA genomico (0,1 µg/µl) 8 µl di enzima di restrizione (10 U/µl) 10 µl buffer enzima di restrizione (10X) 57 µl H2O deionizzata
In un volume totale di 100 µl
La soluzione è stata incubata 2 ore a 37 °C e successivamente agitata vigorosamente per 10 secondi e posta di nuovo a 37 °C per tutta la notte.
- Purificazione
Alla fine della digestione, il DNA è stato purificato aggiungendo 100 µl di fenolo, agitando e centrifugando per 3 minuti a 13.000 x g per separare le due fasi: organica e acquosa. Alla fase acquosa recuperata è stata aggiunto un ugual volume di cloroformio e le due fasi sono state separate di nuovo centrifugando per 3 minuti a 13.000 x g. La fase
acquosa è stata trasferita in una nuova provetta e sono stati aggiunti 2 volumi di etanolo al 95%, 1/10 di volume di 3 M NaOAc (sodio acetato) (pH 4,5) e 1 µl di glicogeno; il DNA digerito è lasciato precipitare a -20 °C per tutta la notte. Il DNA precipitato è stato raccolto con una centrifugazione a 10.000 x g, per 25 minuti a 4 °C, lavato per due volte con etanolo freddo all’80% e lasciato ad asciugare all’aria. Il DNA è stato infine solubilizzato in 15 µl di tampone Tris-EDTA (10M/0,1M pH 7,5). A questo punto 1 µl di campione viene visualizzato su gel di Agarosio (1,5%) per quantizzare il DNA recuperato e controllare la qualità della digestione.
- Legame degli adattatori
Ad ogni campione sono stati legati particolari adattatori caratterizzati da un gruppo amminico all’estremità 3’ in grado di bloccare l’estensione dell’estremità per prevenire la formazione di un sito di attacco del primer AP1, inoltre se l’adattatore viene esteso al 5’ non si crea il sito di legame con il primer AP1 e la reazione si blocca, infine la lunghezza inferiore dei primers che si legano all’adattatore consentono una PCR soppressiva, prevenendo l’amplificazione di stampi con estremità 3’ estesa contenenti il sito per il legame con il primer AP1 (Fig.4.5).
Miscela di legame:
X µl contenenti 0,4 µg di DNA digerito e purificato per ogni digestione 1,9 µl Genome Walker Adaptor (25µM)
1,6 µl Ligation Buffer 10X 0,5 µl T4 DNA Ligasi (6 U/µl) X µl di H2O fino a 8 µl
La miscela è stata incubata per 16 ore e la reazione fermata tramite incubazione a 70 °C, sono stati successivamente aggiunti 72 µl di Tris-EDTA (10M/1M, pH 7,5) e la miscela agitata vigorosamente per 15 secondi.
- Amplificazioni
Sono state effettuate due amplificazioni successive utilizzando come stampo rispettivamente il DNA legato agli adattatori e la miscela della I PCR diluita 1:50.
Miscela I PCR Cicli utilizzati:
1 µl Template 94° 72° 94 ° 67° 67° 0,5 µl dNTP 20 Mm 25’’ 3’ 25’’ 3’ 7’
5 µl Buffer 10X
1 µl Adaptor Primer 1 (10µM) 7cicli 32 cicli x µl Primer specifico 1 (tab. sotto)
1 µl Advantage 2 Polymerase Mix 50 H2O fino a volume finale 50 µl
Miscela II PCR Cicli utilizzati:
1 µl Template 94° 72° 94 ° 67° 67° 0,5 µl dNTP (20 mM) 25’’ 3’ 25’’ 3’ 7’ 5 µl Buffer (10X)
1 µl Adaptor Primer 2 (10µM) 5 cicli 20 cicli 1 µl Advantage 2 Polymerase mix 50X
x µl Primer Specifico 2 (tab. sotto) H2O fino a volume finale 50 µl
I Primer Specifici utilizzati nella miscela di amplificazione I e II sono riportati nelle tabelle sottostanti.
Primer Specifici per WUSCHEL Primer Specifici per HaL1L
I PCR 15 RW per 5’ RAGE
II PCR 25 RW per 5’ RAGE
I PCR In 7 per 5’ RAGE
3 RAGE 3 per 3’RAGE
II PCR 1 RAGE CDS per 5’ RAGE
3 RAGE 2 per 3’RAGE
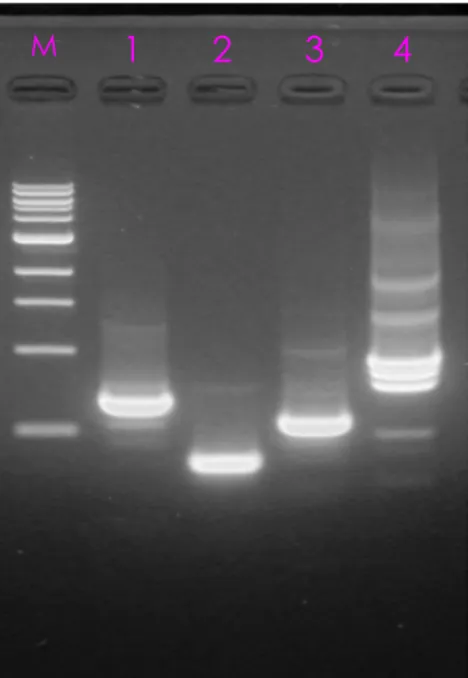
I prodotti delle amplificazioni sono stati controllati su gel di agarosio (1,5%) e i frammenti di dimensione attesa sono stati clonati e sequenziali (Fig. 4.6).
3 4
1 2
M
Fig. 4.6: Elettroforesi su gel di agarosio (1,5%) colorato con Bromuro di Etidio. Corsia 1, DNA amplificato nella reazione di 5’RAGE tagliato con enzima Dra I Corsia 2, DNA amplificato nella reazione di 5’RAGE tagliato con enzima EcoR V Corsia 3, DNA amplificato nella reazione di 5’RAGE tagliato con enzima Pvu II Corsia 4, DNA amplificato nella reazione di 5’RAGE tagliato con enzima Stu I M = Marker Phix 174 digerito con Hae III.
4.13 RACE 3’
Per condurre la RACE al 3’ una serie di oligonucleotidi sono stati selezionati allineando sequenze aminoacidiche di proteine WUSCHEL (Fig 4.4) di specie vegetali diverse e della famiglia delle proteine WOX di
Arabidopsis thaliana presenti in banca dati. L’oligonucleotide FW-ANT, di
22 nucleotidi di lunghezza (ATGGAACCTCAACAACAACA) è stato capace di amplificare un frammento identificato come omologo del gene WUSCHEL.
In questa reazione di amplificazione del cDNA, insieme all’oligonucleotide FW-ANT, è stato utilizzato l’oligonucleotide “Anchor”, complementare ad una porzione dell’oligonucleotide Oligo-dT-Anchor a sua volta impiegato nella reazione di retrotrascrizione.
Le condizioni della reazione di amplificazione sono state le seguenti:
CICLI UTILIZZATI:
temperatura (°C) 94° 94° 53° 72° 72° tempo 5’ 30’’ 30’’ 50’’ 7’ numero cicli 354.14 RACE 5’
Questa tecnica permette l’isolamento e il sequenziamento dell’estremità 5' di un mRNA. L’mRNA estratto da germogli fiorali è stato retrotrascritto, con un oligo-dT per ottenere il primo filamento di cDNA. Dopo digestione con RNasi, per degradare l’RNA residuo, il cDNA è stato purificato dai
nucleotidi non incorporati e dall’oligonucleotide. Alle estremità 3’ del cDNA è stata costituita una coda di poli-C, mediante l’utilizzo di TdT (Terminal Deoxynucleotidyl Transferase), che consente l’appaiamento dell’oligonucleotide Oligo-dG-anchor. Quest’ultimo oligonucleotide è stato utilizzato, in una prima reazione di PCR, in coppia con un oligonucleotide specifico: 15 RW. L’amplificato ottenuto è stato re-amplificato in una seconda reazione di PCR con un terzo oligonucleotide sequenza-specifico, 25 RW, e l’oligonucleotide AUAP. Di seguito è descritta la reazione.
- Sintesi del cDNA
In una provetta da 0,5 ml sono stati aggiunti: • RNA totale 5 µg • Oligo-dT (1 µM) 2,5 µl • H2O DEPC fino a un volume finale di 15,5 µl
La miscela è stata incubata a 70 °C per 15 minuti per denaturare l’RNA e posta in ghiaccio per 1 minuto per bloccare la reazione. Dopodiché alla miscela di reazione sono stati aggiunti:
• PCR buffer 10X 2,5 µl • MgCl2 25 mM 2,5 µl
• dNTP (10 mM ciascuno) 1,0 µl • DTT 0,1 M 2,5 µl
La miscela è stata incubata per 1 minuto a 42 °C, dopodiché è stato aggiunto 1µl di retrotrascrittasi (SuperScript II RT: 200 unità per µl) e mantenuta a 42 °C per 50 minuti.
Il volume finale della reazione è 25 µl.
Infine, la reazione è stata bloccata incubando la miscela a 70 °C per 15 minuti.
- Purificazione del cDNA
Terminata la retrotrascrizione, l’RNA residuo è stato eliminato mediante digestione enzimatica aggiungendo 1µl di RNasi e incubando per 30 minuti a 37 °C. Alla fine la reazione è stata bloccata ponendo la miscela in ghiaccio.
- Attacco di una coda di poli-C in posizione 3'
Alle estremità 3' del cDNA è stata costituita una coda di poli-C utilizzata in seguito come sito di riconoscimento del primer Oligo-dG-anchor. La reazione è stata la seguente:
in un tubo da centrifuga da 0,5 ml sono stati aggiunti • cDNA purificato 6,5 µl
• dCTP 2 mM 2,5 µl • Tailing Buffer 5X 5,0 µl
La miscela è stata incubata 3 minuti a 94 °C e posta in ghiaccio per 1 minuto. È stato aggiunto 1 µl di TdT e la reazione è avvenuta incubando a 37 °C per 10 minuti. Per inattivare la TdT la miscela è stata scaldata a 65 °C per 10 minuti.
- Amplificazione del cDNA
Il cDNA così ottenuto è stato amplificato mediante 2 PCR successive.
I PCR: in una reazione di 50 µl come segue:
cDNA stampo 5 µl PCR buffer 10X 5 µl MgCl2 50 mM 3 µl dNTP (10 mM ciascuno) 4 µl oligonucleotide 15 RW (10 µM) 2 µl
oligonucleotide Oligo-dG-anchor (10 µM) 2 µl
Taq polimerasi 5 U/ µl (Promega) 0,5 µl
H2O 28,5 µl
Le condizioni di reazione sono state le seguenti:
CICLI UTILIZZATI:
temperatura (°C) 95° 94° 69° 72° 72° tempo 5’ 30’’ 30’’ 20’’ 7’
numero cicli 35
II PCR: in una reazione di 50 µl come segue:
I PCR diluita 1/50 2 µl PCR buffer 10X 5 µl MgCl2 50 mM 3 µl dNTP (10 mM ciascuno) 4 µl oligonucleotide 25 RW (10 µM) 2 µl oligonucleotide AUAP (10 µM) 1 µl
Taq polimerasi 5 U/ µl (Promega) 0,5 µl
H2O 32,5 µl
CICLI UTILIZZATI:
temperatura (°C) 95° 94° 70° 72° 72° tempo 5’ 30’’ 30’’ 20’’ 7’
PRIMER SEQUENZA TM CARATTERISTICHE POSIZIONE DNA
SONDA 1 F GAAACATATGGAACGTGGAG 55 senso +36 ; +56
SONDA 1 R CAATGCACTCATTGTCTTCTG 56 antisenso +1023 ; +1044
SONDA 2 F GAGCAAGACCGCTTTATGCC 59,4 senso +1048 ; +1068
SONDA 2 R GTGTATCTTCATCGCTACAG 55,3 antisenso +1294 ; +1314
SONDA 3 F GTGAGCGTGGGTCGATAAGG 62 senso +1329 ; +1349
SONDA 3 R AGTCTTTACACTGAGCATAC 53 antisenso +1531 ; +1551
DHOF AGGGGTGAGCCGCTTGTCA 62 senso -
DHOR AATCGAAACCAGTTAGTCT 52 antisenso
-AGIF AACCCTAACGTCGTCATCGCC 66 senso
-AGIR GACAGGTTCCTGCCGATAGCT 66 antisenso
-TCF GTCGTCAAATGTTCGAGAACAGGAT 72 senso
-TCR TGAGCAAGATGTGCGTTATGATCAG 76 antisenso
-QUEF GCTTTGTGTTTACTGATTCCG 60 senso
-QUER CAAGCAGAGATACAATACAAC 58 antisenso
-CHI-TCDNA CTAGAGAGAGACAATTCC 51 senso +5 ; +13
CHI-REL CAATGCACTCATTGTCTTC 54 antisenso +1025 ; +1044
7RAGEIN ACGAAGACGCCTAGTGGTCAGCGATCAGCA 71 antisenso +332 ; +362
CHI-SF TATGCTCAGTGTAAAGAC 50 senso +1532 ; +1550
CHI-SR GTAGATGGAGAGTGTCAG 54 antisenso +1671 ; +1689
PROMF GATGGCCGAAATCTGAAGTGCAAGCGCTTA 68 senso -1736 ; -1766
PROMR ACGACAACCATTGGTTAGTGGCGCAACTGT 68 antisenso -530 ; -560
ANT GCAAGAACCAGTCGAGGAGTCCG 66 senso -2162 ; -2185
SUX GCGCTTGCACTTCAGATTTCGGCC 66 antisenso -1739 ; -1763
3 RAGE 1 CTGGTTTCGATTTTCGAGCCGTGTGTTGGT 68 senso +1552 ; +1582
PCR
S
C
REENING CLONI SO
NDA
UFFL AATACTAATAATAACGGAACTGTTAAATCTTAAACACTCC 64 antisenso +1732 ; +1772
In7=7RAGEIN
3 RAGE 3 CTCTCTTGTTTTGTCCCCACGAATCTGTCA 67 senso +785 ; +815
1 RAGE CDS GTGGATGGGGAGCCTGTGGTAGCCATGGAA 72 antisenso +62 ; +92
RAGE HaL1L
15 RW GGTAGTGGTGGTGGTGAGATGGAATCTGCAT 69 antisenso + 436 ; + 467
RAGE WUS 25RW GAGTGAACCGTTTCTTCTGGCGTTCACGAGCT 71 antisenso +336 ; + 368
FW-ANT ATGGAACCTCAACAACAACA 53 senso -
AUAP GGCCACGCGTCGACTAGTAC 66 -
-RACE WUS
Oligo-dG-anchor GGCCACGCGTCGACTATACGGGIIGGGIIGGGIIG - -