4. DISCUSSIONE
4.1. La regione 15q11-q14
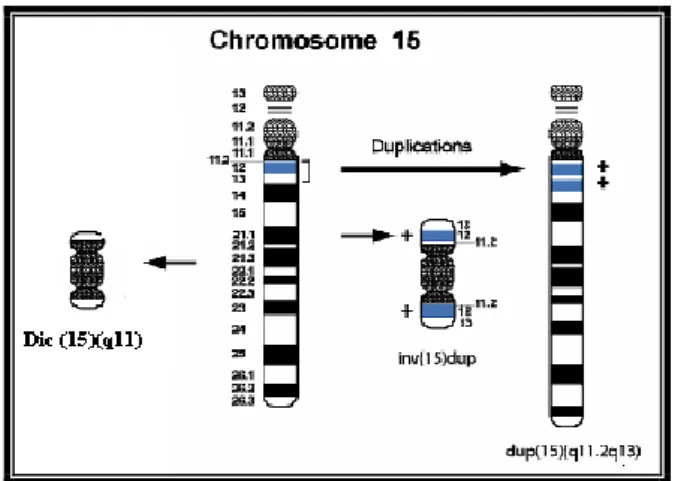
L‟estremità prossimale del cromosoma 15 è caratterizzata dalla presenza di numerose sequenze ripetute che la rendono particolarmente suscettibile al verificarsi di crossing-over tra sequenze omologhe non alleliche (NAHR, non- allelic homologous recombination); sono infatti riportati in letteratura numerosi esempi di riarrangiamenti genomici quali delezioni, duplicazioni e markers sovrannumerari, coinvolgenti il 15q11-q14.
L‟altra interessante peculiarità di questa regione è la presenza di un cluster di geni sottoposti a imprinting, per cui alcuni geni sono espressi solo dal
cromosoma ereditato dal padre, mentre altri da quello ereditato dalla madre. Le alterazioni strutturali coinvolgenti questa regione possono quindi dare origine a fenotipi differenti a seconda dell‟origine parentale del cromosoma coinvolto nel riarrangiamento. L‟esempio più conosciuto è quello della sindrome di Prader-Willi e Angelman in cui è la delezione interstiziale di una stessa regione a determinare fenotipi differenti.
4.2 Imprinting
4.2.1. Meccanismo dell’imprinting
L‟imprinting genomico si evidenzia quando l‟espressione di un gene o di una regione cromosomica risulta differente in relazione al genitore dal quale è stato ereditato. (Per quanto riguarda un gene sottoposto ad imprinting) Nelle cellule somatiche dell‟individuo sono presenti entrambi gli alleli materno e paterno ma uno solo dei due è funzionalmente attivo e l‟altro è silenziato (Hitchins et al., 2002).
L‟imprinting genomico è reversibile da una generazione all‟altra, ossia la
differenziazione tra cromosoma di origine materna o paterna viene cancellata durante la gametogenesi e viene stabilito un nuovo “imprint” a seconda del sesso
dell‟individuo stesso.
L‟imprinting genomico è un fenomeno epigenetico: l‟allele viene contassegnato a seconda del genitore di provenienza con una marcatura che può essere determinata da un pattern di metilazione, una configurazione cromatinica o altri meccanismi non noti.
Il pattern di espressione dei geni soggetti a imprinting può essere complesso:
• alcuni geni soggetti a imprinting sono espressi monoallelicamente solamente in un particolare organo o tessuto, mentre possono essere espressi da entrambi gli alleli parentali in altre parti del corpo: ad esempio UBE3A (ubiquitin protein ligase E3A) ha un‟espressione materna in specifiche regioni del cervello e biallelica in altre. • geni soggetti a imprinting possono anche essere espressi monoallelicamente durante uno specifico stadio dello sviluppo: ad esempio, il gene IGF2 (insulin-like growth
factor 2) localizzato sul cromosoma 11, ha espressione paterna nella maggior parte dei tessuti fetali, ma biallelica durante l‟infanzia e nel fegato adulto.
• esistono anche geni soggetti a imprinting in modo opposto in tessuti diversi, come il gene GRB10 (human growth factor receptor-bound 10) localizzato sul cromosoma 7, che mostra espressione paterna nel cervello fetale, ma di cui è stata ritrovata
un‟isoforma ad espressione materna nel muscolo scheletrico ed ha espressione biallelica in altri tessuti.
4.2.2. Centro di inattivazione
Il raggruppamento in domini dei geni soggetti a imprinting nel genoma, si è
probabilmente evolutivamente selezionato perché l‟espressione o il silenziamento dei geni soggetti a imprinting entro un cluster è regolato in modo coordinato da uno o più elementi che agiscono in cis detti „centro di imprinting‟ (IC) o „imprinting control element‟ (Hitchins et al., 2002).
Il „centro di imprinting‟ è attivo nella linea germinale nello stabilire la marcatura del genitore d‟origine e durante lo sviluppo post-zigotico dove è necessario per il
mantenimento della marcatura (Mann et al., 2000; Bittel et al., 2003).
L‟IC regola l‟instaurarsi delle differenze allele-specifiche nella metilazione del DNA, nella struttura della cromatina, e nell‟espressione. Gli alleli attivi presentano una struttura cromatica simile all‟eucromatina, più accessibile agli enzimi modificatori del DNA, iperacetilazione degli istoni H3 e H4 e mancanza di metilazione delle isole CpG. Le caratteristiche opposte si riscontrano sull‟allele silenziato.
4.2.3. Imprinting della regione 15q
Solamente una parte di questa regione 15q11-q13 (regione critica di
Prader-Willi/Angelman) (mostrata in fig X) è soggetta a imprinting genomico; anomalie a carico di questa sono responsabili di due disturbi neurologici clinicamente distinti, come la Sindrome di Angelman e la Sindrome di Prader-Willi, a seconda che coinvolgano l‟allele di origine materna o quello di origine paterna.
La figura 4.1 mostra l'organizzazione della regione PW/AS, localizzata a livello della banda citogenetica q11-13 del cromosoma 15 umano. All‟interno della regione è presente un Centro bipartito di imprinting (Imprinting Center Region, ICR), ovvero una regione di controllo organizzata in due unità funzionalmente distinte in grado di controllare e mantenere l‟instaurarsi del corretto stato di metilazione e quindi
dell‟effetto parentale sull‟espressione dei geni della regione.
Evidenze sperimentali [Buiting et al. (1995) e (1999); Dittrich et al. (1996, Saitoh et al.1996 ] hanno permesso di individuare per entrambe le unità, PWS e AS, del centro di imprinting bipartito, la più piccola regione di sovrapposizione delle delezioni, definita come “short region of overlap” (SRO). PWS-SRO consta di 4.3 kb ed è localizzato parte nel promotore e parte nel primo esone del gene SNRPN; AS-SRO consta di 880 bp ed è localizzata 35 kb a valle del promotore del gene SNRPN. Raramente a fronte di un pattern di metilazione errato non è possibile identificare la presenza di grosse delezioni, disomia uniparentale, o microdelezioni a livello del centro di imprinting, in questi casi il meccanismo molecolare rimane sconosciuto, potendo essere rappresentato da
mutazioni puntiformi della regione IC come persino da alterazioni dei meccanismi che regolano lo stesso instaurarsi dell‟imprinting.
I geni espressi dal cromosoma di origine paterna sono ZNF127, NDN, SNRPN, PAR-5, IPW, e PAR-1 e sono evidenziati in viola nella figura 4.1; mentre il gene espresso dal cromosoma materno è UBE3A.
Figura 4.1: Regione PW/AS. IC è il centro di imprinting bipartito che agisce in cis. La regione IC che controlla la metilazione del cromosoma paterno è colorato in viola come i geni espressi dall'allele paterno. Al contrario UBE3A espresso dall'allele materno e la regione di IC che lo controlla sono colorati in rosso. I geni di cui non si conosce lo stato di metilazione o che non sono soggetti a imprinting sono rappresentati in verde. Immagine tratta e modificata da Jiang Y. Et al 1999.
4.3. Struttura della regione
Nel 2007 la regione 15q11-q14 è stata nuovamente sequenziata da Makoff et al. i quali hanno realizzato una mappa segmentale mostrata nella figura
sottostante. Un importante conseguenza del sequenziamento di questa regione è che, per la prima volta, è stato possibile mappare con esattezza la localizzazione dei punti di rottura coinvolti nei riarragiamenti.
Figura 4.2 Struttura del cromosoma 15.
Come mostra la figura 4.2, l‟intervallo 15q11-q14 presenta 5 regioni costituite da sequenze ripetute, LCR (low copy-repeats), che presentano tra di loro una elevata somiglianza; ciascuna è costituita da più frammenti che nella figura 4.2 sono riportati come segmenti Y P L E S H C R Q A Z K F U che contengono almeno sette geni / sequenze di pseudogeni, tra cui HERC2 (MIM 605.837), MYLE, e una serie di trascritti sconosciuti. (Makoff et al., 2007)
Queste 5 regioni sono quelle in cui si verificano le rotture nei differenti riarragiamenti e per questo motivo vengono indicate come breakpoints: BP1, BP2, BP3, BP4 e BP5.
La forte omologia dei breakpoint predispone infatti al verificarsi di NAHR. NAHR avviene esattamente con lo stesso meccanismo della ricombinazione omologa (appaiamento filamenti omologhi, formazione del chiasma, risoluzione del crossing-over) con l‟eccezione che, nella NAHR, l‟alta omologia di sequenza dei segmenti cromosomici fa si che il meccanismo cellulare di controllo della ricombinazione non sia in grado di
riconoscere l‟appaiamento di segmenti non perfettamente identici (Lupski e Shaffer 2000).
L‟appaiamento può avvenire fra LCRs appartenenti a cromosomi omologhi (intercromosomico), fra cromatidi fratelli (intracromosomico), all‟interno di un singolo cromatidio (intracromatide). In base al meccanismo di appaiamento,
all‟orientamento e alla complessità delle LCRs, NAHR può dare origine a delezioni, duplicazioni, inversioni, markers o altri riarrangiamenti più complessi che possono anche coinvolgere cromosomi non omologhi. Ricombinazioni tra LCR dirette (con lo stesso orientamento) causano delezioni o duplicazioni, mentre ricombinazioni tra LCR inverse (con orientamento opposto) causano inversioni e markers; I meccanismi di formazione dei markers sono in realtà più complessi in quanto richiedono due serie di eventi mutazionali ossia rotture e non disgiunzione ( Gu et al., 2008).
Figura 4.3: Esempi di ricombinazione omologa non all‟elica. Le Segmental duplication fungono da substrato per la NAHR (a e b) NAHR intercromosomale, intracromosomale, o intracromatidica tra sequenze ripetute orientate nello stesso senso causa delezioni e/o duplicazioni. (c e d) NAHR intercromosomale, intracromosomale, o intracromatidica tra sequenze ripetute orientate in senso opposto causa inversioni. Le sequenze ripetute sono rappresentate dalle box blu con l‟orientamento indicato dalla freccia gialla. L‟evento di ricombinazione è rappresentato da una croce rossa.
Come sottolineato precedentemente, in questa regione sono infatti descritte delezioni, duplicazioni e markers coinvolgenti i differenti breakpoint; in alcuni casi queste alterazioni genomiche sono associate a un quadro fenotipico ben definito ( esempio: PWS/AS ); in altri il quadro clinico è più sfumato e la descrizione di nuovi pazienti può aiutare a definire la presenza di una eventuale nuove sindromi. Inoltre, sempre nella regione sono state descritte duplicazioni e delezioni il cui significato patogenico è ancora da definire e che probabilmente costituiscono delle Copy-Number Variants (CNV) (Itsara et al., 2009) .
Le CNS sono varianti strutturali di dimensioni intermedie (comprese fra 1Kb e 3Mb) individuate recentemente e inaspettatamente numerose nel nostro
genoma. Queste varianti strutturali costituiscono una fonte di variabilità
all‟interno di ogni genoma e quindi contribuiscono in modo preponderante alla diversità umana. Inoltre, questo tipo di varianti possono estendersi anche per milioni di basi e quindi contenere molti geni o intere regioni regolatorie. Capire il significato di una CNV a livello fenotipico è spesso difficile; sebbene alcune di queste varianti strutturali in alcune regioni del genoma non abbiano alcuna conseguenza fenotipica, altre invece possono essere causa di alcune malattie genetiche (Itsara et al., 2009) .
4.3.1 Delezioni
A seconda dei diversi BP coinvolti, il meccanismo di NAHR puo‟ dare origine a differenti tipi di delezioni interstiziali.
NAHR tra sequenze ripetute all‟interno di BP1/BP3 e BP2/BP3 determinano le delezioni riscontrate nei soggetti PWS/AS:
- BP1/BP3 (classe I): le sequenze all‟interno di BP1 e BP3 coinvolte nella NAHR sono complessivamente costituite da ripetizioni dirette molto lunghe (340 kb e 360 Kb rispettivamente) che contengono anche copie complete o troncate del gene HERC2, (HERC2dup)(figX); la delezione coinvolge una regione di circa 5543kb che si estende dal gene HERC2 al gene NIPA1. - BP2/BP3 (classe II): sono le più comuni; i frammenti di BP2 e BP3 in cui si verifica NAHR sono costituiti da ripetizioni dirette lunghe (240 kb)(figX); la delezione coinvolge una regione di circa 57.040 Kb che si estende dal gene
CYFIP1al gene HERC2 (Makoff et al., 2007)
Le sindromi di Angelman e Prader-Willi sono due disordini neuro comportamentali clinicamente distinti. La sindrome di Prader-Willi è caratterizzata da ipotonia neonatale, ipogonadismo, iperfagia che porta ad obesità, bassa statura, mani e piedi piccoli, problemi comportamentali e ritardo mentale (Horsthemke et al., 2003).
La sindrome di Angelman è un disordine multisistemico le cui caratteristiche cliniche includono: severo ritardo dello sviluppo, profonda compromissione del linguaggio, disordini di movimento ed equilibrio, caratteristico profilo comportamentale che include riso frequente e inappropriato, e una personalità facilmente eccitabile. In comune ad alcuni casi, ma non a tutti sono anche: microcefalia, disturbi del sonno, ipopigmentazione e strabismo (Lossie et al., 2001).
Sono state identificate altre delezioni più rare che coinvolgono i breakpoints
BP2/BP4 mentre al momento non sono state ancora descritte delezioni tra BP1/BP4.
4.3.2. Delezioni e duplicazioni BP1/BP2
Nel 2007 è stato descritto (Murphy et al. 2007) un paziente con una delezione di 253kb tra BP1 e BP2 ed un quadro clinico evocativo della sindrome di Angelman, e ciò ha indotto a supporre che la delezione fosse responsabile del fenotipo patologico. La stessa delezione era presente anche nel padre che mostrava caratteristiche cliniche simili, ma più lievi.
Studi successivi (Shaikh et al. 2009) sembrano invece dimostrare la non patogenicità di questa alterazione genomica. Una valutazione del database per le CNVs nella regione (chr15: 20.300.000 -20.800.000, hg17, NCBI build 35) ha infatti identificato
22 CNVs, comprese 15 delezioni e 7 duplicazioni; 12 su 22 CNVs dei controlli comprendevano tutta la regione critica presente nel paziente con fenotipo patologico descritto da Murphy. CNV tra BP1 e BP2 rappresentano un esempio in cui il
significato patogenetico non è ancora chiaro.
4.3.3. Delezioni BP3/BP4/BP5
Sharp et al. 2008 hanno analizzato circa 2000 pazienti con ritardo mentale, epilessia e dismorfismi faciali, per cercare riarrangiamenti gnomici.
I risultati ottenuti suggeriscono che le delezioni che avvengono tra BP4/BP5 sono patogenetiche (6/2082 probandi analizzati vs 0/2062 controlli), invece le duplicazioni che avvengono tra BP3/BP4 e tra BP4/BP5 possono essere varianti benigne oppure essere associate ad un fenotipo patologico più lieve. Sulla base di questi studi, sembrerebbe che, a differenza delle delezioni, le duplicazioni di geni all'interno di questa regione possano non avere conseguenze a livello del fenotipo; ulteriori studi sono però necessari per confermare questa interpretazione.
Le delezioni di 1.5Mb localizzate tra BP4/BP5 (in 15q13.3) sono state associata ad una sindrome caratterizzata da ritardo mentale e convulsioni (Sharp et al. 2008). Questa microdelezione contiene almeno sei geni tra cui il gene CHRNA7(recettore neuronale colinergico nicotinico) un canale ionico che media la trasmissione del segnale neuronale. Vari studi hanno suggerito che CHRNA7 sia implicato
nell‟epilessia mioclonica giovanile e nell‟epilessia benigna dell'infanzia. Questo gene è un eccellente gene candidato in quanto può spiegare il fenotipo osservato nei
genetico per l‟epilessia richiede però ulteriori indagini.
4.3.4. Duplicazioni interstiziali
Le duplicazioni interstiziali della regione 15q11-q13 sono abbastanza frequenti, ma sono difficili da evidenziare con analisi di citogenetica classica; con l'avvento delle analisi molecolari ne e‟ stato descritto un numero crescente e i punti di rottura dei breakpoint di tali duplicazioni sono sono stati quindi descritti con una maggiore precisione.
Le duplicazioni interstiziali della regione prossimale 15q che non includono la regione critica di PWS/AS non hanno effetti clinici, sono generalmente
familiari, e possono essere considerati come normali polimorfismi.
Pazienti che presentano duplicazioni interstiziali che includono PCWACR hanno un fenotipo patologico e presentano ritardo di sviluppo, ritardo mentale, autismo o tratti autistici, difficoltà di coordinamento motorie, e lievi
dimorfismi.
E‟ stato ipotizzato, basandosi su un piccolo numero di casi, che le duplicazioni di origine paterna abbiano un fenotipo più mite rispetto a quelli che sono di origine materna. [Bolton et al., 2001]
4.3.5. Marker cromosoma 15
I Marker cromosomici soprannumerari (SMC) 15 sono stati descritti per la prima volta da Van Dyke et al. (1977) e sono formati da due copie invertite del braccio corto, dal centromero, e dalla porzione prossimale del braccio lungo del cromosoma 15.
L‟architettura dei BP può portare alla formazione di marker cromosomici
sovrannumerari dicentrici. Se le rotture hanno coinvolto i breakpoints BP1 e BP2, ovvero i breakpoints prossimali alla regione critica PWS/AS, allora tali marker non sono associati ad alcun quadro patologico [Maraschio et al. (1988); Cheng et al. (1994)]; quando invece sono coinvolti breakpoints distali BP3, BP4, BP5 ed i marker che si formano contengono quindi copie della PWASR, l‟anomalia cromosomica é patogenetica [Robinson et al. (1993); Blennow et al. (1995)]
I SMC (15) più comuni originano da eventi di ricombinazione che avvengono tra BP4/ BP5, anche se sono stati riportati numerosi marker che originano dalla ricombinazione tra BP3 e BP3. [Makoff J et al. (2007)].
Le ricombinazioni sono mediate da ripetizioni invertite presenti tra BP4/BP5 (500kb) e in BP3 (200kb). Le ripetizioni invertite si trovano anche in BP2, ma i marker che ne derivano non danno fenotipo e quindi la loro incidenza è sicuramente sottostimata. I meccanismi di formazione dei marker sono più complessi rispetto a quelli che portano a delezioni/duplicazioni, ed oltre a comprendere il meccanismo NAHR, sono necessari anche altri eventi mutazionali quali le rotture e la non disgiunzione.
E‟ probabile che la non disgiunzione giochi un ruolo nella formazione di tutti i SMC (15) (Schreck et al. 1977), perché si verifica con una frequenza molto maggiore nelle
donne rispetto i maschi; questo spiega la predominanza di origine materna degli SMC (15) (Robinson et al. 1993). I SMC (15) si possono formare comunque anche durante la spermatogenesi, come è evidente dall‟ osservazione di piccoli SMC (15) di origine paterna su studi effettuati in maschi infertili (Dawson et al. 2002). In alternativa, i grandi SMC (15) potrebbero essere letali se sono ereditati dal padre, anche se triplicazioni interstiziali ereditati dal padre sono stati descritti (Cassidy et al 1996;. Ungaro et al 2001.).
Il misalignement più frequente che porta alla formazione di SMC (15) è uno scambio di tipo U tra cromosomi omologhi durante la meiosi I, seguita da fusione illegittima dei cromatidi e non disgiunzione (Schreck et al 1977.; Martinson et al. 1996). Un'altra teoria propone che, dopo la rottura pre-meiotica, un singolo cromosoma si replichi e che le "sticky" terminali si uniscano per formare i SMC (15) con telomeri identici (Schreck et al. 1977).
Si ipotizza che il misalignement di queste sequenze ripetute, seguita dalla ricombinazione illegittima, sia il risultato del riarrangiamento cromosomico. Il riarrangiamento formato dipenderà dall'orientamento degli elementi di DNA e dal tipo di scambio avvenuto, cioè, se l'evento è inter - o intracromosomico.
Figura 4.4: Anomalie strutturali del cromosoma 15. La regione 15q11-q13 e colorata in azzurro. A sinistra e rappresentato il marker extracromosomico polimorfico (non contiene la regione 15q11-q13) a destra sono rappresentate la anomalia INVDUP15 e una duplicazione della regione.
4.4. I GENI ED IL FENOTIPO
La regione PW/AS è ricca di geni, che sono almeno 20. I geni, come i cromosomi, sono presenti in coppie, (una ereditata dalla madre ed una dal padre). Per la maggioranza dei geni, entrambe le copie sono attive o “espresse” e ciascuna copia del gene produce il prodotto proteico.
Tuttavia, per alcuni geni (e/o regioni del cromosoma), solo una copia è attiva (e perciò produce la proteina), mentre l‟altra non viene espressa.
Per alcuni geni la copia attiva è quella presente sul cromosoma di origine paterna, mentre per altri geni, la copia attiva è quella presente sul cromosoma di origine materna.
La regione PWACR include sia geni che sono espressi su entrambi i cromosomi, quello di origine paterna e quello di origine materna, che quelli espressi su un solo cromosoma, di origine paterna o materna.
Il gene SNRPN (Small Nuclear Ribonucleoprotein Polypeptide N), espresso sul cromosoma paterno ma silente su quello materno rappresenta un punto di riferimento chiave della regione. Tale gene è coinvolto nello splicing dell' pre-mRNA (trascritto primario). Il promotore di SNRPN infatti è localizzato vicino ad una isola CpG completamente metilata sul cromosoma materno e ipometilata su quello paterno; lo stato di metilazione di tale promotore è considerato rappresentativo dello stato di metilazione dell‟intera regione 15q11-q13. Parte del centro bipartito di Imprinting (la porzione che controlla l‟allele di origine paterna) si sovrappone a questa regione. Il gene HECR2 è localizzato vicino al punto di rottura telomerico. Si ritiene che le delezioni interstiziali della regione PW/AS siano originate da crossing-over ineguale fra le copie complete o troncate del gene HERC2, la cui sequenza comprende
numerosi elementi ripetuti in basso numero di copie (low-copy-repeats).
Il gene ATP1OA (noto anche come ATP1OC) produce una proteina, che si pensa sia coinvolta nel movimento delle molecole fuori e dentro le cellule. Tale proteina è espressa nel cervello, ma solo la copia materna è generalmente attiva. La maggior parte delle persone con idic(15) ha quattro copie di questo gene, invece delle solite due. Ci sono due evidenze che indicano che la regione 15q 11q13 sia un buon candidato a contenere un gene, coinvolto nell‟epilessia. La prima evidenza è che la perdita della copia di questa regione ereditata dalla madre causa la sindrome di
Angelman, nella quale è presente epilessia. La seconda evidenza è l‟osservazione che copie in eccesso di questa stessa regione cromosomica, come nell‟idic(15), sono spesso associate a crisi epilettiche.
Altri geni identificati nella regione critica PW/AS sono: IPW, ZNF127 e NDN, espressi dal locus paterno, il “gene Angelman” UBE3A, il cui imprinting materno è
tessuto specifico e il cluster dei geni GABAA, il cui stato di imprinting e ancora incerto.
Il gene UBE3A fornisce informazioni per la produzione di una proteina, che ha la funzione di avere come bersaglio altre proteine all‟interno delle cellule, che devono essere degradate. La degradazione delle proteine è un normale processo, che rimuove le proteine non necessarie o danneggiate ed aiuta a mantenere la normale funzione cellulare. Entrambe le copie del gene UBE3A sono attive nella maggior parte dei tessuti del nostro organismo. Nel cervello, tuttavia, solo la copia di origine materna è generalmente attiva.
Le mutazioni che avvengono invece a livello del gene UBE3A causano la sindrome di Angelman, è noto infatti che mutazioni in questo gene possono interferire con il normale sviluppo del cervello.
Il gene ZNF127 codifica per una proteina zinc-finger. E‟ un gene imprinted ed è espresso esclusivamente dal padre.
Il cluster dei geni GABAA sono geni candidati per l‟autismo i quali codificano per le sub-unità dei recettori GABAA .
Duplicazioni o delezioni dei tre geni che codificano per le sub-unità dei recettori GABA (GABRB3, GABRA5 e GABRG3) possono determinare uno squilibrio nella disponibilità delle subunità di questi recettori GABA e ciò può alterare l'attività dei principali neurotrasmettitori inibitori del cervello.
Il gene (P), la cui delezione è associata alle caratteristiche di ipopigmentazione in soggetti PW o AS, non e' sottoposto ad imprinting.
Un altro interessante gene con un ruolo a livello cerebrale è il gene NDN che codifica per una proteina chiamata NECDIN che è una proteina nucleare la quale
viene espressa solo da alcuni neuroni del cervello (SNC). E‟ stato ipotizzato che la sua funzione sia quella di arrestare la crescita cellulare dopo il periodo embrionale mitotico, durante il periodo di assestamento neuronale. E‟ un gene imprinted ed è espresso esclusivamente dal padre.
Repetto et al. hanno trovato in un paziente, che presentava la duplicazione in questa regione ma di origine parentale sconosciuta, il gene NDN mutato. (controllare) Depienne et al. hanno ipotizzato che il gene CYFIP1 (cytoplasmatic FMR1
Interacting proteine) sia il gene candidato dell‟autismo. Tale gene codifica per una proteina che interagisce con FMR1, il gene responsabile della sindrome dell‟X fragile. Spesso questa sindrome è associata con l‟autismo per questo il gene CYFIP1 è il gene candidato . Oltre a questo la proteina codificata da CYFIP1 interagisce con Rho GTPasi Rac che ha un ruolo nella regolazione della migrazione assonale e la morfologia della colonna vertebrale.
Questi studi forniscono ulteriori prove che uno o più geni all'interno della regione PWACR hanno un effetto deleterio sullo sviluppo mentale e fisico.
Nel corso del nostro studio abbiamo effettuato la caratterizzazione di
riarrangiamenti cromosomici con analisi di citogenetica classica e di genetica molecolare in 3 casi che presentano un fenotipo patologico.
Lo scopo di questo lavoro è stato anche quello di dimostrare l‟importanza e i limiti delle diverse tecniche di citogenetica classica e molecolare, come pure delle
metodologie molecolari, quali CGH, FISH e MS-MLPA, che, solo se combinate insieme, permettono una definizione più chiara e precisa del riarrangiamento cromosomico.
Abbiamo successivamente confrontato il fenotipo del caso in studio con quello dei casi riportata in letteratura che presentano un riarrangiamento simile; questa comparazione ha permesso di stabilire una correlazione tra genotipo e fenotipo, identificando geni potenzialmente coinvolti nel causare il quadro clinico.
CASO I (46XY, dup 15q11.2-q13.1)
Le duplicazioni interstiziali che coinvolgono la regione 15q11-13 sono conosciute fino dal 1974 e da allora sono stati descritti circa 40 casi. E‟ interessante osservare che il fenotipo di questi pazienti sembra essere differente a seconda che la duplicazione sia ereditata dal padre o dalla madre; questo concetto risulta coerente con il fatto che la regione in analisi è
“imprinted” e quindi presenta una differente espressione genica sulla base dell‟origine parentale.
Nella tabella 4.1 sono riportati tutte le caratteristiche dei casi riportati in letteratura con duplicazione materna , nella tabella 4.2 ci sono quelli di duplicazione paterna.
Caratteristiche riscontrate Numero dei pazienti
Percentuale %
Epilessia 5/38 13
Ritardi mentali 5/38 13
Autismo 9/38 24
Ritardo dello sviluppo 18/38 47 Ritardo del linguaggio 13/38 34
Dimorfismi faciali 9/38 24
Ipotonia 8/388 21
Iperattività 6/38 15
Iperlassità dei legamenti 3/38 8 Mancanza di
coordinazione
Caratteristiche riscontrate Numero dei pazienti
Percentuale %
Ritardi mentali 3/12 25
Autismo 5/12 42
Ritardo dello sviluppo 5/12 42 Ritardo del linguaggio 6/12 50 Dimorfismi faciali 1/12 8
Ipotonia 3/12 25
Mancata interazione sociale
3/12 25
Iperlassità dei legamenti 4/12 33 Mancanza di
coordinazione
2/12 17
Come può essere evidenziato dalla tabella 4.1 il ritardo dello sviluppo è la caratteristica più frequente riscontrata nei pazienti( 42%). Altri aspetti comuni sono il ritardo del linguaggio (39%), l‟autismo (32%), ipotonia (28.6%) e l‟epilessia (17.8%) ( Schroer et al., 1997; Roberts et al., 2002; Bolton et al., 2001; Thomas et al., 1999; Mao et al., 1999; Repetto et al., 1998; Browne et al., 1997; Cook et al., 1997; Depienne et al., 2009) .
In alcuni casi il fenotipo di alcuni probandi riportati in letteratura non viene descritto o viene descritto molto sommariamente, per cui è possibile che le percentuali riportate nella tabella siano in realtà sottostimate.
Nella tabella 4.2 sono invece riportati i casi di duplicazione di origine paterna che presentano un fenotipo patologico; il loro numero è molto più basso rispetto ai soggetti con la duplicazione di origine materna. La maggior parte di questi pazienti (6 soggetti descritti in letteratura) ha un fenotipo normale e l‟assenza di un fenotipo patologico potrebbe rappresentare un bias per cui questi soggetti non vengono identificati e siano quindi in realtà più numerosi di quanto stimato.
Sono descritti in letteratura solo 12 casi con un fenotipo patologico; le
caratteristiche più comuni sono il ritardo del linguaggio (50%), l‟autismo (42%) e il ritardo di sviluppo (42%) (Veltman et al., 2005; Bolton et al., 2002;
Depienne te al., 2009; Mohandas et al., 1999; Engelen et al., 1999) . Nel complesso i pazienti con duplicazioni di questa regione condividono dimorfismi faciali, ipotonia, legamenti lassi, ritardo di sviluppo, epilessia, ritardo del linguaggio, ritardo mentale, mancanza di coordinazione, mancata interazione sociale, e autismo.
Il grado di compromissione risulta però estremamente variabile con un range che va dal fenotipo perfettamente normale (spesso se la duplicazione è di origine paterna), al borderline, fino a quadri clinici fortemente compromessi. Sarebbe molto interessante correlare questi fenotipi con le diverse estensioni di duplicazione (e quindi al coinvolgimento di geni diversi), ma essendo le descrizioni piuttosto datate e mancando casi analizzati con tecniche quali CGH array, questa correlazione risulta per ora di difficile applicazione.
Il paziente da noi studiato presenta una duplicazione interstiziale del cromosoma 15, a livello della banda q11-q13, di origine materna.
Le caratteristiche cliniche del nostro caso sono state confrontate con quelle dei casi di duplicazione interstiziale (tabella 4.1), con particolare attenzione per i casi che presentano una duplicazione nella stessa regione (15q11-q13). Il caso da noi analizzato non presenta anomalie gravi alla nascita. Le prime analisi fatte al bambino all‟età di 2 anni e 3 mesi mostrano autismo, crisi
epilettiche, ipotonia assiale, deterioramento cognitivo, ritardo psicomotorio con movimenti stereopati (dondolamento antero posteriore del tronco, iactatio
capitis e parossismi di iperpnea), movimento delle mani (batteva le mani), ritardo del linguaggio, mancanza di risposta agli stimoli ambientali con scarsa interazione sociale; il bambino presentava anche lievi note dismorfiche come plagiocefalia, orbite infossate, impianto basso dei padiglioni auricolari e pectus excavatum. Alcune di queste caratteristiche sono comuni ai casi che presentano una duplicazione di questa regione.
La risonanza magnetica evidenzia un aumento degli spazi liquorali pericerebrali soprattutto a livello frontale. Questo tipo di analisi in letteratura era riportata solo per due casi, di cui una risultava normale.
Al bambino è stata diagnosticata una forma di epilessia (caratteristica clinica presente in 13% dei casi riportati in letteratura) che risulta ben controllata dai farmaci.
Esiste poi un gruppo di caratteristiche, quali la presenza dei lignamenti lassi e l‟iperattività, che sono relativamente frequenti in pazienti con duplicazione 15q11-q13 ma che non si riscontrano nel nostro paziente.
La variabilità dei tratti fenotipici tra i differenti casi di duplicazione 15q11-q13, compreso il nostro paziente, può essere attribuibile a svariate motivazioni. 1) Geni modulari background genetico
Un‟ipotesi alternativa che può spiegare una variabilità fenotipica associata ad una regione duplicata è il differente background genetico di ciascun individuo. L‟entità del danno derivante dalla duplicazione di un gene può infatti variare in relazione alla presenza di eventuali genocopie e può essere influenzata dalla differente attività di proteine polimorfiche facenti parte dello stesso pathway funzionale.
2) microRNA
I microRNA sono un tipo di RNA regolatori. Essi sono in grado di riconoscere mRNA a sequenza complementare e impedirne la traduzione guidandoli verso la degradazione. Sebbene i microRNA siano conosciuti da tempo e siano usati anche a scopo sperimentale per il silenziamento genico, recentemente una nuova scoperta ha reso il quadro ancora più complesso. Alcuni scienziati del Medical Center di Boston, infatti, hanno scoperto che il ruolo di microRNA e mRNA può essere ribaltato: RNA trascritti, ma non codificanti, possono legarsi ai microRNA impedendo che essi possano operare il silenziamento genico, aggiungendo così una altro punto alla lista dei possibili meccanismi di regolazione.
4) Stato di metilazione nella regione duplicata.
L‟ alterazione della metilazione dà origine a fenotipi patologici. La metilazione di questa regione è stata molto studiata, ma non ci sono evidenze in letteratura sullo stato di metilazioni di frammenti duplicati; ovvero secondo le nostre conoscenze non è noto se la metilazione del frammento duplicato è identica a quella della regione in singola copia. E‟ infatti possibile che qualche gene possa essere metilato diversamente e ciò potrebbe rendere fuorvianti i risultati delle tecniche basate sullo stato di metilazione per stabilire l‟origine parentale del frammento duplicato.
Inoltre una variazione nello stato di metilazione dei geni del frammento duplicato potrebbe avere conseguenze sul fenotipo e quindi spiegare la variabilità fenotipica dei soggetti con la stessa alterazione cromosomica, ereditata dallo stesso genitore.
infatti ambigui. L‟origine della duplicazione pare di origine paterna, ma questo risultato non è stato confermato con le altre tecniche. Questo può essere dovuto ad una metilazione alterata del frammento duplicato cioè ad una variazione di metilazione dei geni del frammento duplicato.
CASO II
Il primo caso di SMC (15) è stato descritto da Schreck et al nel 1977. Da allora sono stati riportati in letteratura più di 100 casi di SMC (15) che coinvolgono varie regioni del cromosoma. Nella maggior parte dei casi gli SMC (15) risultano essere de novo mentre solo in alcuni sono familiari (nella maggior parte di origine materna). Nella tabella 4.3 sono riportati le caratteristiche dei casi con SMC descritti in letteratura. CARATTERISTICHE RISCONTRATE NUMERO DEI PAZIENTI PERCENTUALE % Ritardo mentale 64/89 72
Ritardo dello sviluppo 68/89 76
Ipotonia 38/89 43 Obesità 2/89 2 Infertilità 17/89 19 Dimorfismi 22/89 25 Bassa statura 20/89 23 Problemi comportamentali 35/89 39 Convulsioni 31/89 35 Autismo 10/89 11
Ritardo del linguaggio 3/89 3
Epilessia 1/89 1
Come può essere evidenziato dalla tabella il ritardo dello sviluppo è la
sono: ritardo mentale di vario grado (72%), ipotonia (43%), problemi
comportamentali (39%), convulsioni (35%) e infertilità (19%). (Roberts et al., 2003, crolla et al., 2004; Koochek et al., 2005; Ungaro et al., 2001; Battaglia et al., 2008; Webb 1994; Warburton 1991; Roberts et al., 2003; Wolpert et al., 2000; Hogart et al., 2009; Leana-Cox et al., 1994Long et al., 1996)
I due pazienti da noi analizzati non presentano un fenotipo anomalo con l‟esclusione di un problema legato all‟infertilita‟.
Il primo paziente è una donna di 36 anni; le tappe dello sviluppo sono risultati normali. All'età di 27 anni ha avuto una normale gravidanza e a 35 anni ha avuto amenorrea asintomatica brusca. Il profilo ormonale non mostrava alterazioni di rilievo.
Il termine POF (Premature Ovarian Failure) viene utilizzato per indicare un gruppo eterogeneo di patologie caratterizzate da amenorrea che insorge prima dei quaranta anni, da bassi livelli di estrogeni e da alti livelli di gonadotropina. I casi più critici possono presentare sia un‟amenorrea primaria nella quale il menarca non appare mai, che un‟amenorrea secondaria nella quale il menarca appare ma poi si interrompe, scomparendo intorno ai 20 anni.
Il 10% dei casi che presentano la POF ha una eziologia notevolmente eterogenea, comprendente fattori autoimmuni, ambientali (come radiazioni, chemioterapici, infezioni) e fattori genetici (Valetto et al., )
Sono stati identificati vari geni candidati che vengono considerati come fattori di rischio per questa patologia di cui uno è l‟estensione delle ripetizioni CGG al 5‟UTR del gene FMR1, il gene responsabile della sindrome dell‟X-fragile. Studi effettuati sul cromosoma X hanno identificato 2 regioni uno chiamato CR1 (critical
region 1) e l‟altro CR2 (critical region 2) i quali sono essenziali per la normale funzione ovarica e il normale reproductive lifespan. Nessuno dei geni localizzati in questa regione ha mostrato una chiara funzione nella fisiologia ovarian e le analisi delle mutazioni in pazienti che presentano la POF non confermano il loro ruolo causativo.
4.5. Confronto tecniche utilizzate
Se consideriamo i casi riportati in letteratura, si nota come spesso i reports risalgano a qualche decennio fa: ovviamente in questi casi come prima analisi genetica è stato fatto il cariotipo, il quale però non sempre è informativo (ovvero non sempre rileva la regione duplicata, in quanto inferiore al potere risolutivo del bandeggio).
Il cariotipo permette infatti di evidenziare la presenza di marker (caso2) , anche se non permette di definirne, l‟estensione, mentre non è assolutamente in grado di evidenziare CNV. Per ciò che riguarda le duplicazioni, solo in tre casi il cariotipo le ha rilevate; negli altri casi invece solo l‟utilizzo di tecniche quali FISH, l‟analisi dei micro satelliti o l‟MLPA hanno permesso di rilevare l‟anomalia genetica.
La tecnica della FISH (ibridazione fluorescente in situ) permette tramite l‟uso di sonde specifiche di determinare non solo la presenza, ma anche l‟ estensione e l‟orientamento della duplicazione (invdup o tandem). Ovviamente questo è possibile utilizzando un pannello di sonde in esperimenti successivi, quindi con
dispendio di tempo e denaro, e con l‟analisi non solo di cromosomi metafasici, ma anche di nuclei interfasici, in quanto i segnali risultano più distanziati e di migliore leggibilità.
Nella maggior parte dei casi in letteratura, per confermare i dati ottenuti da questa tecnica viene fatta anche l‟analisi dei microsatelliti: questa tecnica non solo serve da conferma ma permette anche di determinare l‟origine parentale della duplicazione. Tecniche di più recente introduzione come l‟ MLPA sono state utilizzate solo in un caso recentemente descritto (Depienne et. Al, 2009): come precedentemente sottolineato, questa tecnica ha il vantaggio di permettere in un singolo esperimento il rilevamento della duplicazione, la sua precisa estensione, e la determinazione dell‟origine parentale.
Nella nostra esperienza di laboratorio, questa tecnica fornisce risultati molto chiari nel caso di delezioni o di disomie uni parentali (sindromi
PW/Angelmann); nel caso di duplicazioni, invece, l‟analisi quantitative e qualitativa dei picchi deve essere fatta con grandissima attenzione e i risultati possono essere di più difficile interpretazione per quanto riguarda ereditarietà. Dai dati di letteratura, l‟analisi dell‟origine parentale è stata affrontata anche con metodiche come il Southern blot, dopo digestione con enzimi di restrizione sensibili alla metilazione, oppure con PCR metilazione specifica.
Il CGH array, una tecnica di recente introduzione, non ci risulta che sia mai stato usato per determinare in modo più corretto e specifico l‟estensione della
regione di duplicazione; soltanto una volta è stata utilizzata la tecnica del CGH su metafasi, con risultati ovviamente molto più approssimativi.
Nel nostro caso, abbiamo voluto utilizzare tutte le metodiche di normale uso nel nostro laboratorio, al fine di stabilire in maniera esatta l’estensione della
regione duplicata, in quanto l‟ associazione fenotipo patologico/genotipo risulta tanto più interessante quanto più esatta è la definizione della porzione genomica interessata.
Nel nostro caso abbiamo sottoposto il DNA del nostro probando al CGH array, che ha stabilito in modo inequivocabile che la duplicazione si estende dal nucleotide 20335887 al 26199055, come si nota dai valori medi dei rapporti di fluorescenza (circa 0.6). Questa tecnica ci fornisce inoltre i geni contenuti nella regione e duplicati. La tecnica di MLPA ha confermato la duplicazione nel probando, mentre l‟ MS-MLPA( Methylation-specific MLPA), che permette di stabilire l‟origine parentale, ha fornito risultati dubbi. Per questo motivo, per confermare l‟estensione della duplicazione e verificarne l‟origine parentale, abbiamo effettuato una analisi molecolare della regione 15q11-q13 mediante tipizzazione di marcatori altamente polimorfici (STRs). Per ciascun
microsatellite è stata verificata la segregazione allelica da genitore a figlio. La duplicazione dell‟allele materno è stata riscontrata per vari micro satelliti.
Tale risultato è stato ulteriormente confermato quando sono stati analizzati i genitori del probando con CGH ed è stato visto che la madre presentava la stessa duplicazione
L‟uso di tradizionali tecniche di colorazione per caratterizzazione dei cromosomi marker spesso non è di ausilio a causa di un insufficiente pattern di bandeggio. Per questo, molti markers diagnosticati negli anni precedenti la scorsa decade sono rimasti non caratterizzati. L‟introduzione della Fluorescence In Situ Hybridization (FISH) usando probes centromero, cromosoma e locus specifici ha reso possibile definire l‟origine cromosomica. Più recentemente l‟uso del Reverse Chromosome Painting (micro-FISH) ha dato un aiuto tradizionale nel determinare l‟origine
cromosomica più in dettaglio (Corter et al. 1992; Thangavelu et al. 1994; Viersbach et al. 1994; Blennow et al. 1995; Muller et al. 1995; Anderlid et al. 2001).
L‟applicazione combinata di queste tecniche permette di ottenere una buona
caratterizzazione dei cromosomi marker e mira a incrementare la conoscenza relativa al Prima dell‟avvento degli studi di FISH il rischio di anormalità fenotipica associata alla presenza di un cromosoma marker de novo aveva una stima del 13% (10.9% satellitari, 14.7% non satellitati) e si basava sul follow up di diagnosi prenatali (Warburton et al. 1991). A tutt‟oggi, solo parte dei marker è stata ben caratterizzata citogenetica mene permettendo una precisione correlazione genotipo-fenotipo. Una dettagliata caratterizzazione molecolare dei markers in individui normali ed anormali assieme ad un follow-up a lungo termine possono aiutare ad identificare le aree del genoma che sono più sensibili ad uno sbilanciamento.