21
3. GLI IMMINOGLICALI E LA REAZIONE DI FERRIER
Gli imminoglicali sono azapiranosi caratterizzati da un doppio legame adiacente all’azoto endociclico. Tali composti risultano essere particolarmente interessanti perché possono partecipare, data la presenza dell’insaturazione, a varie reazioni (cicloaddizioni, cross-couplings metallo catalizzati), e perché possono costituire intermedi utili per la sintesi di azazuccheri e di oligosaccaridi di azazuccheri.
In realtà, però, sono pochi, in letteratura, gli studi effettuati su tale classe di composti. Lo stato dell’arte riguardante gli imminoglicali è limitato, infatti, al loro utilizzo, tra l’altro parziale, nella reazione di Ferrier (Schema 3.1).
In questa reazione, utilizzata normalmente per i glicali e già citata precedentemente ( pag. 10), viene introdotto, in presenza di un acido di Lewis, un nucleofilo in posizione α rispetto all’eteroatomo endociclico e viene contemporaneamente eliminato il sostituente presente in C(3) per ottenere un glicoside 2,3 insaturo.
Schema 3.1. Reazione di Ferrier sugli imminoglicali
I primi studi che hanno portato ad applicare la reazione di Ferrier agli imminoglicali e che hanno contribuito a spiegarne il meccanismo risalgono alla fine degli anni ‘80.9,10
Comins e coll. avevano infatti intrapreso uno studio sulle α-alchilazioni delle piridine9 un substrato, quindi, diverso dagli imminoglicali- ed erano poi passati ad utilizzare, sempre per α-alchilazioni, composti con un atomo di azoto endociclico adiacente ad un’insaturazione.10
Gli studi di Comins avevano inizialmente indicato come l’attacco di un nucleofilo effettuato su un sale di acil piridinio non sostituito avvenisse in maniera non selettiva su C(2) e C(4), le posizioni più reattive dell’anello piridinico e del corrispondente sale di N-acil piridinio per l’addizione nucleofila.8
O OAc AcO AcO Nu Acido di Lewis (LA) O AcO AcO Nu
22
Comins aveva esaminato l’addizione di ioduri di organozinco a piridine in presenza di un acil cloruro. Aggiungendo, per esempio, ioduro di organozinco ad un sale di 1-(fenossicarbonil)piridinio 3.2 aveva ottenuto l’1-(fenossicarbonil)-1,4-diidropiridina 3.3 e la
1-(fenossicarbonil)-1,2-diidropiridina 3.4 in rapporto 68:32 (Schema 3.2).
Per ottenere l’alchilazione regioselettiva in posizione 2, Comins e coll. avevano così pensato di utilizzare un sale di acil piridinio sostituito in C(4) (Schema 3.3).
Schema 3.3 Alchilazione regio selettiva in posizione 2con generica sostituzione in C(4).
Aggiungendo ioduro di clorobutilzinco al sale di fenossicarbonile della 4-cloropiridina si era ottenuto selettivamente 3.8 con una resa del 66% (Schema 3.4).
Cl ZnI 3.3 3.4 PhOCOCl 3.2 3.1 N N COOPh N N COOPh COOPh Cl Cl 1) PhOCOCl 2) R'ZnI 3.5 3.6 N R N R COOPh R' 1) PhOCOCl 2) Cl ZnI 3.8 3.7 N N Cl Cl COOPh Cl Schema 3.2. Reazione di alchilazione di piridine
23
Era stato poi ipotizzato che maggiori quantità di α-addizione in substrati simili potessero essere ottenute utilizzando, come intermedio reattivo, invece di un sale di N-acil piridinio, un sale formato a partire dall’1-acil-4-idrossi-1,2,3,4-tetraidropiridina 3.9.
Tale composto, come si può notare, presenta proprio un’insaturazione adiacente all’azoto endociclico.10
Comins e Kozikowski avevano mostrato che i γ-idrossi enecarbammati sono precursori di ioni N-acil imminio, e che le 1-acil-4-idrossi-1,2,3,4-tetraidropiridine 3.9 potevano essere trasformate in sali di 1-acil-2,3-diidropiridinio 3.10 in situ per trattamento con un acido di Lewis. La successiva addizione di un nucleofilo avveniva in posizione α e conduceva alla corrispondente piperidina 2,3 insatura 3.11 (Schema 3.5).
Schema 3.5. Addizione di nucleofili sul sale di 1-acil-2,3-diidropiridinio
L’impiego del composto 3.12 con un gruppo ossidrilico protetto aveva permesso l’addizione di un reagente organometallico come nucleofilo, altrimenti non utilizzabile in presenza di un gruppo ossidrilico libero. L’aggiunta a 3.12 di un acido di Lewis quale BF3.OEt2 aveva portato alla formazione, in situ, di una specie reattiva di N-acil imminio 3.10
e l’aggiunta di un reagente di organo-zinco, in benzene e a temperatura ambiente, aveva portato alla formazione della 2-alchil-∆3-piperidina 3.13 (Schema 3.6). Si era avuta, quindi,
nuovamente un’addizione in
α.
Schema 3.6. Addizione di un reagente di organo-zinco al sale di 1-acil-2,3-didropiridinio.
3.9 3.10 N OH COOR N COOR LA Nu 3.11 N COOR Nu N OMe COOR N BF3.OEt 2 R'ZnI N 3.12 3.10 3.13 COOR COOR R' 3.9 N OH COOR 1 2 3 4
24
Anche le reazioni di 3.12 con nucleofili soft come l’alliltrimetilsilano e i silil enol eteri avevano portato principalmente all’addizione in α dello ione imminio coniugato.
Un ulteriore passo effettuato era stato quello di applicare tale procedura ad una 1-acil-4-metossi-1,2,3,4-tetraidropiridina con un gruppo alchilico con una ben definita stereochimica in posizione 2.
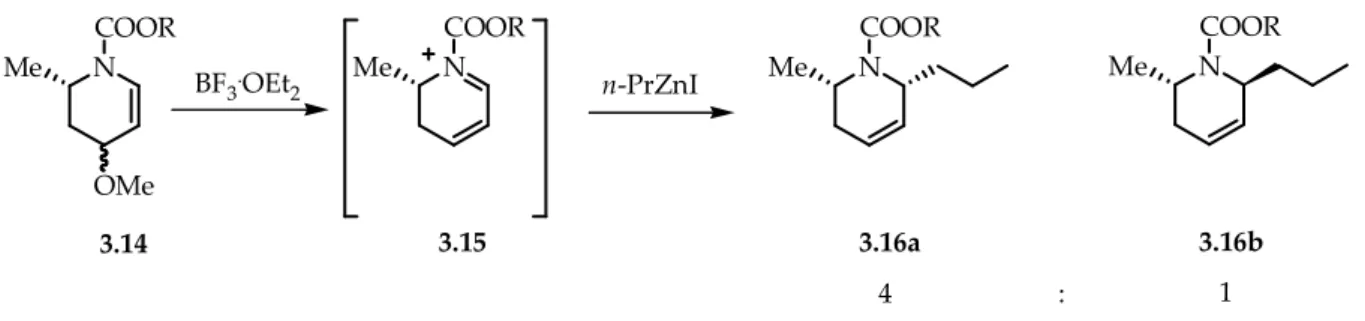
La reazione del composto 3.14 con un reagente di alchil zinco in presenza di un acido di Lewis aveva portato all’ottenimento della cis-2,6-dialchil-∆3-piperidina 3.16a e della trans-2,6-dialchil-∆3-piperidina 3.16b in rapporto 4:1 (Schema 3.7). La stereochimica relativa dei composti era stata assegnata in base all’analisi 1H NMR. Il maggiore diastereomero ottenuto era stato confermato essere proprio il cis.
Schema 3.7. Sintesi di 2,6-dialchil-∆3-piperidine diastereoisomere.
La stereoselettività della reazione era stata spiegata sulla base di effetti stereoelettronici.
A causa della forte tensione 1,3 allilica presente tra il gruppo metilico in C(2) e il gruppo N-acilico dello ione imminio che si forma nel mezzo di reazione, il gruppo metilico di
3.15 va ad occupare una posizione assiale e la specie assume la conformazione 3.15b
(Fig.3.1).
Il successivo attacco pseudoassiale stereoelettronicamente favorito su 3.15b da parte del reagente organometallico conduce preferenzialmente alla formazione del composto cis.
Figura 3. 1. Attacco pseudo assiale stereo elettronicamente favorito del reagente organometallico su 3.15b. N COOR N n -PrZnI N 3.14 3.15 3.16a BF3.OEt 2 N 3.16b 4 : 1 OMe Me COOR Me COOR COOR Me Me N COOR Nu Me 3.16a N PhO O Me PhO N O Me Nu -3.15a 3.15b
25
Con questi studi era così iniziato l’esame del comportamento di composti caratterizzati dalla presenza di un’insaturazione e di un atomo di azoto endociclico protetto in presenza di un acido di Lewis e di un nucleofilo. Il meccanismo delle reazioni appena esaminate vedeva quindi la formazione di uno ione N-acil imminio coniugato come intermedio.
Nel 1998 Graig e coll. hanno effettuato uno studio su composti che presentano le stesse caratteristiche strutturali di 3.14.11
I composti presi in considerazione erano infatti le 1,4-bis(arilsulfonil)-1,2,3,4-tetraidropiridine 3.17 con gruppi di varia natura in C(2) e con stereochimica non definita in C(4). Queste tetraidropiridine venivano fatte reagire con C-nucleofili aventi anche funzione di acidi di Lewis.
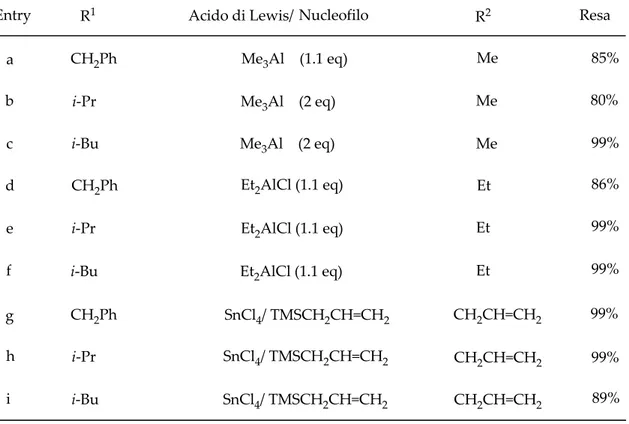
Le reazioni di 3.17 con trimetilalluminio cloruro, a temperatura ambiente e in toluene, avevano portato all’ottenimento, in breve tempo (0.5-2 ore), di composti di sostituzione nucleofila 3.19 (entry a-c, Tabella 3.1) con rese eccellenti e formazione di un unico diastereomero quale prodotto di reazione (Schema 3.8). La natura 2,6-cis di questi composti era stata inequivocabilmente stabilita da studi NOE e dall’analisi cristallografica a raggi X.
Anche le reazioni di 3.17 con cloruro di dietilalluminio avevano condotto agli analoghi etilici (entry d-f) come diastereomeri cis singoli.
Erano state poi effettuate reazioni di 3.17 con SnCl4 e alliltrimetilsilano e, anche in
questo caso, erano stati ottenuti esclusivamente gli isomeri 2,6 cis (entry g-i).
N Ts Ts R1 N Ts R1 R2 3.17 3.19 (Vedere Tabella 3.1) N Ts R1 3.18 LA/Nu N Ts Ts R1 3.17 1 2 3 4
26
Le reazioni sopra-elencate procedevano attraverso uno meccanismo di SN1’. Si otteneva
difatti un unico prodotto con completa diastereoselettività, indipendentemente dalla configurazione in C(4) di 3.17.
Era stato ipotizzato anche in questo caso che, grazie all’acido di Lewis, si formasse uno ione imminio coniugato (Schema 3.8), e che il successivo attacco nucleofilo avvenisse esclusivamente in posizione 2 sulla faccia α. In questo modo si verificava un’esclusiva addizione sin con ottenimento del solo diastereomero 2,6-cis disostituito.
Tabella 3.1. Reazioni di 3.17 in presenza di acidi di Lewis
Il fatto che l’attacco nucleofilo sullo ione imminio avvenisse sulla faccia α, apparentemente più impedita per la presenza del gruppo R1, era stato spiegato come una conseguenza della preferenza stereoelettronica per un attacco pseudoassiale che permette alla reazione di procedere attraverso uno stato di transizione a sedia.
Inoltre la struttura ai raggi X del composto 3.17 aveva rivelato che il gruppo N-tosilico
è orientato in maniera da minimizzare le interazioni repulsive con il sostituente assiale in C(2)
Entry R1 Acido di Lewis/ Nucleofilo R2 Resa
a CH2Ph Me3Al (1.1 eq) Me 85% b i-Pr Me 3Al (2 eq) Me 80% c i-Bu Me 3Al (2 eq) Me 99% d e CH2Ph Et2AlCl (1.1 eq) Et 86% f i-Pr Et 2AlCl (1.1 eq) Et 99% g i-Bu Et 2AlCl (1.1 eq) Et 99% h i CH2Ph SnCl4/ TMSCH2CH=CH2 i-Pr SnCl4/ TMSCH2CH=CH2 i-Bu SnCl 4/ TMSCH2CH=CH2 CH2CH=CH2 CH2CH=CH2 CH2CH=CH2 99% 99% 89%
27 N R1 S O O attacco β attacco α Nu Nu 3.18 3.19 17
Figura 3.2. Faccia α dell’intermedio 3.18 stericamente meno ingombrata.
e il trasferimento ragionevole di questa situazione all’intermedio 3.18 indicava come la faccia α di questo intermedio fosse, per un attacco su C(6), stericamente meno ingombrata.
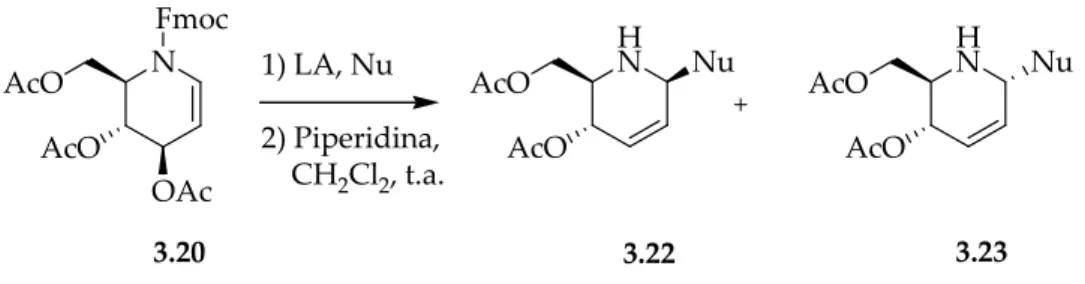
Nel 2003 Dransfield e coll. hanno applicato la reazione di Ferrier all’imminoglicale
3.2012, che non solo presenta l’insaturazione adiacente all’atomo di azoto, ma è caratterizzato da tre gruppi ossidrilici protetti. Ciò rende questo composto un analogo azotato del 3,4,6-tri-O-acetil D-glucale 3.21.
Gli imminoglicali presentano infatti analogie con i glicali, carboidrati caratterizzati dalla presenza di un doppio legame tra C(1) e C(2) e importanti building blocks per la sintesi di monosaccaridi, oligosaccaridi ed altre molecole organiche enantiomericamente arricchite.
Il glucale 3.21, per esempio, per la presenza del doppio legame adiacente all’ossigeno endociclico e di un buon gruppo uscente in C(3), è suscettibile, come abbiamo precedentemente detto, a reazioni che permettono l’introduzione di un’ampia gamma di nucleofili sul C(1) del nucleo zuccherino con concomitante migrazione del doppio legame (riarrangiamento di Ferrier). N Fmoc OAc AcO 3.20 AcO 1 2 5 O OAc AcO 3.21 AcO 1 2 5
28
Analogamente l’imminoglicale 3.20 può reagire con nucleofili in posizione α, formando nuovi legami C-C grazie alla presenza di un acido di Lewis con spostamento del doppio legame in posizione C(2) e C(3) ed uscita del gruppo acetato in posizione allilica C(3) (Schema 3.9).
Come si può notare dai dati riportati nella Tabella 3.2, la reazione di 3.20 con allil trimetil silano e BF3.Et2O (entry 1) effettuata a -50°C, aveva condotto, dopo deprotezione di
Fmoc, al composto 3.22a con una resa del 78%. Era presente, in piccola percentuale, anche l’anomero α 3.23a che risultava essere facilmente separabile.
In maniera analoga era stata effettuata su 3.20 l’addizione di dietil zinco a -20°C in presenza di BF3.Et2O (entry 2) e dell’1-fenil-1-(trimetilsilossi)etilene a -45°C (entry 3). Come
prodotti principali della reazione erano stati ottenuti 3.22b e 3.22c. Inoltre, l’addizione di tipo Prins del metilencicloesano mediata da SnBr4 a temperatura ambiente (entry 4), aveva portato
alla formazione di 3.22d con resa eccellente.
Tabella 3.2. Addizioni all’imminoglicale 3.20 catalizzate da un acido di Lewis
In tutti questi esempi il prodotto maggiormente formato era la 1,5-cis piperidina 3.22, ovvero l’anomero β. È interessante notare come l’andamento stereochimico sia esattamente
Entry Condizioni β : α Prodotti (% resa) 1 2 3 4 H2C=CHCH2SiMe3, BF3.Et 2O 79 : 21 3.22a (78) ; 3.23a (18) Et2Zn, BF3.Et2O 67 : 33 3.22b (63) ; 3.23b (27) H2C=C(OSiMe3)Ph, BF3.Et2O 62 : 38 3.22c (64) ; 3.23c (31) metilencicloesano, SnBr4 86 : 14 3.22d (80) ; 3.23d (10) N Fmoc OAc AcO H N AcO H N AcO Nu Nu 3.22 3.23 3.20 1) LA, Nu 2) Piperidina, CH2Cl2, t.a.
AcO AcO AcO
29
contrario a quello osservato quando gli stessi nucleofili sono aggiunti in condizioni analoghe al 3,4,6-tri-O-acetil D-glucale 3.1. Nei corrispondenti composti ossigenati, infatti, il prodotto presente in maggiore quantità è l’1,5-trans, ovvero l’anomero α.
La reazione dell’imminoglicale 3.24, epimero di 3.20, con dietil zinco in presenza di BF3.OEt2, aveva portato, dopo rimozione del gruppo Fmoc con piperidina in CH2Cl2, alla
formazione delle due tetraidropiridine 3.25 e 3.26 in rapporto 58:42 (1H NMR) con una resa complessiva dell’80% (Schema 3.10).
Schema 3.10 Reazione dell’imminoglicale 3.24 con dietil zinco per dare le tetraidripiridine diasteroisomere 3.25 e 3.26.
Quindi, in maniera analoga a quanto visto per l’epimero 3.20, anche l’imminoglicale
3.24 forniva in prevalenza l’addotto 1,5-cis.
L’assegnazione della stereochimica dei composti 3.22, 3.23, 3.25, 3.26 era stata effettuata in base ad esperimenti NOE.
Anche in questo caso era stato ipotizzato che l’intermedio chiave nella reazione di formazione di legami C-C dell’imminoglicale fosse uno ione N-acil imminio.
Per quanto riguarda la regioselettività, in tutte le reazioni mostrate nella Tabella 3.2 e nella reazione rappresentata nello Schema 3.10, l’attacco nucleofilo avveniva completamente in modo regioselettivo sul C(1) dello ione imminio 3.27b (Schema 3.11), mentre non erano stati isolati prodotti derivati dall’attacco sul C(3). Tale risultato regiochimico veniva razionalizzato sulla base di una preferenza cinetica per l’attacco sulla zona di più bassa densità elettronica dello ione coniugato N-acil imminio.
Per quanto riguarda la diastereoselettività, si formavano preferenzialmente i composti 1,5-cis disostituiti, fatto già riscontrato negli studi prima riportati.
Come di consueto l’addizione del nucleofilo a quella che sembra essere la faccia più impedita del substrato veniva razionalizzata proponendo la presenza in soluzione di due conformazioni, 3.27a e 3.27b, dell’intermedio ione N-acil imminio (Schema 3.11). La
N Fmoc OAc AcO H N AcO H N AcO Et Et 3.25 (58%) 3.26 (42%) 3.24 a) Et2Zn, BF3.Et 2O b) piperidina, CH2Cl2, t.a.
30
repulsione sterica tra il gruppo acetossimetilico in C(5) e il gruppo N-Fmoc sfavorisce il conformero 3.27a rispetto a 3.27b. Il successivo attacco del nucleofilo su 3.27b é ancora una volta controllato da fattori stereoelettronici ed avviene dalla faccia β del sistema che corrisponde ad un più favorevole attacco pseudoassiale, con ottenimento, dopo deprotezione del gruppo Fmoc, del diastereomero cis 3.22.
Dransfield e coll. hanno applicato la procedura descritta nello Schema 3.8 per sintetizzare un prodotto naturale, la (+)-desossiprosofillina, e C-glicosidi di imminozuccheri.
La (+)-desossiprosofillina 3.29, un derivato della (+)-prosofillina, prodotto naturale isolato da Prosopis africana, è infatti sintetizzabile facendo uso della reazione di allilazione in
N OAc OAc OAc O OR BF3.Et 20 (R = 9-fluorenilmetil) Nu -AcO -N OAc RO O OAc N RO O OAc OAc Nu -N OAc OAc O OR Nu -3.27a 3.27b Nu 5 N H AcO AcO 1) BF3.Et 2O, CH2Cl2, -60°C 0°C H2C=CHCH(SiMe3)(CH2)8CH3, 2) piperidina, CH2Cl2, t.a. N H AcO AcO 3.28 3.29(+)-desossiprosofillina N Fmoc OAc AcO 3.20 AcO
Schema 3.11. Conformazioni dello ione N-acil imminio 3.27 e 3.28.
31
C(1) dell’imminoglicale 3.20 con trimetil-(1-nonil-allil)-silano e BF3.OEt2 a -60°C (Schema
3.12).
La sintesi di C-glicosidi di imminozuccheri può avvenire grazie al fatto che le piperidine sostituite sul C(1) formate a partire da 3.20 (3.30 e 3.31 nello Schema 3.13) mediante la procedura descritta, possono essere facilmente trasformate in imminozuccheri C-glicosidici per diidrossilazione stereocontrollata del doppio legame C-C (Schema 3.13).
Un altro studio simile a questo ha previsto l’utilizzo di eterocicli azotati contenenti un carbonile, come gli N-tosil-6-alcossi-2,6-diidro-1H-piridin-3-oni 3.35, preparati a partire dal composto 3.34 (Schema 3.14).13
Anche i composti 3.35a e 3.35b reagivano con allil-silani sul C(6) in presenza di un acido di Lewis (BF3.OEt2) con rese quasi quantitative, grazie alla formazione di uno ione
N-N O n-Bu OR2 Ts N O n-Bu Ts OH R2OH H2SO4 (cat) 3.34 3.35a: R2 = Et 35b: R2 = iPr 2 6 N OAc AcO AcO Fmoc BF3Et20, Et2Zn, CH2Cl2, -20°C N AcO AcO Fmoc Et N AcO AcO Fmoc Et N AcO AcO H Et N AcO AcO H Et OAc OAc OAc OAc 3.20 3.30 3.31 3.32 (43%) 3.33(19%)
Schema 3.13. Sintesi di C-glicosidi di imminozuccheri a partire da 3.20.
32
sulfonilimminio come intermedio. Le reazioni portavano all’ottenimento del solo diastereomero 2,6 cis disostituito.
Tabella 3.3. Reazioni di 3.35 con allil-silani in presenza di un acido di Lewis
Come si può osservare dalla Tabella 3.3, il prodotto ciclopentenil sostituito 3.36b (entry 1) si formava in rapporto 9:1 in favore del diastereomero cis in relazione al nuovo stereocentro introdotto, mentre il cicloesenil diidropiridinone 3.36c veniva ottenuto esclusivamente come unico diastereomero (entry 3). Dalla reazione di 3.35a con propargiltrimetilsilano, veniva ottenuto il prodotto allenil sostituito 3.36d con un’ottima resa (entry 4). Anche il composto 3.36b reagiva in maniera analoga.
La stereochimica osservata era stata determinata grazie all’esame della struttura cristallografica di 3.36c. Dall’analisi ai raggi X era stato determinato che tale composto ha stereochimica cis e, per analogia dei dati spettrali di questo composto con gli altri, anche a tutti i prodotti 3.36a-d della Tabella 3.3 era stata attribuita la stessa configurazione relativa cis tra i sostituenti in C(2) e in C(6).
Entry Nucleofilo Prodotto
1 3.35a TMS N O n-Bu 3.36a (99%) 2 3.35a TMS 3.36b (82%) N O n-Bu Ts Ts H H 3 3.35a 3.36c (79%) TMS N O n-Bu Ts H H 4 3.35a 3.36d (98%) N O n-Bu Ts TMS 5 3.35b TMS 3.36a (98%) Composto Resa
33
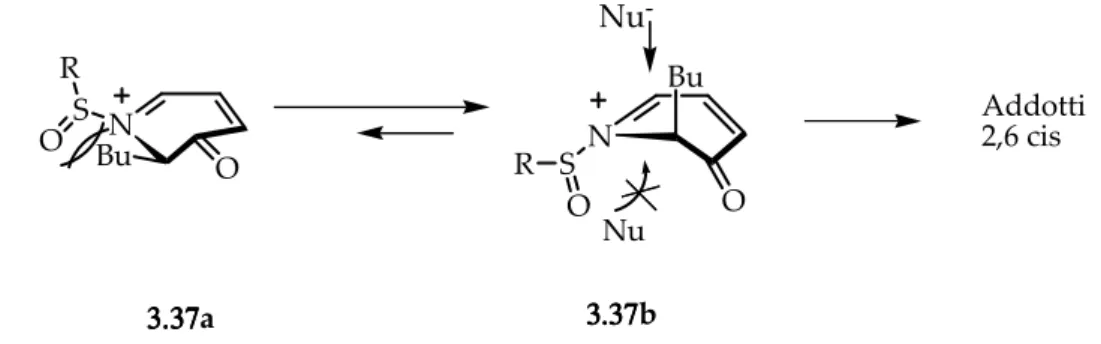
Per razionalizzare la preferenza dei prodotti 2,6-cis-disostituiti veniva considerata nuovamente la tensione 1,3 allilica generata tra il gruppo tosilico e il gruppo butilico. A causa di questa tensione, il butile deve adottare un orientamento pseudoassiale con il gruppo tosilico che va a schermare la faccia opposta della molecola. L’attacco nucleofilo sull’intermedio
3.37, lo ione N-sulfonilimminio nella conformazione 3.37b , non può avvenire dalla faccia
verso cui si dirige il gruppo tosile, a causa dell’ingombro sterico di quest’ultimo. Per questo l’attacco avviene dalla parte del gruppo butilico, e ciò porta alla formazione dei prodotti cis.
.
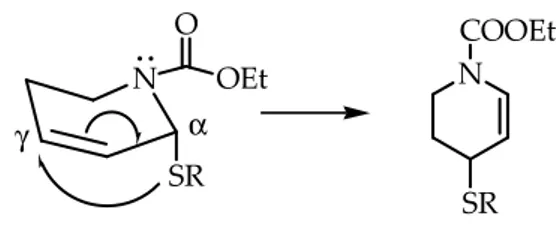
Da ricordare, infine, anche il lavoro di Kozikowski che nel 1990 ha esaminato il comportamento del composto 3.38 con diversi nucleofili (C-, O- ed S-nucleofili) in presenza di acidi di Lewis, variando anche la temperatura e il solvente della reazione stessa.14
Le reazioni effettuate conducevano essenzialmente alla tetraidropiridina α-sostituita 3.39 accompagnata da una piccola percentuale dell’isomero γ-sostituito 3.40 (Schema 3.15), la cui presenza era ben evidente dall’esame 1H NMR del grezzo di reazione.
Utilizzando diversi C-nucleofili il prodotto maggiormente o unicamente ottenuto era quello α-sostituito e ciò poteva essere razionalizzato pensando che un attacco in posizione α
N OH COOEt LA, N COOEt Nu N COOEt Nu Nu 3.38 3.39 3.40 3333....33337777a 3333....33337777b Addotti 2,6 cis N S R O Bu N S R O Bu Nu -Nu O O
Figura 3. 4. Attacco pseudo assiale, stereoelettronicamente favorito, del nucleofilo sulla conformazione 3.37b dello ione N-sulfonilimminio.
34
fosse cineticamente favorito in quanto coinvolgeva la zona di minore densità elettronica di uno ione imminio intermedio di reazione, in analogia ad un corrispondente attacco nucleofilo su uno ione piridinio.
I prodotti γ-sostituiti venivano isolati solo quando veniva fatto uso di nucleofili, quali il tiofenolo e l’etanolo, che sono caratterizzati da un eteroatomo.
In questi casi, infatti, era stato notato che pur potendo avvenire un attacco in α, alcuni fattori favorivano la migrazione dell’S- e dell’O- nucleofilo sulla posizione γ, quali, ad esempio, a) l’indebolimento dell’effetto anomerico causato dell’interazione del lone pair dell’azoto endociclico con il sostituente etossicarbonilico, b) l’intervento di possibili sfavorevoli interazioni di non legame dei sostituenti in relazione 1,2 tra di loro, c) una generale stabilizzazione del sistema enammidico risultante dal processo di riarrangiamento.
L’insieme di questi fattori contribuiva alla formazione del prodotto γ-sostituito.
Figura 3. 5. Migrazione dell’S-nucleofilo sulla posizione γ.
È interessante notare come, mentre negli altri studi sull’addizione di nucleofili ad imminoglicali presenti in letteratura e sopra riportati, gli unici nucleofili usati siano stati i C-nucleofili, in quest’ultimo caso siano state effettuate reazioni anche con O- ed S-nucleofili.
N SR COOEt N SR α γ O OEt