CHAPTER 1
Introduction
The use of radiolabelled bioactive molecules and Molecular Imaging (MI) for diagnostic purposes and clinical or preclinical research is gaining importance in the field of medicine and pharmaceutical sciences. PET (Positron Emission Tomography) is an emerging MI modality widely adopted in Nuclear Medicine to detect in vivo biodistribution of positron emitting tracers and relate their fate to diagnostic conclusion; in particular a wide range of applications has been developed for the diagnosis, staging and therapy response of cancer.
18F, with its half-life of approximately 110 minutes, has gained a prominent role among the positron-emitting radionuclides in many imaging studies and diagnostic procedures. This radionuclide needs to be incorporated into bioactive molecules rapidly, efficiently, and in such a way that the 18F-labelled product retains biological activity. These issues may pose different problems ranging from available chemistry routes, to adequate substrates and/or precursors, to the obvious necessity of adopting technical solutions to prevent radiation exposure of personnel involved in the production and risk of radioactive pollution
Computer-controlled automation of the synthetic process of PET radiopharmaceuticals is then strongly desirable, in particular during scale-up of activity and routine operation, when the level of radioactivity is too high to be compatible with direct equipment handling or source manipulation. Automation is also expected to improve process reliability and performance optimization.
Specific expertise and know-how on radiochemistry practice, chemical step automation and on available technologies and methodological approaches have been acquired during this work. Thiey have developed from learning how to handle unsealed positron-emitting sources, produced at the IFC-CNR Cyclotron, to application of basic radiochemistry of short-lived
radionuclides and finally to the use, maintenance and exploitation of a full automated processing unit, that is used for the routine production of the radiopharmaceutical 18F-2-fluoro-2-deoxyglucose (FDG). Furthermore, due the fact that such an automated unit is installed in a GMP (Good Manufacturing Practice) licensed site for radiopharmaceutical manufacturing at the IFC CNR all the above has to comply with quality assurance principles in the form of validated procedures and Standard Operative Procedures (SOPs).
The experience with the fully automate unit (TRACERlab FxFDG) has been functional to learn the features that are necessary for any automated component to conform with, official standard for radiopharmaceutical production as well as the strong or weak points of such an approach.
This experience is being transferred to a radiochemistry procedure set up during the earlier period of the doctorate school, and still developed at the PET-Cyclotron and Radiopharmaceutical Chemistry Dept of the Institute of Clinical Phisiology of the CNR in Pisa. This procedure is aimed at converting the manual radiosynthesis of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) and its conjugation to a di-Boc-protected insulin to a PC-operated, remote-controlled process.
This part of the project will require further investment allocation and, although still ongoing, it should be considered outside the scope of the present work
1.1. Positron Emission Tomography
Positron emission tomography (PET) is a nuclear medicine imaging modality that employs labeled molecules to examine metabolic functions and their disorders and to probe biological targets in living subjects. A recently developed application focus on the use of PET to unravel interactions between, radioctive probes, namely novel drugs or bioactive molecules, and their biological targets. A major expectation is also the use
morphological changes in humans or animal models of disease (Steven R. Meikle et al, 2006; H. Herzog, 2006) so as to be able to earlier detection and more effective treatment.
The method is absed on the use of unstable (radioactive) nuclides that decay a stable non-radioactive state by emission of a positron (positive electron) from the nucleus. Radionuclides are used because the detection occurs at extremely high efficiency and sensitivity; therfore, the fate of radiolabelled compounds (molecules that have been incorporated into their chemical structure with the radionuclide) can be followed in very small quantities as low as being able to avoid any risk of toxicity or pharmacological effect. They are then known as radiotracers or radiopharmaceuticals, although no drug effect is expected from their use. PET data are represented in the form of images, similar in the appearennce to radiological images. They may display bidimensional (2D) or tridimensional (3D) pictures of the tracer distribution throughout the body. The physical signal used to create those images are represented by the two 511 keV gamma rays released from each event of radioactive decay of a positron emitting nucleus (annihilation radiation)(Fig. 1). These photon couples are measured by simultaneous activation of two opposite detectors (Z.H. Cho et al, 1975). Beside the peculiary in the physical principle, PET has some further advantages over the other imaging techniques: (a) increased sensitivity, which leads to improved temporal resolution, and hence the opportunity to acquire dynamic data (b) accurate and straightforward methods of attenuation and scatter correction, making the technique quantitative (c) higher spatial resolution, in the range of 3–10 mm) (Azeem Saleem et al, 2000).
Fig.1 : Annihilation
A PET study begins with the administration of the radiopharmaceutical, usually by intra venous injection, seldom by subcutaneous injection or inhalation. The scan may start with the administration (dynamic study) or begin after a delay ranging from few minutes to about one hour when a delay time has to be cosidered, according to the biochemical processes involved, to allow transport to and uptake by the organ of interest.
Upon decay, the radioisotope emits a positron, which travels a short distance in the matter before loosing its kinetic energy (thermalisation) and annihilating with an electron. This annihilation converts the two masses of the electron in the corrisponding amount of energy and two high-energy photons (511 keV) leave the point of annihilaition and propagates in nearly opposite directions. This two photons can be detected by two opposite detectors that are operated using a short (~10 ns) time window (coincidence detection) and recognised as a positron decay event (called a true coincidence if neither photon is scattered) that has occurred along the line connecting the two detectors (sometimes referred to as a line-of-response (LOR)).
Summing up many such events results in quantities that approximate line integrals and that can be processed mathematically to calculate the map (intensity vs topography) of the radionuclide distribution. The validity of this approximation depends, of course, on the number of counts collected. PET scanners are usually in the shape of an extensive numeber of detectors arranged in multiring devices and the subject to be scanned layes on a bed sliding inside the rings and through the portion of space where the detectors operates (Field of View, FOV).
During the scanning the subject is surrounded by a cylindrical ring of detectors with a diameter of 80-100 cm and an axial extent of 10-20 cm. The detectors are shielded from radiation from outside the field of view by relatively thick, lead end-shields. The most critical components of a PET camera are the detectors (M. Dahlbom et al, 1988). A more commonly used configuration is shown in figure 2. In these detectors a rectangular bundle of crystals, a block, is optically coupled to several PMTs. When a photon interacts in the crystal, electrons are moved from the valence band to the conduction band. These electrons return to the valence band at impurities in the crystal, emitting light in the process. Since the impurities usually have metastable excited states, the light output decays exponentially at a rate characteristic of the crystal. The ideal crystal has high density so that a large fraction of incident photons scintillate, high light output for positioning accuracy, fast rise-time for accurate timing, and a short decay time so that high counting rates can be handled.
PET scanners may be properly calibrated, using sources having known activity and spatial geometry, and PET images produced in those circumstances give quantitative measurements of the concentration of the radionuclide within the FOV. Most current scanners use bismuth-germanate (BGO), which generates approximately 2500 light photons per 511 keV photon, and has a decay time (i.e., time-constant) of 300 ns. One such block, for example, couples a 7x8 array of BGO crystals to four PMTs where each crystal is 3.3 mm wide in the transverse plane, 6.25 mm wide in the axial dimension, and 30 mm deep. The block is fabricated in such a way that the amount of light collected by each PMT varies uniquely depending on the crystal in which the scintillation occurred. Hence integrals of the PMT outputs can be decoded to yield the position of each scintillation. The sum of the integrated PMT outputs is proportional to the energy deposited in the crystal. The goal of image reconstruction is to recover the radiotracer concentration from the measurements. Retroprojection of all detected coincidence lines resulting from the radioactive events permits to build a 3D volume from where are extracted tomographic plans, for the whole body or studied organ (brain or heart, for example).
The dynamic response capability of the PET scanner can be exploited to yield in vivo pharmacological data. In fact, the values of radioactive concentration per image pixel can further processed considering that the kinetics of the pharmaceutical can be modeled as a linear dynamic system with the arterial concentration of radio-isotope in the blood as the input and the PET measurement as the output. The state variables are the concentrations in different compartments of the tissue, where examples of compartments would be blood, the interstitial space between cells, and the intracellular space. In general, compartments are virtual spaces and need not be related to physical spaces; they may represent, for example, bound and unbound states of the radiopharmaceutical.
The exchange rates between the compartments are parameters of the models. Acquiring a series of images sequentially after injection yields a time-course of the sum of the quantity of tracer in each compartment, i.e., of the output of the model, which can be used to estimate the model’s parameters. These parameters can then be used to calculate physiological parameters of interest, such as blood flow, glucose metabolism, receptor binding characteristics, etc.
Thus, PET can be used for precise quantitative measurements of specific physiological quantities and complex mathematical models and image processing can be incorporated in the form of user-friendly software that converts raw data into quantitative data of units more familiar to end users such as: regional blood flow, regional substrate consumption, maximum binding, distribution volume, affinity and/or density of receptors. (Didier Le Bars, 2006).
1.2. Selection of a radiopharmaceutical
One prerequisite to performing an investigation with positron emission tomography (PET) is the availability of a suitable radiopharmaceutical. The selection, preparation, and preclinical evaluation of a new radiopharmaceutical is addressed by the field of radiopharmaceutical chemistry. The molecular mechanisms underlying PET measurements are receptor binding (either peripheral or cerebral, intracellular, or at the membrane surface), metabolism (enzyme activity or measurement of the resulting trapping or clearance of the radioactivity), transport across the cell membrane (carrier systems, efflux pumps), and binding to several kinds of macromolecules (DNA, RNA, proteins). In the ideal case, radiopharmaceuticals interact only with the target molecules of interest and no nonspecific accumulation is observed. However the ideal radiopharmaceutical does not exist. The development of any radiotracer for a PET study begins with the selection of an appropriate radionuclide. This becomes especially difficult when there exists multiple synthetic
precursors that can allow chemists to produce the same, or nearly the same biomolecule, but with a different radioactive tag. Several factors can influence one's decision in this regard. The first is whether the physical half-life of the radioisotope matches the biological half-life of the process under investigation. Thus, the time necessary to prepare the synthetic precursor, and manipulate it through the subsequent synthetic pathways, and/or purifications can weigh heavily on one's decision. Finally, the nature of the information one is seeking from the PET measurement also plays an important role in the selection of the radioisotope. Whether one is seeking spatial distribution and regional concentrations of a target substance or neurotransmitter binding or uptake site, or whether one is seeking to assess bioactivity relying on metabolitic breakdown of the tracer could impact on this selection. ( Philip H. Elsinga, Radiopharmaceutical chemistry for positron emission tomography). Some common criteria, not dependent on the PET application of the radiotracer, are:
<A high affinity for its target is required to obtain high-contrast PET images. The interaction between the radiopharmaceutical and its target molecule must be the major mechanism underlying accumulation of the radioactivity in the target tissue.
<A high specificity for the target molecule is necessary because interaction of the radiotracer with other types of molecules will interfere with the desired radioactive signal detected by the PET camera.
<Radiolabeling opportunities are required. It is obvious that a selected compound should be amenable to radiolabeling. The labeling procedure needs to generate a radiopharmaceutical with favorable radiochemical yield. Sufficient amounts for human PET studies should reliably be prepared. Parameters that have to be considered are: position of labeling within the molecule (dependent on its metabolic behavior), expected
radiochemical yield and reaction time, specific activity, and choice of radioisotope.
<The distribution of the radiopharmaceutical within the body must be related to the physiological response to measure functionality of the biochemical process under investigation
<Rapid metabolism of a radiopharmaceutical is generally undesirable. Radiolabeled metabolites can bind to other molecules or take part in unknown biochemical processes and result in nonspecific accumulation of radioactivity. Furthermore metabolism complicates the assessment of the arterial input function that is used in tracer kinetic models. If it can be anticipated that extensive metabolism will occur, the contribution of metabolites to the PET signal in various tissues should be determined. In some cases, metabolism of the radiotracer is the mechanism underlying the accumulation of radioactivity in tissue. Radiotracers like [2-18
F]fluoro-2-deoxy--glucose (FDG), [3-18F]fluoro-3-deoxy--thymidine (FLT) (A.F.
Shields et al, 1998), and 9-[(1-hydroxy [3-18
F]fluoro-2-propoxy)methyl]guanine (FHPG) (S.S. Gambhir et al, 1998), (E.F. de Vries et al, 2000) enter the cell and are phosphorylated by kinases. The corresponding monophosphates are irreversibly trapped in the cytoplasm. Radioactivity uptake reflects enzyme activity, which is the rate-limiting step of the biochemical process of interest. ( E.K. Pauwels et al, 1998), (J. Krivokapich et al, 1987).
<The lipophilicity of a radiotracer is an important parameter, since it determines the ability of the molecule to cross cell membranes and barriers like the blood–brain barrier (BBB). The lipophilicity is usually expressed as the partition coefficient between n-octanol and water (log P). For optimal passage of lipid bilayers a log P >1.5–2 is required. Higher
values of log P result in high nonspecific binding caused by hydrophobic interactions with lipids and proteins.
<Clearance of nonspecifically bound radioactivity within the time scale of measurement for PET is necessary to discriminate between specific and nonspecific uptake. This parameter is especially relevant for radiolabeled macromolecules like proteins or antisense oligonucleotides. These large molecules slowly diffuse into the cell and only a small portion bind to the target of interest. The radiolabeled molecules that do not bind specifically must leave the cell again and be cleared from the circulation.
1.3. Traditional PET isotopes.
There are four positron-emitting radioisotopes which are used more than any others. These are fluorine-18, carbon-11, nitrogen-13 and oxygen- 15. The reason these are so commonly used is that they can be easily substituted directly onto biomolecules. C-11, N- 13 and 0-15 are the "elements of life". Substitution of carbon-1 I for carbon- 12 does not significantly alter the reaction time or mechanisms of a molecule. A similar situation exists for nitrogen- 13 and oxygen-15: they can be substituted for their stable counterparts without influencing the bioactivity of the molecule. While fluorine-18 is not a significant element in living systems, its half-life and properties makes its use in labeling of considerable value, so fluorine-18 can often be substituted for a hydroxyl group on a molecule or placed in a position where its presence does not significantly alter the biological behavior of the molecule.
Another topic of importance in the preparation of radioisotopes is that of specific activity. It is important in several applications and particularly important in PET where the radionuclide is incorporated into a radiotracer which is used to probe some physiological process in which very small amounts of the biomolecule are being used. PET is basically a tracer
process without perturbing that process. If the amount of radiotracer is very small in comparison to the amount of the native compound or its competitor, then the process will be perturbed very little. When carrying out such studies as probing the number of receptors or of the concentration of an enzyme, these considerations become even more important (Dannals RF et al, 1991).
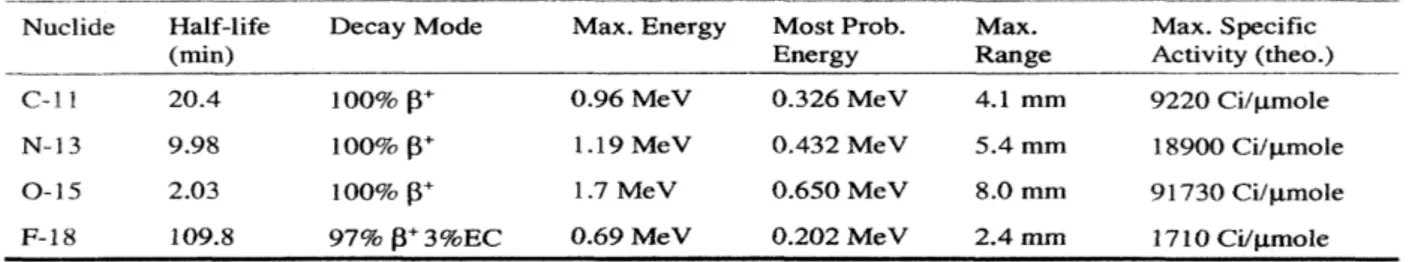
The usual way to express the concept of specific activity is in term of the amount of radioactivity per mole of compound. There is, of course, an ultimate limit and this is when there are nothing but the radioactive atoms or radiolabeled molecules and there are no unlabeled compounds. This condition of the radionuclide (or radiotracer) is defined as carrier-free status; similarly, carrier-added means a known amount of the unradioactive element or product has been added and no-carrier-added refers to a situation where no intentionally addition has been performed. A table of the characteristics of the four PET isotopes is given in table 1: (Fowler JS and Wolf AP, 1982).
Table 1: Characteristics of C-11, N-13, O-15, F-18
1.4. Fluorine-18 radiopharmaceuticals
The element fluorine is in many ways inique, both in chemical characteristics and usefulness in the pharmaceutical and chemical industries. Fluorine has a very small steric size and exhibits very high carbon-fluorine bond energies; as fluorine is also extremely electronegative, such substitution can often produce significant and useful changes in physiochemical and biological properties of organic compounds. In some cases, substitution with fluorine produces a derivative with improved pharmacological properties. Although sometimes considered as
an isosteric replacement for hydrogen, the differences in electronegativity and hydrogen bonding capability of fluorine often make it more like a substitution with a hydroxyl group. Many polyfluorinated organic compounds exhibit unusual and important properties (i.e. Teflon and Freons), and the substitution of organic pharmaceuticals with fluorine has a long and successful history, and is currently widely practiced.
Fluorine-18 is no less a remarkable and versatile positron-emitting radionuclide and is readily available form both particle accelerators and nuclear reactors using a wide variety of nuclear reactions. Decay of fluorine-18 is largely by positron emission (97%), and the relative long half-life of 109.7 minutes permits centralized synthesis of fluorine-18 labeled radiopharmaceuticals and provides opportunities for extended imaging protocols, sometimes continuing as much as 6 to 10 hours.
Although discovered as early as 1937, synthetic applications of fluorine-18 for many years lagged behind radiochemical applications of carbon-11, largely due to the difficulties in fluorination of organic molecules. Most of the methods for labelling with fluorine-18 have thus been developed and improved in the last two decades, along with fluorine-18 labeled radiopharmaceuticals used for human studies. A list of fluorine radiotracers reported for human use is given in table 2 below along with the most used method of synthesis.
The list of fluorinated radiotracers/radiopharmaceuticals is continuosly increasing and new compounds are adding year after year none of them has paralleled the success and use of 2-fluoro-2-deoxyglucose FDG. FDG is the mostly used tracer worldwide due to its application in cancer detection; however, it is also a fundamental tracer in the assessment of disregulation of energetic substrate utilisation such as in diabetes and insuline resistance. Production, availability and use of this tracer are hence key issues in the overall economy of the research project that includes also the synthesis and the use of FB-insulin.
1.5. Design and synthesis of 2-deoxy-2-[18F]fluoro-D-glucose (18FDG)
18F-FDG was modeled after carbon-14 labeled 2-deoxyglucose (14C-2DG). 2
Deoxy-D-glucose (2-DG) is a derivative of glucose in which the hydroxyl group (-OH) on C-2 is replaced by a hydrogen atom (Fig. 3)
Fig.3: Comparison between D-glucose and its derivatives
The biological behavior of 2-DG is remarkably similar to glucose, with a few important differences. Like glucose, 2-DG undergoes facilitated transport into the brain followed by phosphorylation by hexokinase because the hydroxyl group on C-2 is not a critical element for either of these processes. In contrast to glucose, however, metabolism does not proceed beyond phosphorylation because the hydroxyl group on C-2 is crucial in the next step, phosphohexose isomerase. As a result, 2-deoxy-D-glucose-6-phosphate is trapped in the cell providing a record of metabolism. In essence, removal of the hydroxyl on C-2 isolates the hexokinase reaction, This property of 2-DG was noted in 1954 by Sols and Crane (Sols A and Crane RA, 1954). The translation of the 14C-2-DG
method to humans required that 2-DG be labeled with an isotope which decayed by body penetrating radiation and that the chemical properties of the isotope and its position on the deoxyglucose skeleton would not significantly perturb its biochemical and transport properties. Of course, this could be achieved by isotopic substitution of stable carbon in the 2-deoxyglucose structure with carbon-11, and this synthesis was accomplished shortly after the development of 18FDG (MacGregor RR,
the C-F bond is a strong bond and because its 110 minute half-life was sufficiently long for transport from Long Island to Philadelphia where the first human studies were carried out on the Mark IV scanner (Kuhl DE, 1976). The design of an F-18 labeled version of 2-deoxyglucose aimed at substituting the F-18 on a carbon atom which would preserve the properties of the parent molecule. The choice of C-2 for the fluorine substitution was an obvious one. C-2, unlike other carbon atoms in the molecule, can be modified without interfering with either facilitated transport required to bring the molecule across the blood-brain-barrier (BBB) or the hexokinase reaction. It was also reasonable to assume that 2 deoxy-2-fluoro-D-glucose (FDG) would not be a substrate for phosphohexoseisomerase. Thus, it was predicted that 2-deoxy-2-[18F]fluoro-D-glucose would be a good substrate for hexokinase and that,
with the absence of a hydroxyl group on C-2, the phoshorylated product would be intracellularly trapped at the site of metabolism providing a record of metabolic activity which could be imaged externally. The development of 18FDG was further supported by the fact that FDG had
been synthesized in unlabeled form and shown to be a good substrate for hexokinase (Bessell EM, 1972) (Fig.4).
The importance of substituting the fluorine atom on C-2 is illustrated by the dramatic high reduction in affinity for hexokinase with 3-deoxy-3-fluoro-D-glucose and 4-deoxy-4-fluoro-D glucose. (Tab.3)
Tab.3: Hexokinase affinity for glucose and its derivatives
In order to test the hypothesis that FDG would be a good model for 2-DG, FDG was labeled with C-14 (Ido T, 1978). Autoradiographic studies with [14C]FDG in the rat gave similar results as those obtained for [14C]2-DG
and phosphorylation by hexokinase also proceeded as predicted (Reivich M, 1979). These studies formed the groundwork for developing a synthesis for 2-deoxy-2-[18F]fluoro D glucose (18FDG) for studies of brain glucose
metabolism in humans. 18FDG showed to have a high uptake in rapidly
growing tumors (Som P, 1980) as a result of enhanced tumor glycolysis (Weber G, 1977) coupled with its low body background resulted in a very high signal to noise ratio to detect tumors in the body. The low body background from 18FDG is due in part to the fact that 18FDG which is not
phosphorylated by hexokinase is excreted (Gallagher BM, 1978). This contrasts to the behavior of glucose which is not excreted due to resorption from urine to plasma via active transport across the renal tubule. The presence of a hydroxyl group on C-2 which occurs in glucose but not 18FDG is required for active transport (Silverman M, 1970). This
was not anticipated in the initial design of 18FDG for brain studies has
elevated it to the forefront as a tracer for managing the cancer patient (Coleman RE, 2000).
1.6 First synthesis of 18F-FDG for animal and human studies
The chemical reactions used for labelling of organic molecules with fluorine-18 are divided into two categories: nucleophilic and electrophilic reactions. This division is based on the use of a nucleophilic form of fluorine-18 (18F-fluoride ion) or an electrophilic form (18F-F2). Hystorically,
many important fluorine-18 labeled radiopharmaceuticals were initially prepared using electrophilic fluorination and all over the years higher yielding, higher specific activity nucleophilic routes of synthesis developed. As shown in figure 5, electrophilic fluorination refers to the addition of fluorine atoms across a double bond, producing a difluoro derivative of the parent compound.
Fig.5: Electrophilic fluorination
The first synthesis of 18F-FDG was carried out in Brookhaven National
Laboratory by Wolf et al in 1976 by electrophilic fluorination (Fowler JS, 2002). (Fig. 6)
Several improvements to the electrophilic fluorination described above were made thereafter. One of the most useful modifications was the use of acetylhypofluorite 18F-CH3CO2F. The acetylhypofluorite can be produced in
situ from 18F-F2 (Chyng Yann Sciue et al, 1982).
Fortunately, there were two syntheses for unlabeled 2-deoxy-2-fluoro-D-glucose in the chemical literature at the time of these successful studies. One of these involved the electrophilic fluorination of 3,4,6-triacetylglucal with the electrophilic fluorination reagent trifluoromethylhypofluorite (CF3OF) (Adamson J, 1970) which was used in the synthesis of [14C]FDG.
The other synthetic approach to unlabeled FDG involved the use of potassium bifluoride (KHF2) in a nucleophilic displacement reaction (Pacak
J et al, 1969). Though these synthetic schemes was not directly applicable to the synthesis of 18FDG, it was likely that elemental fluorine could be
substituted for CF3OF. This approach was successful and the fluorination
of 3,4,6-tri-O-acetylglucal with elemental fluorine represented a new synthetic route to unlabeled FDG (Ido T et al, 1977). Fortunately the methodology for producing [18F]F2 by the irradiation of a neon target
containing F2 via the 20Ne(d,α)18F reaction using specially prepared nickel
irradiation vessel had already been developed (Lambrecht R et al, 1973). Thus electrophilic fluorination of 3,4,6-tri-O-acetyl-D-glucal with [18F]F2
produced a 3:1 mixture of the F-18 labeled 1,2-difluoroglucose isomer and the 1,2-di fluoro-mannose isomers which were separated by preparative gas chromatography. The 1,2 difluoroglucose isomer was hydrolyzed in HCI to give 18FDG. The yield was about 8%, the purity was >98% and the
Fig.7: electrophilic fluorination of 3,4,6-tri-O-acetyl-D-glucal with [18F]F2
However 18FDG had never been administered to a human subject either in
labeled or in unlabeled form, so there was no adequate safety data to support administration to humans. Therefore toxicity studies were performed in mice and dogs with unlabeled FDG (Som P et al, 1980). Neither mice nor dogs that received FDG showed any gross or microscopic differences with their respective control groups. These results indicated that the anticipated dose of 1 mg of 18FDG (0.014 mg/kg) could be safely
administered to human volunteers. This was a factor of 150 times less than that administered to dogs and 3000 times less than that administered to mice without any evidence of acute or chronic toxicity. Many attempts have been made to develop a nucleophilic substitution for the synthesis of 18F-FDG. This included the use of 18F-CsF, 18F-Et4NF, and 18F-KHF (Fowler JS et al, 2002), (Levy S et al, 1982), (Beeley PA et al,
1984). But the major breakthrough was reported in 1986 by Hamacher et al who had used Kryptofix 222TM as a catalyst (Hamacher K et al,1986).
The reaction had a consistent yield of over 50% and the reaction time was shortened to 50 min. Nucleophilic substitution is a chemical reaction involving the addition of a nucleophilic molecule (highly negatively charged molecule) into a molecule with a leaving group (electron drawing group attached to the parent molecule through an unstable chemical bond). Figure 8 is a general scheme for a SN2 nucleophilic substitution reaction.
Fig.8: nucleophilic substitution
The nucleophilic molecule has a high affinity towards the relatively electron deficient centre in the parent molecule created by the electron pulling leaving group. As a result, the nucleophilic molecule forms a covalent bond with the parent molecule and displaces the leaving group. The stereo-configuration of the parent molecule is also changed. In the synthesis of 18F-FDG, 18F ion is the nucleophile. The precursor is mannose
triflate in which the 1,3,4,6 position carbons of a mannose molecule are protected with an acetyl group and triflate is the leaving group at the 2-carbon. In the presence of Kryptofix 222TM as catalyst and acetonitrile as
solvent, 18F ion approaches the mannose triflate at the 2-carbon, while the
triflate group leaves the protected mannose molecule to form 18F-FDG.
Fig.9: 18F-FDG nucleophilic synthesis
Although synthesis of 18F-FDG can be carried out in different computer
controlled automatic synthesizers, the nucleophilic process proceeds in roughly the same stages. (S Yu, 2006)
1.7 Improvements
After all these achievements, 18F-FDG utility as a radiotracer in the
neurosciences and in the diagnosis of heart disease and cancer grew. This stimulated the investigations of different synthetic methods to improve yields and to increase availability. Other electrophilic routes were developed and nucleophilic routes were sought. The most useful of the electrophilic routes was labeled acetylhypofluorite CH3CO2[18F] which
offered advantages over [18F]F2 in terms of yield and
experimental simplicity. However, it was subsequently found that the stereospecificity of acetylhypofluorite was dependent on reaction conditions and solvent with one of the most commonly used methods giving ca. 15% of 2-deoxy-2-[18F]fluoro-D-mannose, an isomer with the
fluorine atom occupying the axial position. A reinvestigation and analysis of the product distribution from other fluorination reagents derived from elemental fluorine showed that they all produce the mannose isomer in varying amounts (Bida GT et al, 1984). The synthesis producing the most acceptable product purity involved the gaseous CH3CO2[18F] fluorination of
3,4,6-tri-O-acetyl-D-glucal in freon-11. In this synthesis, the ratio of 18FDG:18FDM was 95:5.
Tab.4: 18F-FDG synthesis routes
In addition to the production of the F-18 labeled mannose isomer, there were other limitations to the electrophilic route to 18FDG. The nuclear
reaction commonly used to produce [18F]F2 was the 20Ne(d.α)18F
reaction in a high pressure neon gas target to which a small amount of F2
gas was added (Casella V et al, 1980). At that time handling elemental fluorine, the most reactive of all elements, required special precautions. However, the major limitation was that under the best circumstances, only 50% of the label is incorporated into the product. This is also the case in the use of CH3CO2[18F] because half of the label is lost in the conversion of
[18F)F2 to CH3CO2[18F]. In terms of F-18 yield, another nuclear reaction the 18O(p,n)18F reaction, was far superior as can be seen when the cross
Fig.10: Comparison between 18O(p,n)18F and 20Ne(d,α)18F reaction
With the availability of high yields of [18F]fluoride, the development of a
high yield nucleophilic route to 18FDG became even more compelling. A
number of approaches were tried prior to 1986 and reported in the previous table. All of these had to face difficult steps including low incorporation of F-18 and difficulty in removing protective groups. Thus the electrophilic route, with its limitations, remained the method of choice through 1985. A major advance in the synthesis of 18FDG from
[18F]fluoride was reported in 1986 when it was discovered that kryptofix
[2.2.2] could be used to increase the reactivity of [18F]fluoride (Hamacher
er al., 1986). In essence, kryptofix masks the potassium ion which is the counterion of the [18F]fluoride. The reaction of [18F]fluoride with
1,3,4,6-tetra-0-acetyl -2-O trifluoromethane suIfonyl-β-D-manno-pyranose to give 1,3,4,6-tetra-O-acetyl-2-[18F]fluoro-(3-D-gluco-pyranose gives a 95% incorporation of F-18 and the overall synthesis including purification proceeds in about 60% yield. The synthesis involves 2 steps, displacement with [18F]fluoride and deprotection with HCl. This was an almost perfect
Fig.11: 18F-FDG synthesis with Kriptofix
Thus, this new method served an increasing need in the nuclear medicine and the neuroscience communities which were discovering new uses for
18FDG. It is also simple and amenable to automation and, in the 15 years
since it was reported, a number of automated synthesis modules have become commercially available (Satyamurthy N et al, 1999).
1.8 18F-FDG synthesis (1986 to present)
No major new developments have been made following this simple, high-yield nucleophilic route. However, a number of variants have been investigated to improve the displacement and the deprotection steps and considerable effort has been put into fine-tuning the reaction and to identifying impurities and contaminants which are carried through to the final product. This has become more critical with the increasing use of
18FDG in clinical practice where a pharmaceutical quality product is
required. One of the goals has been to optimize the removal of kryptofix 2.2.2 which is used to facilitate the displacement reaction. Methods have been reported for both the removal (Moerlein SM et al, 1989), ( Alexoff DL et al, 1991) and the detection (Ferrieri RA et al, 1993), ( Chaly T and Dahl JR, 1989) of kryptofix. Alternatives to the use of kryptofix 2.2.2 have been investigated in order to avoid its appearance as a contaminant in the final product. These include the use of tetrabutylammonium as the counterion (Yuasa M et al, 1997), (Brodack JW et al, 1988). Another improving method is synergistic with the use of an anion exchange resin to recover 0-18 enriched water for reuse. Alternatives to deprotection with HCl have
and reported to efficiently hydrolyze the acetylated labeled precursor in 10-15 minutes at 100 degrees thereby eliminating the need for a neutralization step in the synthesis (Mulholland GK, 1995) and also serving to remove kryptofix 2.2.2. The use of base hydrolysis in the deprotection step has also been investigated as an approach to reduce the need for high temperatures and to decrease the synthesis time. Though epimerization at C-2 is a known reaction of aldoses under basic conditions and in this case would produce 18FDM as a radiochemical impurity
(Varelis P and Barnes RK, 1996), a systematic study of the reaction conditions for basic hydrolysis determined that epimerization could be limited to 0.5% using 0.33 M sodium hydroxide below 40 degrees for about 5 minutes to avoid the neutralization step in the synthesis (Meyer G-J et al, 1999). 2-Deoxy-2-chloro-D-glucose (CIDG) was identified as an impurity during ion chromatographicdetermination of the specific activity of 18FDG preparations from the nucleophilic route (Alexoff DL et al,
1992). CIDG is produced as a competing displacement reaction with chloride ion which comes from different sources including HCl used in the hydrolysis step. The amount of CIDG can be reduced by using sulfuric acid instead of HCl for hydrolysis. The reduction of the amount of ClDG has also been an impetus for avoiding HCl in the hydrolysis step. Though CIDG does not present a toxicity problem, its presence is not desireable for the standards of pharmaceutical quality. Advances in chemistry and the remarkable properties of 18FDG have largely overcome the limitations of
the 110 minute half-life of fluorine-18 so as to avoid the need for an on-site cyclotron and chemistry laboratory. Currently 18FDG is used by many
hospitals as an `off the shelf' radiopharmaceutical for clinical diagnosis in heart disease, in seizure disorders and in oncology, the area of most rapid growth. However, its ready availability has opened the possibility to also use it in more widespread applications in the human neurosciences including drug research and development (Fowler IS et al, 1999). This is an important application because with 18FDG it is possible to determine
which brain regions are most sensitive to the effects of a given drug.
1.9 Mathematical 18F-FDG model
The autoradiographic method for measuring regional metabolic rate of glucose in the brain of rat using [14C]deoxyglucose (Sokoloff L et al, 1977)
has been modified for human studies using positron emission tomography (PET) and [18F]2-fluoro-2-deoxy-D-glucose ([18F]FDG) (Phelps et al., 1979),
(Reivich et al., 1979). The three-compartment model with four rate constants K1*, k2*, k3* and k4* is often simplified by assuming that the
dephosphorylation rate of FDG-6-phosphate in brain tissue is small enough that it can be ignored (k4*=0). At least a part of the observed k4*
may be explained by tissue heterogeneity (Schmidt K et al., 1992). Metabolic rate of glucose (MRglu) can be calculated from the equation
, where Cglu is the concentration of glucose in plasma and LC is the
lumped constant. Originally FDG studies were always analyzed using compartment model with the three or four rate constants. Only later it was found out that a graphical method, s.c. Gjedde-Patlak plot (Patlak CS et al, 1985) can be used to directly estimate the combination of the model rate constants. This combined term, uptake rate for FDG (Ki*=K1*k3*/(k2*+k3*))
is robust and very fast to calculate, and is therefore also suitable for computation of parametric images.
1.10 Lumped constant (LC)
The lumped constant of Sokoloff et al. (1977) is based on the biochemical principles of competitive substrate kinetics. It accounts for the differences in transport and phosphorylation rates between D-glucose and
2-fluoro-2-uptake rate. A common assumption in brain FDG studies is that lumped constant is uniform over the whole brain and in all subject and patient groups. Hexokinase favours glucose over FDG, and transport favours FDG over glucose. Although there are several different estimates on the normal value of LC in the brain, the estimates are always less than 1.0, representing that in normal condition the phosphorylation is the rate-limiting step in glucose uptake. In supply limited conditions (hypoglycemia and ischemia) LC increases (Crane PD et al, 1981), (Hawkins RA et al, 1981). Lumped constant is a function of the rate constants (Sokoloff et al. 1977). The variability of LC results primarily from changes in k3/k2 for
glucose and for FDG (k3*/k2*) as is expressed in below (Phelps ME et al,
1983):
Phelps et al. (1983) used values p=0.50 and q=1.67 (q/p=3.34) to estimate the changes in LC between normal and ischemic brain regions. This equation was used (Sasaki H et al, 1986) to estimate whether LC is uniform over the whole brain. This is the derived equation for LC which is independent of the model used:
, where Ki* is the unidirectional clearance from the circulation to the
metabolic compartment (Ki*=K1*k3*/(k2*+k3*)), τ is the ratio between FDG
and glucose clearances (K1*/K1), and φ is the phosphorylation ratio
between FDG and glucose (k3*/k3). The recommended LC for brain
[18F]FDG studies is 0.65, if irreversible uptake is assumed (3-parameter
model or Gjedde-Patlak plot without kLOSS), and LC=0.81, if
with kLOSS) (Wu H-M et al, 2003). In rat brain studies, lumped constant of
0.71 is recommended (Tokugawa J et al, 2007), ( Krohn KA et al, 2007).
1.11 Applications of 18F-FDG in PET
2-[18F]-Fluoro-2-deoxy-d-glucose has a wide range of possible clinical
imaging applications in neurology, oncology and cardiology. FDG PET has been widely used for research for more than 25 years (T. Ido et al, 1978). More recently, FDG has become the most widely used PET radiopharmaceutical in the clinical imaging of oncologic, cardiac and neurologic applications.
Current oncologic FDG PET applications include the following: evaluation of solitary pulmonary nodules, staging of lung cancer, staging of malignant melanoma, staging or restaging of colorectal carcinoma, staging of lymphomas, staging of head and neck cancers, staging of gastroesophageal cancer, staging of recurrent breast cancer, evaluation of thyroid cancer metastases and evaluation of response to therapy in breast cancer. Not all tumors have avid uptake of FDG; for example, prostate cancer, well-differentiated thyroid cancer and bronchioaveolar lung carcinoma may have only mild or no visible FDG uptake. Benign lesions may also have high FDG uptake such as infection, inflammation and granulomatous diseases.
The current cardiac FDG PET application is for the evaluation of myocardial viability. The clinical question of cardiac viability may occur when there is decreased left ventricular wall motion. This lack of regional myocardial contraction may be due to an area of previously infarcted myocardium, hibernating myocardium or stunned myocardium. In the case of infarcted myocardium, the tissue is scarred and reversible dysfunction is lacking. In the cases of hibernating myocardium and stunned myocardium, this tissue is still “viable” in the sense that the wall motion abnormality will resolve after successful coronary
stunned myocardium is that hibernating myocardium has severe ischemia even during the resting state, whereas stunned myocardium has normal perfusion at rest but suffers from episodes of severe ischemia during stress, conditioning it into a state of decreased function. FDG PET myocardial metabolism imaging is typically performed in conjunction with a perfusion image. This can be performed with a myocardial perfusion radiopharmaceutical such as N-13 ammonia. The purpose of the resting perfusion image is to show the regions of normal myocardial perfusion, which should also show normal myocardial glucose metabolism and uptake of FDG. More important, this perfusion scan may show a region of decreased perfusion that can represent myocardial infarction or viable tissue. The presence or absence of FDG uptake in this region of decreased perfusion determines whether the myocardium is viable or infarcted, respectively. In addition to identifying the myocardium that is still viable for revascularization, the detection of a significant region of viable myocardium has prognostic significance in that when left untreated, such patients with this condition will have increased unfavorable clinical events.
Current indications for FDG PET brain imaging include the evaluation of complex partial epilepsy and the evaluation of Alzheimer's dementia (AD) (K. Herholz and W.D. Heiss, 2004). The role of PET in the evaluation of complex partial epilepsy is to identify which temporal lobe the seizure focus is residing. The localization of the seizure origin is clinically important for the small number of patients who fail treatment with antiepileptic medications and who may be candidates for surgical resection of a portion of their temporal lobe. These patients are typically children who are under 7 years old and who are able to tolerate a brain resection with minimal residual neurologic deficit. For such an aggressive therapy, the concordance of multiple tests is important prior to brain resection. FDG PET imaging is only one of many diagnostic studies (SPECT, EEG, MRI, MEG and grid and depth electrodes) used to help
confirm the site of the seizure origin. FDG PET does not detect the actual anatomical lesion; rather, it detects a relative hypometabolism in the affected temporal lobe while the patient is in the interictal state (Carl K. Hoh, 2007).