RISULTATI E DISCUSSIONE
Estrazione di cerio(III) e lantanio(III) come
N,N
-dibutilcarbammati
Secondo il protocollo messo a punto in questi stessi laboratori per altri centri metallici (Nd, Eu, Tb, Sm)1 sono state effettuate le estrazioni dei cationi trivalenti La3+ e Ce3+ da fase
acquosa in solvente idrocarburico come N,N-dibutilcarbammati metallici.
Il legante N,N-dibutilcarbammato viene preparato in situ, facendo gorgogliare fino a saturazione (circa 30 minuti) CO2 in una soluzione di dibutilammina in eptano secondo
l'equazione (1):
CO2 + 2NHBu2 [NH2Bu2][O2CNBu2] (1)
La fase organica così ottenuta viene aggiunta a 0 °C ad una soluzione acquosa che contiene il
centro metallico all'interno di un imbuto separatore. La fase metallica è presente come LnCl3
acquoso. Anche se in studi precedenti2 era stato visto che a partire dai solfati di Cu(II) e Zn(II)
veniva ottenuta una resa di estrazione migliore rispetto ai cloruri, probabilmente per la migliore affinità dello ione solfato per la fase acquosa, nel caso dei lantanidi questa opzione non era possibile per la bassa solubilità in fase acquosa dei solfati3. Sembra rilevante far
notare che al contrario di quanto accade per altri centri lantanidici, per il cerio non si passa facilmente dall'ossido, prodotto commerciale a basso costo, al cloruro. In effetti la ceria commerciale CeO2, come riportato in letteratura4, è una specie relativamente inerte, e non è
stato possibile discioglierla nemmeno in acido concentrato a caldo. La soluzione acquosa di CeCl3 è stata preparata in questo caso direttamente a partire dal cloruro eptaidrato mentre la
soluzione acquosa di LaCl3 è stata preparata per clorurazione dell'ossido.
Le due fasi vengono quindi agitate vigorosamente per pochi secondi per garantire la formazione dell'N,N-dibutilcarbammato metallico ed il suo passaggio nella fase organica, mentre il cloruro di dibutilammonio rimane completamente in fase acquosa, secondo quanto descritto dalla reazione (2) e dallo schema in figura 1:
nLnCl3(aq) + 3n[(NH2Bu2)(O2CNBu2)](org/aq) 3n[NH2Bu2]Cl(aq) + [Ln(O2CNBu2)3]n(org) (2)
Le due fasi vengono lasciate separare e, la fase acquosa sottostante (torbida per la presenza dei sali di dibutilammonio) viene scartata, mentre la fase organica surnatante, incolore e limpida, viene trasferita attraverso il collo dell'imbuto in un pallone ed infine evaporata a pressione ridotta e temperatura ambiente.
L'estrazione viene condotta da una soluzione circa 0,33 M di LnCl3 usando un largo eccesso
di ammina (rapporto molare 1:10) rispetto al rapporto molare di 1:6 richiesto per la formazione di Ln(O2CNBu2)3 (4) ed un rapporto in volume eptano/H2O di circa 2,5. Lavorare
in eccesso di ammina permette di minimizzare le reazioni di idrolisi dell'N,N-dibutilcarbammato durante il mescolamento della fase acquosa e quella organica, in quanto per azione di massa sia l'equibrio di carbonatazione (1) che quello di idrolisi (3) favoriscono la formazione del legante dibutilcarbammato, ed inoltre permette di migliorare l'efficienza del
processo di estrazione perchè in presenza di ammina e CO2 si instaurano equilibri del tipo
riportato sotto (5). In questo modo si formano specie ioniche in genere a nuclearità ridotta e più solubili nel solvente organico rispetto ai complessi neutri.
[O2CNBu2]- + H2O NHBu2 + HCO3- (3)
Ln3+ + 6NHBu
2 + 3CO2 1/n[Ln(O2CNBu2)3]n + 3[NH2Bu2]+(4)
1/n[Ln(O2CNBu2)3]n + 2x/mNHBu2 + x/mCO2 1/m[NH2Bu2]x[Lnm(O2CNBu2)3m+x] (5)
Questi derivati, in accordo con l’equilibrio (5), si trasformano facilmente nelle specie neutre per riduzione della pressione parziale di biossido di carbonio o per trattamento sotto vuoto a caldo.
Figura 1: reazioni di estrazione di cationi Ln3+ da fase acquosa a fase organica come
N,N-dialchilcarbammati.
Il solvente organico usato per l'estrazione deve presentare ovviamente bassa solubilità in acqua ed avere un punto di ebollizione non troppo elevato, in quanto il prodotto viene recuperato per evaporazione a pressione ridotta della fase organica. L'eptano presenta entrambe le caratteristiche, in particolare la solubilità in acqua dell'eptano5 è di 2,93 ± 0,20 g
eptano/106 g H2O a 20 °C.
La quantità di metallo non estratto è stata valutata per analisi gravimetrica. La fase acquosa viene trattata con HCl in modo da disciogliere completamente i sali di ammonio presenti e portata a volume in un matraccio tarato. Un’aliquota della soluzione viene prelevata a trasferita in un crogiolo tarato di Pt e calcinata a 850°C fino a peso costante. Dal peso degli ossidi ottenuti (La2O3 e CeO2) è possibile risalire alle rese di estrazione. Per il cerio la
percentuale di metallo non estratto è risultata riproducibilmente di circa il 2 %, in linea con quanto riportato per gli altri lantanidi e per Cu(II) e Zn(II)6, mentre l'estrazione del lantanio è
risultata meno efficiente; riproducibilmente circa il 35 % di metallo rimane in fase acquosa. Dopo l'estrazione la fase organica trasferita in un pallone viene evaporata sotto agitazione a temperatura ambiente e pressione ridotta abbastanza velocemente per allontanare completamente l'acqua rimasta disciolta in eptano. Sia per La che per Ce dopo l'evaporazione viene ottenuto un residuo gelatinoso incolore, in cui è presente dibutilammina, utilizzata in eccesso stechiometrico durante l'estrazione, che si allontana difficilmente per il suo alto punto di ebollizione (159°C). Il processo è velocizzato mediante lavaggi con eptano anidro e successivo trattamento sotto vuoto. Il progressivo cambiamento di consistenza del residuo, da gelatinoso a solido, in seguito ai trattamenti di stripping dell'ammina rende consistente l'ipotesi dell'esistenza di equilibri diversi tra diverse specie, ioniche e neutre (equilibrio 5),
così come verificato per altre specie carbammiche7. Ad esempio, l’equilibrio (6) è stato
M2(O2CNR2)6 + [O2CNR2]- [M2(O2CNR2)7]- M = Ti, V; R = iPr (6)
Le specie anioniche, favorite dalle basse temperature e da pressioni elevate di CO2,
presentano nello spettro IR una banda intorno a 1610-1620 cm-1, tipica di un carbammato
monodentato che non è presente per la specie neutra.
Come ulteriore esempio si può citare che la specie Mg8(CO3)2(O2CNMe2)129 risulta meno
solubile in solventi organici rispetto alla specie ionica [Me2NH2]3[Mg8(CO3)2(O2CNMe2)15],
caratterizzata cristallograficamente. L'interconversione tra le due specie dipende dalla presenza di ammina e CO2 nell’ambiente di reazione. In soluzione sono probabilmente
presenti più specie secondo gli equilibri 7, 8 e 9.
[Me2NH2]3[Mg8(CO3)2(O2CNMe2)15] [Me2NH2]2[Mg8(CO3)2(O2CNMe2)14] + [Me2NH2][O2CNMe2] (7)
[Me2NH2]2[Mg8(CO3)2(O2CNMe2)14] [Me2NH2][Mg8(CO3)2(O2CNMe2)13] + [Me2NH2][O2CNMe2] (8)
[Me2NH2][Mg8(CO3)2(O2CNMe2)13] [Mg8(CO3)2(O2CNMe2)12] + [Me2NH2][O2CNMe2] (9)
Considerata l’esperienza descritta per gli altri sistemi è possibile che la specie estratta sia una specie ionica che in soluzione può mettersi in equilibrio con una specie neutra. Questo potrebbe aiutare a spiegare la trasformazione di un residuo oleoso in un solido facilmente recuperabile per successivi trattamenti di solubilizzazione in eptano seguiti da rimozione della fase volatile.
L' N,N-dibutilcarbammato metallico così ottenuto può essere conservato a lungo all'interno di fiale chiuse in atmosfera inerte.
I prodotti recuperati sono incolori e mostrano una moderata sensibilità nei confronti di una breve eposizione all'umidità atmosferica. Lo spettro IR delle specie rimane invariato per tempi brevi, segno che l'idrolisi è lenta allo stato solido ma risultano prontamente reattivi nei confronti di acqua e acidi protici.
I prodotti recuperati sono molto solubili in eptano (e negli altri comuni solventi organici) e non cristallizzano neanche a -30°C da soluzioni concentrate (circa 1,2 M). Le specie sono state quindi caratterizzate tramite analisi elementare di metallo e CO2. Il contenuto di metallo
è determinato per analisi gravimetrica, il contenuto di CO2 viene valutato facendo reagire
un'aliquota accuratamente pesata della specie con una miscela di acido acetico/toluene e misurando lo sviluppo di gas con una buretta gas-volumetrica, come descritto precedentemente da Cotton e Calderazzo10, il cui schema è riportato nella sezione Parte
Sperimentale. In ambiente acido infatti la specie libera biossido di carbonio ed il numero di leganti carbammato coordinati è uguale al numero di molecole di CO2 liberate (10).
M(O2CNR2)n + 2nH+ → Mn+ + nH2NR2+ + nCO2 (10)
Le estrazioni sono state ripetute almeno tre volte in modo da dimostrare la loro riproducibilità sia per quanto riguarda le rese di estrazione che per la natura delle specie ottenute. Le analisi indicano per le specie recuperate le composizioni La4(CO3)(O2CNBu2)10, e Ce(O2CNBu2)3.
Per il cerio si ottiene la specie omolettica come per altri lantanidi precedentemente preparati in questi laboratori (Nd, Eu, Tb)1 11. Anche le rese di estrazione e le rese di sintesi del nostro
Tabella 1: rese di estrazione di lantanidi da fase acquosa a fase organica come specie omolettiche di formula Ln(O2CNBu2)3 .
Metallo in fase acquosa Resa di estrazione
Ce(O2CNBu2)3 2.5 % 63.5 %
Nd(O2CNBu2)3 4.0 % 67.3 %
Eu(O2CNBu2)3 1.0 % 60.8 %
Tb(O2CNBu2)3 4.0 % 75.2 %
Per quanto riguarda il lantanio la specie parzialmente idrolizzata carbonato-carbammato di composizione La4(CO3)(O2CNBu2)10 ha un precedente nella specie Sm4(CO3)(O2CNBu2)10 12.
Il lantanio ha il raggio ionico più grande della serie e normalmente numeri di coordinazione maggiori; è possibile che il numero di leganti carbammato usato nel protocollo di estrazione non sia sufficiente per la sua estrazione e condizioni la natura del prodotto. Se infatti rimane acqua nella sfera di coordinazione del metallo della specie estratta questa ragionevolmente porta ad un prodotto di parziale idrolisi.
Il fatto che il prodotto di estrazione abbia ripetutamente la stessa composizione La4(CO3)
(O2CNBu2)10, potrebbe far propendere per questa ipotesi, piuttosto che per l'ipotesi di
un'idrolisi durante il processo di estrazione, che porterebbe più probabilmente ad una serie di composti diversi e quindi ad un prodotto non a stechiometria definita.
L'estrazione dovrebbe in questo caso essere ripetuta con un rapporto più alto ammina metallo per riuscire a spostare tutta l'acqua nella sfera di coordinazione del metallo.
Questa interpretazione non sembra applicabile al centro di samario per cui altri studi sono necessari per riuscire a comprendere i risultati ottenuti.
Al fine di ottenere ulteriori informazioni sulla loro nuclearità i complessi ottenuti sono stati caratterizzati tramite HRSM (spettrometria di massa ad alta risoluzione), utilizzando una sorgente di ionizzazione APPI (fotoionizzazione a pressione atmosferica). La fotoionizzazione a pressione atmosferica è stata scelta in quanto è una tecnica soft che produce poca frammentazione, ed è quindi adatta per cercare di ottenere informazioni sulla nuclearità di un composto di coordinazione. Una soluzione in toluene e acetonitrile 1:1 del campione allo 0.02 % viene nebulizzata e ionizzata con una lampada UV (al kripton); il toluene agisce come dopante nel processo di fotoionizzazione, viene cioè ionizzato dai fotoni emessi dalla lampada UV e trasferisce la carica all'analita (ionizzazione chimica).
In realtà, nonostante la tecnica di ionizzazione APPI sia soft, non è stato comunque possibile osservare il picco dello ione molecolare. Lo spettro assomiglia notevolmente a quello degli altri N,N-dibutilcarbammati dei lantanidi1. Il picco molecolare è assente, e lo spettro è
dominato da due specie principali (figura 2): il catione [La2(O2CNBu2)5]+ (specie 1); ed il
catione [La4(O)(O2CNBu2)9]+(specie 2). La loro identità è stata confermata mediante
confronto con la massa accurata teorica (ad alta risoluzione) e con le distribuzioni isotopiche simulate. Il segnale principale è la specie 2, [La4(O)(O2CNBu2)9]+; che potrebbe essersi
generato a partire dalla specie La4(CO3)(O2CNBu2)10 per perdita di un legante carbammato e
successiva decarbossilazione del carbonato, spiegabile con la temperatura relativamente alta della sorgente (350°C), necessaria per la volatilizzazione del solvente (12).
Il catione [La2(O2CNBu2)5]+ (specie 1) potrebbe essersi formato o per perdita di carbammato
[Bu2NCO2]- dalla specie dinucleare La2(O2CNBu2)6, o ancora più probabilmente per rottura
della specie tetranucleare La4(O2CNBu2)12.
Figura 2: spettro di massa di La4(CO3)(O2CNBu2)10 ottenuto con ionizzazione APPI.
L'attribuzione dei picchi è la seguente: m/z 1138,51 [La2(O2CNBu2)5]+, m/z 2120 [La4(O)
(O2CNBu2)9]+ e m/z 2315,96 [La4(O)(O2CNBu2)10]Na+.
Non è stato possibile ottenere spettri di massa del carbammato di cerio perchè la specie è risultata poco solubile nel solvente utilizzato (acetonitrile) probabilmente per un'estesa idrolisi del prodotto dovuta a solventi non anidri comportando intasamento del capillare in fase di analisi. Lo studio deve quindi essere ripetuto.
Anche in assenza del picco molecolare appare probabile per le nostre specie una struttura tetranucleare. Questo è ragionevole perchè gli N,N.diisopropilcarbammati di lantanidi(III), precedentemente preparati in questi stessi laboratori13 mediante il metodo anidro e
caratterizzati cristallograficamente, hanno tutti nuclearità 4 (Ce, Pr, Nd, Eu, Gd, Ho, Er, Yb, Lu)14; nel caso di Tb è stato identificato nello spettro di massa un picco a bassa abbondanza
relativa corrispondente alla specie tetranucleare [Tb4(O2CNBu2)12Na]+ ed inoltre in tutti gli
spettri è presente il frammento tetranucleare [Ln4(O)(O2CNBu2)9]+ come specie principale.
Inoltre la specie neutra tetranucleare La4(O)(O2CNiPr2)10 è nota ed è stata anche caratterizzata
strutturalmente (figura 3).
Anche se la caratterizzazione IR non è molto informativa per questi complessi può essere usata come metodo veloce per controllare che le caratteristiche dei prodotti ottenuti nelle varie estrazioni abbiano le stesse caratteristiche. Gli N,N-dialchilcarbammati presentano assorbimenti molto intensi nella regione 1700-1300 cm-1 6, ma non ci sono dei criteri generali
per stabilire la struttura in base al numero e alla natura delle bande. Il numero di modi di coordinazione del legante N,N-dialchilcarbammato sono molti e generalmente in uno stesso complesso coesistono differenti modi di coordinazione: monodentati, bidentati a ponte o terminali (vedi Introduzione). Si può dire al massimo che complessi contenenti solo gruppi
carbammato a ponte o terminali bidentati non hanno bande al di sopra di 1600 cm-1, che sono
caratteristiche dei leganti monodentati7.
Sono stati registrati spettri ATR-IR per ogni campione; i risultati ottenuti sono stati confrontati con gli spettri di letteratura per le specie N,N-dibutilcarbammiche di neodimio, europio, terbio e samario, ottenute per via anidra ed estrazione (tabella 2).
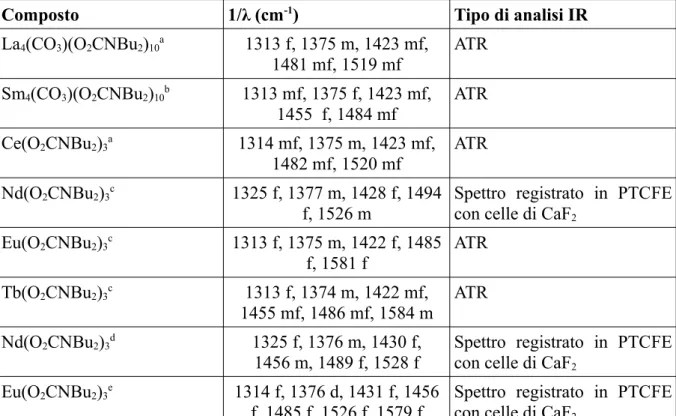
Tabella 2: principali bande IR per gli N,N-dibutilcarbammati di lantanidi noti in letteratura.
Composto 1/λ (cm-1) Tipo di analisi IR
La4(CO3)(O2CNBu2)10a 1313 f, 1375 m, 1423 mf,
1481 mf, 1519 mf
ATR
Sm4(CO3)(O2CNBu2)10b 1313 mf, 1375 f, 1423 mf,
1455 f, 1484 mf ATR Ce(O2CNBu2)3a 1314 mf, 1375 m, 1423 mf, 1482 mf, 1520 mf ATR Nd(O2CNBu2)3c 1325 f, 1377 m, 1428 f, 1494
f, 1526 m Spettro registrato in PTCFEcon celle di CaF2
Eu(O2CNBu2)3c 1313 f, 1375 m, 1422 f, 1485 f, 1581 f ATR Tb(O2CNBu2)3c 1313 f, 1374 m, 1422 mf, 1455 mf, 1486 mf, 1584 m ATR Nd(O2CNBu2)3d 1325 f, 1376 m, 1430 f, 1456 m, 1489 f, 1528 f
Spettro registrato in PTCFE con celle di CaF2
Eu(O2CNBu2)3e 1314 f, 1376 d, 1431 f, 1456
f, 1485 f, 1526 f, 1579 f Spettro registrato in PTCFEcon celle di CaF2
a)Questa tesi (estrazione). b)A. Di Giacomo, Tesi di Laurea, A.A. 2012-2013 (estrazione). c)P. Falvo, Tesi di Laurea, A.A. 2012-2013 (estrazione). d)C. Della Porta, Tesi di Laurea, A.A. 1992-1993 (via anidra). e)A. Merigo, Tesi di Laurea, A.A. 1992-1993 (via anidra).
Anche se gli spettri IR non riescono a dare informazioni sulla composizione analitica delle specie, dall'esame dei segnali in tabella si notano forti somiglianze nell'intervallo 1300-1600
cm-1. Possiamo escludere la presenza di gruppi carbammati monodentati perchè le bande si
trovano tutte ben al di sotto di 1600 cm-1, la banda più alta per la specie di lantanio è a 1519
cm-1,mentre quella più alta per la specie di cerio si trova a 1520 cm-1. D'altra parte il modo di
coordinazione monodentato appare poco probabile per complessi lantanidici in quanto non soddisfa le esigenze di ottenere alti numeri di coordinazione, tipici della serie f, ed infatti non è mai stato riscontrato in nessuna determinazione strutturale RX per le specie lantanidiche,
mentre è presente in specie con numeri di coordinazione più bassi, come Cu(O2CNBz2)2(NHBz2)2 6, con coordinazione quadrato planare e Si(O2CNR2)415(con R = Me,
Et, iPr), tetraedrico, che presenta solo carbammati monodentati ed ha una banda di
assorbimento IR intorno ai 1700 cm-1, uno dei più alti valori noti in letteratura per questa serie
di composti.
Sono stati acquisiti spettri ir lasciando il composto La4(CO3)(O2CNBu2)10 esposto all'aria per
diversi tempi (20 e 45 minuti) senza avere significativi cambiamenti nelle bande indicando che il prodotto non mostra un'elevata reattività allo stato solido per breve esposizione all'aria. Risulta difficile attribuire le bande dovute al gruppo carbonato, in quanto cadono nella zona occupata dalle bande del carbammato.
Gli spettri NMR registrati sui derivati diamagnetici di lantanio La4(CO3)(O2CNBu2)10 non
danno informazioni sul numero di leganti dialchilcarbammato diversi né sui loro modi di coordinazione, anche perchè in generale si assiste ad un rapido scambio in soluzione tra leganti terminali e a ponte19. Sfortunatamente nello spettro al 13C non si riesce a distinguere il
segnale del carbonio del gruppo carbonato. Si ottiene infatti un segnale largo centrato a 164 ppm attribuibile sia al carbonio del gruppo carbossilico che a quello del carbonato. Quindi anche la caratterizzazione NMR, quando possibile, ha offerto ben poche informazioni analitiche.
L'N,N-dibutilcarbammato di cerio(III), di composizione Ce(O2CNBu2)3, reagisce con
diossigeno anidro sia allo stato solido che in soluzione (toluene). Introducendo un'atmosfera di O2 nel recipiente di reazione dove è contenuto il composto si assiste ad un rapido
cambiamento di colore: se il derivato è allo stato solido si assiste al viraggio da incolore a giallo senape e, se si trova in soluzione, da incolore a giallo acceso.
Il derivato isopropilico Ce4(O2CNiPr2)12 descritto precedentemente in letteratura14, si ossida in
modo del tutto analogo a Ce4(O)2(O2CNiPr2)12 (figura 4).
Figura 4: strutture molecolari di A) Ce4(O2CNiPr2)12 e B) Ce4(O)2(O2CNiPr2)12, da essa
ottenuta per reazione con diossigeno.
Le due specie sono state caratterizzate strutturalmente. La specie di Ce(III) Ce4(O2CNiPr2)12 è
incolore ed ha una struttura tetranucleare lievemente diversa rispetto a quella degli N,N-diisopropilcarbammati degli altri lantanidi: è infatti meno simmetrica e leggermente più complessa. Nel caso dei lantanidi da Nd a Lu i quattro atomi metallici sono disposti ai vertici di un tetraedro distorto che possiede due assi di simmetria, nel caso del cerio invece il
tetraedro è schiacciato. Poichè Ce3+ ha un raggio ionico maggiore, le distanze Ce-O sono più
lunghe di quelle osservate ad esempio per Nd4(O2CNiPr2)12 ed il numero di coordinazione, che
nel composto di neodimio è 7 per tutti gli atomi metallici, è 7 per tre degli atomi di cerio e 8 per il quarto. Le lunghezze di legame Ce-O risultano più corte per gli atomi eptacoordinati, con un range che va da 2.32 a 2.69 Å, rispetto al cerio ottacoordinato (2.40 – 2.75 Å). Gli atomi di cerio eptacoordinati, come per gli altri carbammati dei lantanidi, hanno una geometria di una bipiramide pentagonale distorta, mentre quello ottacoordinato è un antiprisma quadrato distorto.
Il prodotto di ossidazione è il μ-osso derivato di Ce(IV), di formula Ce4(μ3-O)2(O2CNiPr2)12 in
cui il gruppo osso è tricoordinato. La sua struttura ha la stessa architettura di U4(μ3
-O)2(O2CNEt2)12. La molecola ha un centro di simmetria così che sono presenti due coppie di
atomi di Ce equivalenti, formando una losanga perfettamente planare. Tutti gli atomi di cerio della molecola hanno numero di coordinazione 8, ma le geometrie di coordinazione delle due coppie sono leggermente diverse: una coppia ha la geometria di un antiprisma quadrato distorto, mentre l'altra coppia ha una geometria che può essere difficilmente ricondotta ad un poliedro regolare o semiregolare.
Nel nostro caso il prodotto di ossidazione è così solubile nei solventi idrocarburici saturi che non è stato possibile cristallizzarlo nemmeno da soluzioni concentrate a freddo.
Dopo ossidazione gli spettri NMR possiedono segnali più stretti rispetto a quelli del precursore, in accordo con la formazione di una specie diamagnetica.
Si ritiene che avvenga la reazione (13).
Ce4(O2CNBu2)12 + O2 Ce4(O)2(O2CNBu2)12 (13)
Il processo è possibile perché le grandi molecole tetranucleari nella struttura cristallina di Ce4(O2CNiPr2)12 lasciano canali e cavità attraverso le quali può diffondere il diossigeno
all'interno della struttura cristallina; la reazione infatti può avvenire, oltre che in soluzione anche allo stato solido. L'ossidazione da Ce4(O2CNiPr2)12 a Ce4(μ3-O)2(O2CNiPr2)12 avviene
soltanto con moderati riarrangiamenti strutturali. Si pensa che il diossigeno per prima cosa reagisca sulla superficie del solido per poi diffondere verso l'interno guidato da un gradiente di potenziale chimico.
La caratterizzazione di questo nuovo composto merita ulteriori approfondimenti, in particolare appare interessante una sua caratterizzazione strutturale.
Aggraffaggio di cerio su matrici inorganiche
N,N-dibutilcarbammati di cerio possono essere usati come precursori di catalizzatori
eterogenei16.
Come detto nell'introduzione l'ossido di cerio supportato è un valido catalizzatore di ossidazione che trova attualmente innumerevoli applicazioni nell'industria e nella vita di tutti i giorni. Metodi classici di deposizione di metalli su supporti inorganici sono la coprecipitazione e l'impregnazione. Entrambi i metodi si basano sull'assorbimento fisico: il supporto viene immerso nella soluzione del composto metallico da depositare e il metallo viene fatto assorbire o facendo precipitare il composto oppure per semplice prolungato contatto tra la soluzione e i pori della matrice. Un'altro metodo prevede l'aggraffaggio di metalli sulla matrice con formazione di legami covalenti. L'ancoraggio chimico che viene così ottenuto permette di ridurre le possibilità di lisciviazione.
Negli ossidi inorganici ExOy i centri E superficiali presentano numero di coordinazione più
basso rispetto a quelli interni. È facile prevedere che se esposti all'umidità atmosferica siano in grado di assorbire chimicamente molecole di acqua, cioè di reagire con formazione di
gruppi ossidrile superficiali.
In generale la superficie degli ossidi presenta gruppi -OH e acqua assorbita fisicamente che
può essere rimossa sotto vuoto a temperatura prossima ai 100°C17 18. Per rimuovere l'acqua
assorbita fisicamente, in modo da liberare gli ossidrili sulla superficie e renderli disponibili per l'aggraffaggio, le matrici utilizzate in questo lavoro sono state trattate sotto vuoto a lungo prima a temperatura ambiente e poi a caldo (vedere parte sperimentale).
Si possono distinguere vari tipi di ossidrili superficiali (figura 5): -gruppi ossidrili isolati (a).
-gruppi ossidrili vicinali (b). -gruppi ossidrili geminali (c).
-gruppi ossidrili a ponte μ2 (d) e μ3 (e)
Figura 5: tipi di ossidrili superficiale per ossidi inorganici ExOy.
Se l'acqua assorbita viene allontanata a temperature relativamente basse, la rimozione dei gruppi ossidrilici inizia invece a temperature alte e viene completata solo intorno ai 1000°C. La calcinazione dell'ossido ne provoca una perdita di peso dovuta alla condensazione degli ossidrili superficiali con la formazione di legami M-O-M e liberazione di acqua. Appare così possibile stimare dalla diminuzione di peso il numero di ossidrili superficiali per le varie matrici usate. Questo dato è interessante perchè dà informazioni sul numero massimo di siti reattivi per una matrice. È ragionevole però pensare che la distribuzione degli ossidrili sulla superficie possa comportare un numero di siti attivi più basso perchè alcuni ossidrili sono troppo interni o troppo vicini tra loro.
Tabella 3: contenuto in moli di OH per grammo per le matrici utilizzate in questo lavoro (per le lore specifiche vedere la parte sperimentale).
Matrice SiO2 γ-Al2O3
mol OH / g matrice 2,8 – 3,1 x 10-3 3 x 10-3
Come anticipato nell'introduzione gli N,N-dialchilcarbammati metallici sono precursori convenienti per la facilità della loro preparazione, la loro stabilità in atmosfera controllata e la loro reattività verso reagenti protici.
Gli ossidrili presenti sulla superficie della silice sono sufficientemente acidi da riuscire a
protonare i leganti carbammato legati al cerio, con formazione di CO2 e ammina, così che il
metallo si lega alla matrice inorganica attraverso la formazione di legami covalenti E-O-Ce (14).
≡E-O-H + Ce(O2CNBu2)3 → ≡E-O-Ce(O2CNBu2)2 + CO2 + NHBu2 (14)
Come precursore è stato utilizzato il complesso Ce(O2CNBu2)3, precedentemente preparato e
Recentemente, in lavori di tesi paralleli al mio, è stato studiato l'aggraffaggio del terbio su varie matrici inorganiche (silice19, allumina, titania e ceria20) con l'obiettivo di preparare
precursori per materiali fotoluminescenti.
Lavorando in atmosfera di diazoto soluzioni di Ce(O2CNBu2)3, in eptano anidro sono state
gocciolate su una sospensione del supporto (SiO2). Durante l'aggiunta la velocità di
gocciolamento deve essere mantenuta lenta e regolare, e la sospensione sottoposta ad agitazione meccanica, in modo da garantire un aggraffaggio il più omogeneo possibile. Poichè durante la reazione di aggraffaggio si libera biossido di carbonio, periodicamente sono stati fatti nell'apparecchiatura cicli di vuoto-azoto, con lo scopo di allontanare la CO2 e
spostare la reazione verso destra21 (14).
Nell'aggraffaggio su γ-allumina, al contrario di quello che succede per la silice, durante il gocciolamento non è stato osservato sviluppo di gas all'interno del sistema.
Questa osservazione è in linea con i risultati ottenuti nell'aggraffaggio di terbio su γ-allumina. Per questa reazione seguita per via gas-volumetrica, è stato osservato un piccolo assorbimento di CO2 invece dell'atteso sviluppo di gas. Poichè l'esperimento era stato condotto saturando
l'ambiente di reazione di biossido di carbonio a temperatura costante un assorbimento dovuto alla matrice22 23 24 25 è stato escluso. Nel caso dell'aggraffaggio di terbio il mancato
assorbimento era stato spiegato ipotizzando la presenza di meccanismi diversi. In effetti la superficie della γ-allumina è complessa e non ben definita. Un modello comunemente accettato prevede la presenza di una molteplicità di ossidrili diversi per numero di coordinazione del catione e per il numero di cationi a cui sono coordinati2627 (figura 6).
Figura 6: ossidrili superficiali della γ-Al2O3.
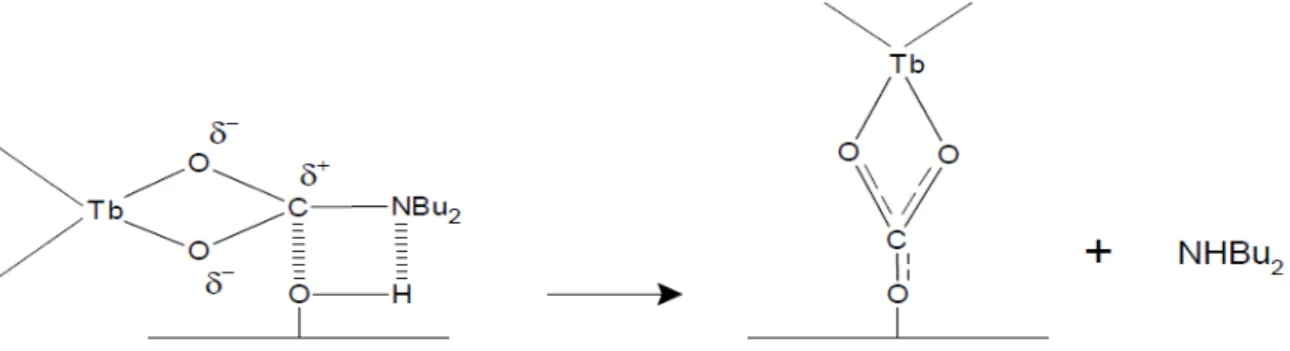
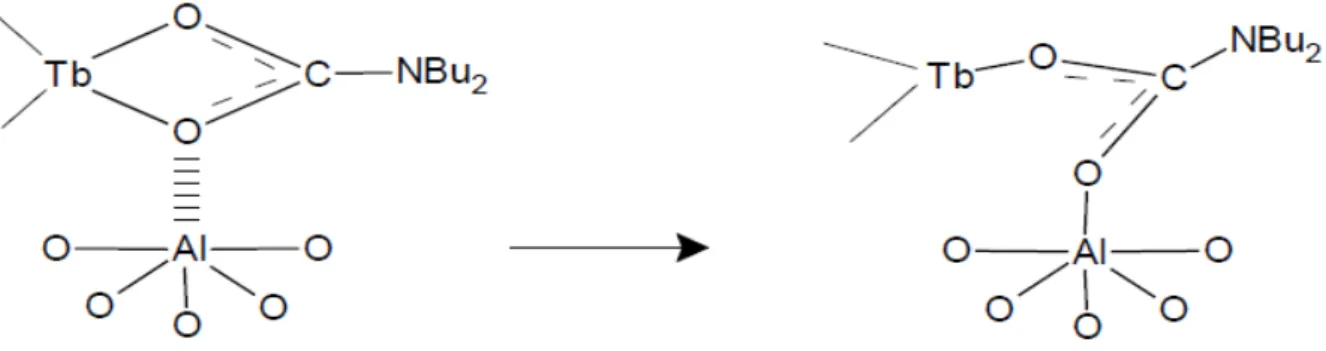
Da tutto ciò appare possibile che ci siano ossidrili (o in generale atomi di ossigeno) nucleofilici o anche centri acidi di Lewis coinvolti nell'aggraffaggio oltre ad ossidrili con l'usuale acidità di Broensted (figure 7 e 8).
Figura 7: ipotesi per l'aggraffaggio di Tb(O2CNBu2)3, su γ-allumina con formazione di
Figura 8: ipotesi per l'aggraffaggio di Tb(O2CNBu2)3, su γ-allumina senza formazione di
ammina.
Da quanto detto l'assenza di sviluppo di CO2 osservato sperimentalmente durante
l'aggraffaggio del cerio sulla γ-allumina può essere spiegato da vari fattori, a partire dalla capacità della matrice di assorbire biossido di carbonio, fino alla presenza parallela di più meccanismi di ancoraggio del metallo al supporto.
Dopo aver lasciato la sospensione in agitazione per 12 ore, il solido è stato recuperato per filtrazione, lavato con aliquote di eptano anidro in modo da solubilizzare il complesso non legato covalentemente, seccato a pressione ridotta e temperatura ambiente ed infine infialato e conservato in atmosfera di diazoto.
Il contenuto di metallo ancorato sulla matrice è stato stimato titolando il contenuto di cerio rimasto nel filtrato28. I carichi di metallo aggraffato sono dello stesso ordine di grandezza per
i due supporti.
Nel caso dell'allumina questo valore corrisponde al carico massimo (0,27 mmol/g) come è come è possibile dedurre della presenza di cerio nel filtrato.
Lo stesso carico (0,28 mmol/g) in assenza di metallo nel filtrato ci fa prevedere per la silice un carico massimo più alto.
In effetti, dai dati ottenuti per il terbio, il carico massimo di aggraffaggio sulla silice è più del doppio di quello ottenuto dall'allumina: 0,34 mmol Tb/g SiO2 contro 0,12 mmol Tb/g Al2O3.
Lo studio tramite spettroscopia infrarossa in riflettanza è stato condotto solo nel caso della
silicea. Sono presenti bande anche se poco intense attribuibili alla presenza di carbammati
(1314, 1375, 1423, 1482 e 1520 cm-1). Segue che la reazione di aggraffaggio non riguarda la
totalità dei raggruppamenti carbammato. Studi in gas-volumetria per l'aggraffaggio del terbio su silice20 hanno permesso di stimare che abbia reagito circa il 45 % dei leganti carbammato.
Ciò è interpretabile sulla base di requisiti geometrici immaginando che, nel momento in cui il metallo si ancora alla matrice, i leganti che puntano in direzione opposta non vengano coinvolti. È inoltre da sottolineare che per una specie polinucleare è possibile in principio che un solo legante dell'aggregato permetta di ancorare più centri metallici. Il processo di aggraffaggio appare dipendente anche dalle caratteristiche della matrice: la distribuzione degli ossidrili superficiali può presumibilmente portare ad una molteplicità di specie metalliche ancorate sulla matrice. In generale non possiamo escludere la presenza di oligomeri aggraffati o anche di specie con un singolo metallo legato a più atomi della matrice. Nella descrizione del sistema l'unica cosa che possiamo affermare con sicurezza è la presenza di leganti carbammato residui.
Frazioni dei prodotti ottenuti sono state calcinate a 400°C ottenendo un prodotto giallo dovuto al CeO2 formato sulla superficie. I composti ottenuti sono stati infialati e conservati in
atmosfera inerte. Studi IR non hanno evidenziato, come era presumibile, bande dovute ai
leganti carbammato.
Dall'osservazione al microscopio elettronico a scansione (SEM) dei prodotti ottenuti per calcinazione si osserva che la distribuzione del cerio sulle matrici è piuttosto omogenea (figura 9). Le misure SEM-EDX sulle superfici dei prodotti confermano che i rapporti molari Ce / M (dove M = Al, Si) sono pressoché costanti al variare dell'area analizzata.
Figura 9: immagini SEM dei prodotti di aggraffaggio di cerio su A) allumina e B) silice seguito da calcinazione.
Sintesi di ceria e ceria drogata con lantanio mediante idrolisi degli
N,N
-
dibutilcarbammati
metallici
Studi di idrolisi totale di N,N-dialchilcarbammati metallici sono scarsi. Si possono citare solo studi preliminari sul rame e sul titanio, condotti in questi laboratori e non ancora pubblicati. L'idrolisi totale dei carbammati di Cu(II)29 di formula Cu
2(O2CNEt2)4(HNEt2)2 e
Cu(O2CNiPr2)2 porta alla formazione di Cu2(OH)2(CO3)∙(HNR2).
Sono stati condotti inoltre studi30 di idrolisi totale di carbammati di Ti(IV) Ti(O
2CNR2)4 (R =
Me, Bu, Et, iPr). In questi casi la reazione, anziché ai carbonati, porta verso la formazione di
ossidi.
Si ottengono in realtà titanati di ammonio di formula generale (NH2R2)xTiO(2+x/2) con x piccolo
ma variabile e dipendente da R. In questi casi non si è ottenuto l'ossido di titanio neanche dopo prolungato riscaldamento a 120-160 °C a pressione ridotta.
La basicità dell'idrossido del metallo, specie intermedia nell'idrolisi del legante carbammato può spiegare la formazione di un carbonato in presenza di biossido di carbonio in alternativa alla formazione dell'ossido (vedi figura 6 Introduzione). Questo permette di razionalizzare i risultati della reazione di idrolisi per il carbammato di Cu(II), centro basico, e di Ti(IV), centro acido.
Un parametro importante è anche la pressione parziale di CO2 nel mezzo di reazione. In
effetti, in reazioni di idrolisi parziale è possibile che un carbammato di un metallo che ha un
idrossido basico, in atmosfera di N2, possa fornire una specie μ-osso anziché un carbonato,
come ad esempio nel caso del μ-osso derivato di La, descritto poco sopra, di formula La4(O)
(O2CNiPr2)1031.
In questo lavoro di tesi è stata studiata l'idrolisi completa di Ce(O2CNBu2)3 disciolto in
toluene per aggiunta di un largo eccesso di acqua (soluzione H2O/THF con rapporto molare
H2O/Ce = 50). Sono state condotte varie prove di idrolisi ottenendo diversi prodotti al variare
rapidità di aggiunta della soluzione di H2O/THF. In linea generale il procedimento è stato il
seguente: il composto Ce(O2CNBu2)3 è stato disciolto in toluene anidro e sotto vigorosa
agitazione è stata aggiunta una soluzione di H2O/THF (rapporto 1:5). Il THF, solubile sia in
acqua che in un solvente organico rende più rapido il processo di idrolisi. Mano a mano che procedeva l'idrolisi si osservava un iniziale intorbidimento della soluzione, poi la formazione di specie colloidali incapaci di decantare prontamente, in seguito la formazione di un residuo voluminoso e la finale precipitazione di una specie semisolida che sembrava appiccicarsi alle pareti del pallone.
Il residuo dopo ripetuti lavaggi con una soluzione di toluene e THF in modo da allontanare eventuali impurezze di dibutilammina, è stato essiccato a pressione ridotta (alla stessa temperatura di quella mantenuta durante il processo di idrolisi). Si otteneva a questo punto un solido di consistenza polverosa ben trasferibile. I prodotti ottenuti sono stati caratterizzati per calcinazione a 850 °C, per RX su polveri e per spettroscopia IR allo stato solido.
I dati dell'analisi elementare di metallo sono in linea con le formule individuate per identificazione del composto tramite RX su polveri.
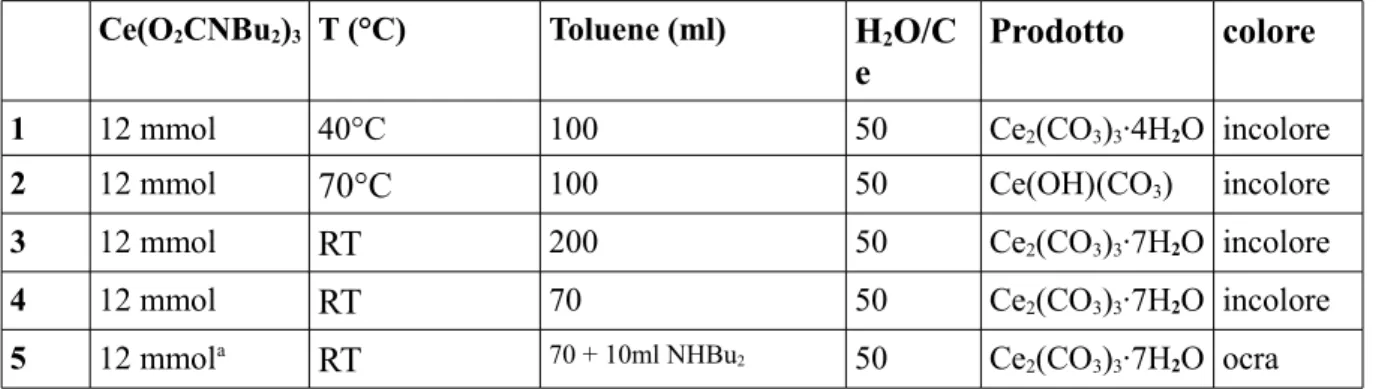
In tabella 4 vengono riassunte le condizioni ed i prodotti ottenuti per le varie prove.
Tabella 4: carbonati di cerio isolati e caratterizzati in questo lavoro. a)Idrolisi condotta in eccesso di dibutilammina.
Ce(O2CNBu2)3 T (°C) Toluene (ml) H2O/C
e
Prodotto colore
1 12 mmol 40°C 100 50 Ce2(CO3)3∙4H2O incolore
2 12 mmol 70°C 100 50 Ce(OH)(CO3) incolore
3 12 mmol RT 200 50 Ce2(CO3)3∙7H2O incolore
4 12 mmol RT 70 50 Ce2(CO3)3∙7H2O incolore
5 12 mmola RT 70 + 10ml NHBu2 50 Ce
2(CO3)3∙7H2O ocra
Le diverse concentrazioni della soluzione di precursore e i tempi di aggiunta della soluzione
di H2O/THF non influiscono sulla composizione del prodotto ottenuto.
La natura del prodotto formato dipende dalla temperatura del processo di idrolisi e di essiccamento. Operando a bassa temperatura (da temperatura ambiente fino a 40°C) si ottengono carbonati; cambia il tipo di fase e il diverso contenuto di acqua di cristallizzazione. Nell'idrolisi a temperatura ambiente si forma la specie Ce2(CO3)3∙7H2O, che analizzata
mediante RX su polveri ha mostrato di avere la struttura del minerale lantanite (figura 11). In realtà la lantanite contiene 8 molecole di acqua: piccole quantità di una specie amorfa a cui è stata allontanata anche solo parte dell'acqua di cristallizzazione durante il trattamento di essiccamento sotto vuoto del campione possono spiegare il contenuto di acqua da noi trovato.
Figura 11: pattern XRD di Ce2(CO3)3∙7H2O, ottenuto per idrolisi a temperatura
ambiente.
Figura 12: struttura cristallina di Ce2(CO3)3 ·8H2O.
Esiste in letteratura la struttura cristallina della lantanite determinata per diffrazione ai raggi X su cristallo singolo3233. Come si vede dalla figura 12, ogni atomo di Ce è ottacoordinato. Ce1
ha una geometria ad antiprisma quadrato distorto con 4 atomi di ossigeno dei gruppi carbonato e 4 atomi di ossigeno appartenenti a acqua coordinata. Ce2 ha geometria dodecaedrica distorta con 6 atomi di ossigeno dei gruppi carbonato e 2 atomi di ossigeno appartenenti a acqua coordinata.
La struttura complessiva è formata da strati paralleli in cui i gruppi carbonato che fungono da leganti bidentati per Ce2 puntano verso lo spazio tra i piani, che è riempito da acqua di
cristallizzazione che si mette a ponte tra gli strati formando legami a idrogeno con i gruppi carbonato.
Nell'idrolisi a 40°C invece è stato ottenuto il composto di formula Ce2(CO3)3∙4H2O, con un
numero più basso di molecole di acqua nel reticolo cristallino e struttura del minerale calchinsite (figura 13).
Figura 13: pattern XRD di Ce2(CO3)3∙4H2O, ottenuto per idrolisi a 40 °C.
Il minerale è stato scoperto solo in tempi recenti34 (Big Sandy Creek, Montana, USA) e in
letteratura non sono riportati nè metodi di preparazione nè uno studio strutturale per diffrazione RX su cristallo singolo. Esiste solo uno studio di RX su polveri sul minerale, con parametri di cella tali da fare ipotizzare una struttura a strati simile a quella della lantanite35.
A temperatura relativamente più alta (70°C) viene ottenuta la specie idrosso-carbonato di formula Ce(OH)(CO3). A questa temperatura viene persa tutta l'acqua di cristallizzazione e si
può ipotizzare la formazione di un idrosso-carbonato a partire da carbonato per perdita di CO2
associata alla reazione con acqua di cristallizzazione.
Figura 14: pattern XRD di Ce(OH)(CO3), ottenuto per idrolisi a 70 °C.
Il diffrattogramma di polveri (figura 14) della specie è pressoché sovrapponibile a quello del minerale ancylite36 (di formula generale Sr
2-x(Ce,La,Nd)x(CO3)2(OH)x·(2-x)H2O) del quale
rappresenta la composizione limite per x = 237. Nella struttura dell'ancylite è presente per il
metallo una sola posizione cristallografica. Lo stronzio ed il lantanide la occupano alternativamente con una distribuzione disordinata nel cristallo. Nel nostro caso la presenza del solo lantanide rimuove questo tipo di disordine. Inoltre l'aumento di carica positiva (cerio
invece che stonzio) richiede che tutti gli atomi di ossigeno non appartenenti a gruppi carbonato appartengano a gruppi ossidrile. Nella struttura del minerale, il metallo è circondato da 8 atomi di ossigeno appartenenti a gruppi carbonato e a 2 ossidrili o molecole di acqua (figura 15). In questo caso si riconoscono nello spettro segnali dovuti a tracce di ceria. In particolare è evidente il segnale largo a circa 30°, valore di 2θ per cui si ha il segnale più intenso nello spettro della ceria.
Figura 15: proiezione del poliedro di coordinazione del catione M nell'ancilite.
La prova 3 è stata condotta lavorando a pressione ridotta. Nello schlenk contenente la soluzione di Ce(O2CNBu2)3 in toluene è stato fatto il vuoto mediante una pompa meccanica e
mediante un'imbuto gocciolatore dotato di equilibratore di pressione la soluzione di H2O/THF
è stata aggiunta molto lentamente alla miscela di reazione. Durante l'aggiunta, ad intervalli regolari, veniva fatto il vuoto nell'apparecchiatura in modo tale da allontanare la CO2
sviluppata durante l'idrolisi del carbammato, con l'intento di ottenere direttamente ossido di cerio anziché carbonato. La natura del prodotto ottenuto chiarisce che durante l'idrolisi il carbonato si forma facilmente a temperatura ambiente anche a bassa pressione parziale di biossido di carbonio.
La prova in eccesso di ammina è stata fatta per favorire la formazione di particelle di piccole dimensioni (nanoparticelle)38. In letteratura è infatti riportato che usando un surfattante
(idrossido di ammonio39, urea40, idrogeno carbonato di ammonio41) è possibile condizionare la
forma e la dimensione delle particelle di carbonato ottenuto, che sono mantenute nell'ossido dopo trattamento termico42. Quest'affermazione ha precedenti in letteratura sia per Ce(OH)
(CO3)43 che per Ce2(CO3)3∙8H2O44.
Le nanoparticelle hanno un rapporto superficie-volume molto alto, il liquido in cui sono immerse, a causa della tensione superficiale esistente all'interfaccia con la particella tende a ridurre la superficie di contatto promuovendo l'aggregazione delle particelle.
L'aggiunta di un surfattante (tensioattivo) che ricopre la superficie delle particelle diminuisce la tendenza all'aggregazione a causa delle forze repulsive tra le molecole dei tensioattivi che circondano le nanoparticelle.
Ammine alchiliche quali appunto la dibutilammina si comportano come tensioattivi45. Il
solido ottenuto dopo la precipitazione per idrolisi era molto fine e decantava in tempi molto lunghi (ore). A causa della presenza di ammina in eccesso il colore non era bianco ma ocra. Nonostante la differenza di colore il solido ottenuto dopo essiccamento a pressione ridotta ha un'analisi di metallo consistente con la specie Ce2(CO3)3∙7H2O, che è normalmente ottenuta
In tabella 5 vengono riportate le principali bande IR per i carbonati di cerio ottenuti. Le bande attribuibili al carbonato di cerio sono tutte molto intense e larghe probabilmente per la presenza di più modi di coordinazione.
Tabella 5: principali segnali ATR-IR in cm-1 per i composti Ce
2(CO3)3∙4H2O, Ce2(CO3)3∙7H2O e
Ce(OH)(CO3).
Ce2(CO3)3∙4H2O 1456 1360 846
Ce2(CO3)3∙7H2O 1471 1369 e 1338 848
Ce(OH)(CO3) 1488 1415 858
Gli spettri IR dei prodotti ottenuti sono molto simili, con una banda intorno a 1470-1460 cm-1,
una banda (singola o sdoppiata) intorno a 1350 cm-1 ed una banda singola intorno a 845 cm-1.
Per Ce(OH)(CO3), oltre alle bande attribuibili al gruppo carbonato, si osserva una intensa
banda del gruppo OH a 3456 cm-1 46.
In letteratura è riportata una certa variabilità nel trattamento termico necessario per convertire i carbonati a ceria con temperature che variano dai 300 ai 700 °C4748.
Per chiarire se era possibile evidenziare una decomposizione termica a ceria in condizioni più blande per una delle fasi da noi preparate, sono stati fatti studi di termogravimetria e calorimetria differenziale a scansione termica (TG-DSC).
Trattamenti temici condotti a temperature più basse dovrebbero fornire composti con cristalliti più piccoli e meno aggregati, potenzialmente preferibili per usi in catalisi.
Gli studi TG-DSC da noi effettuati sono stati condotti sotto flusso di azoto, con un gradiente termico di 10 °C/min, cercando di applicare esattamente le medesime condizioni per tutti i campioni, in modo tale da poter paragonare il loro comportamento sotto trattamento termico e valutare quale di essi decarbonatasse nelle condizioni più blande.
Per la conversione completa dei carbonati a CeO2 si può prevedere prima la perdita di
molecole di H2O di cristallizzazione e poi di molecole di CO2 con ossidazione del metallo.
Nel seguito vengono discussi caso per caso i dati sperimentali ottenuti e confrontati con le informazioni di letteratura:
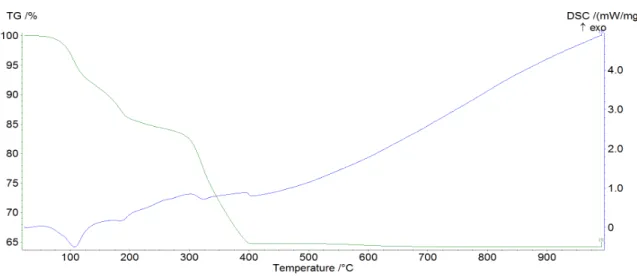
1) Ce2(CO3)3·4H2O
Le perdita di peso totale ottenuta è stata del 35,5%, in buon accordo con il valore teorico di 35,3%.
Nella parte iniziale del termogramma (fino a 250°C) si assiste alla perdita delle 4 molecole di H2O di cristallizzazione: perdita sperimentale -13,8 %, in accordo con la perdita teorica di
13.5%.
Quello che può essere osservato dal termogramma è che le 4 molecole di H2O non vengono
perse tutte contemporaneamente; il processo avviene almeno in due fasi distinte: prima
vengono perse 2 molecole di H2O entro i 100°C, con una perdita di peso di 6.3 % e poi
successivamente le ultime due con una perdita di peso sperimentale di 7,2 % entro i 200°C. I due picchi nella DSC corrispondenti a queste due perdite appaiono sdoppiati, da cui possiamo dedurre che le quattro perdite di acqua sono distinte ed avvengono in successione. Inoltre integrando le aree nel grafico calorimetrico è possibile vedere che i due processi richiedono diverse quantità di energie: sono entrambi processi endotermici, ma la perdita delle prime due molecole richiede molta più energia della perdita delle seconde (-125 J/g della prima contro -15 J/g della seconda); quindi la perdita delle ultime due molecole di acqua potrebbe essere in parte concomitante all'inizio dell'ossidazione del composto (processo esotermico).
Nella zona tra i 250 e i 600 °C è possibile notare dai dati ottenuti che il processo di decarbonatazione e quello di ossidazione del metallo da Ce(III) a Ce(IV) avvengono in contemporanea.
In questa zona nel grafico TG si assiste ad una perdita di peso (-22%), che corrisponde alla perdita di CO2; nel grafico DSC si assiste però ad una forma irregolare del grafico, segno che
c'è una sovrapposizione tra il picco endotermico della decarbonatazione e quello esotermico di ossidazione.
Dai dati ottenuti il processo a cui si assiste può essere così schematizzato (15):
Ce2(CO3)3 ·4H2O Ce2(CO3)3 ·2H2O Ce2(CO3)3 2CeO2 (15) 2) Ce2(CO3)3 ·7H2O
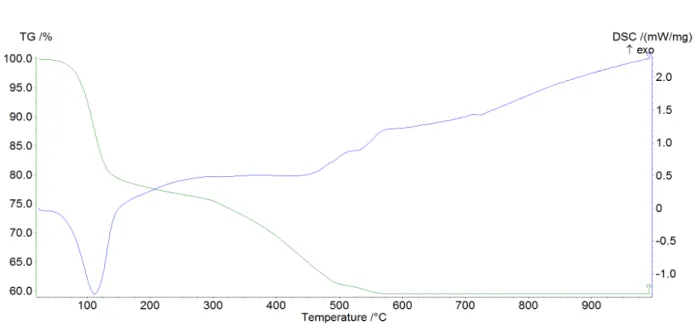
Figura 17: grafico TG-DSC per Ce2(CO3)3·7H2O.
La perdita totale di peso è stata di -40.5%, in accordo con il dato teorico di 41,3%.
Entro i 250°C si assiste ad una perdita di peso di 21.50%, corrispondente alla perdita delle 7 molecole di H2O di cristallizzazione (perdita teorica -23.8%).
A differenza di Ce2(CO3)3·4H2O le 7 molecole di acqua sembrano allontanarsi praticamente in
contemporanea e nel grafico DSC appare un unico picco endotermico.
Anche in questo caso la reazione di decarbonatazione e quella di ossidazione avvengono in contemporanea nel range 250-900°C. Nel grafico DSC si assiste ad una situazione analoga al caso di Ce2(CO3)3·4H2O, in cui il picco esotermico di ossidazione e quelli endotermici di
decarbonatazione sono sovrapposti creando una forma irregolare del grafico.
È possibile confrontare i grafici ottenuti per i due carbonati di cerio preparati con quello riportato in letteratura49 per il composto Ce
2(CO3)3·8H2O, tenendo presente che il differente
gradiente termico usato durante le misurazioni, 10 °C/min nel nostro caso e 5 °C/min per i giapponesi, potrebbero far sì che le transizioni da noi misurate avvengano a temperature leggermente superiori.
Figura 18: grafici TG-DSC di letteratura per Ce2(CO3)3·8H2O A) in flusso di aria e B) in
flusso di N2.
La perdita di acqua da noi registrata per gli studi sulla lantanite avviene in condizioni apparentemente simili a quelle riscontrate in letteratura sia per le misurazione all'aria (18A)
che sotto N2 (18B). Nel range successivo il nostro termogramma appare intermedio tra i due
riportati, anche se molto più vicino a quello registrato sotto N2 . Nel nostro caso non si
osserva una separazione netta tra i due processi (decarbonatazione e ossidazione) ma si riesce comunque ad evidenziarli.
Nella misurazione di letteratura condotta in flusso di N2 (Fig 18 B) i due processi si
differenziano più marcatamente. Intorno ai 400-500°C si ha la perdita di due molecole di CO2
(decarbonatazione) con un picco endotermico netto nella DSC e poi nel range 500-600°C la perdita dell'ultima molecola di CO2 e ossidazione (acquisto di mezza molecola di O2) con un
picco solo lievemente endotermico dovuto alla sovrapposizione di un processo endotermico e di uno esotermico. Il range di temperatura da noi registrato è lo stesso riportato in letteratura per il termogramma sotto azoto.
Il processo è stato schematizzato secondo l'espressione 16:
3) Ce(OH)(CO3)
Figura 19: grafico TG-DSC per Ce(OH)(CO3).
La perdita totale di peso per la specie, in accordo con il dato sperimentale gravimetrico ottenuto per calcinazione del campione a 850°C, è stata di -19%, minore del dato teorico di 20.7%; questo perchè nel campione sono già presenti tracce di ceria, visibili nello spettro RX su polveri (figura 13), che si erano formate durante il processo di idrolisi condotto a 70°C. All'inizio (entro i 40°C) si assiste ad una perdita di peso di -1.65% con un processo endotermico, che dovrebbe corrispondere a una perdita di acqua igroscopica, che infatti viene allontanata a temperature molto basse, suggerendo che si tratti di acqua adsorbita e non di cristallizzazione.
Nel range 120-700 °C si assiste ad un'unica perdita netta di -19%, ed anche in questo caso il processo di decarbonatazione e quello di ossidazione sono concomitanti.
Confrontando i termogrammi possiamo vedere che il processo di decarbonatazione inizia a temperature leggermente più basse per il composto Ce(OH)(CO3) rispetto ai carbonati
Ce2(CO3)3·nH2O, ma la perdita di peso rallenta e non è completa fino ai 600-700°C.
Per il composto Ce2(CO3)3 ·7H2O il processo è invece molto lento e graduale e procede dai
250 ai 600-700°C.
Per il composto Ce2(CO3)3 ·4H2O si osserva un processo più netto e rapido con una perdita di
peso quasi completa intorno ai 300°C che si completa però come negli altri casi alla temperatura di 600-700°C.
Questi studi, come già detto, sono stati condotti nelle stesse condizioni in atmosfera di azoto. Nonostante questo è possibile che tracce di aria possano modificare sensibilmente il termogramma perchè rendono più facile il processo ossidativo, e tenendo questo in considerazione ed esaminando gli studi di letteratura esistenti non abbiamo riscontrato grosse differenze nella reazione di decarbonatazione delle tre fasi da noi preparate.
Questi risultati indicavano che era possibile ottenere la ceria per trattamento termico in condizioni relativamente blande.
In letteratura esistono vari metodi per ottenere ceria in dimensioni nanoparticellari. Sono generalmente basati su sintesi in soluzione in solventi acquosi e non acquosi50. Tra questi
possiamo ricordare la precipitazione, la sintesi solvotermale, il metodo sol-gel, la sintesi per combustione o decomposizione termica.
La precipitazione51 viene generalmente condotta innalzando il pH di una soluzione acquosa di
bassa, questi vengono precipitati da soluzioni di sali di cerio per azione di basi. Per ottenere particelle di piccole dimensioni è possibile utilizzare agenti che subiscono una lenta idrolisi con liberazione di ioni idrossido in soluzione, ad esempio esametilentetrammina o urea. Questo permette di controllare il grado di sovrassaturazione della soluzione e di evitare gradienti di concentrazione. Il precipitato ottenuto (in genere idrossocarbonati se si usa esametilentetrammina52 o ossocarbonati se si usa urea53) viene quindi trattato termicamente
(300-900 °C) fino ad ottenere ceria di dimensioni che variano da 6 a 40 nm54. Le dimensione
aumentano con la temperatura e il tempo di invecchiamento del precipitato.
In genere non si ottengono però particelle di dimensioni uniformi. Per ovviare a questo inconveniente è possibile condurre la precipitazione in presenza di agenti templanti, ad esempio polietilen glicol55. Utilizzando vari templanti è possibile preparare particelle di
Ce(OH)(CO3) di forme che vanno dall'ellitticca all'ottaedrica alla rombica ecc., ed è stato
dimostrato che per trattamento termico si mantiene la stessa forma nelle particelle di ceria56.
Per trattamento termico si può avere aggregazione delle particelle per processi di sinterizzazione.
Anche nel metodo solvo/idrotermale57 i precursori vengono disciolti in acqua o in altro
solvente. Le specie possono idrolizzarsi innalzando il pH ed il precipitato che si forma viene scaldato in autoclave per alcune ore. Le alte pressioni permettono al solvente di raggiungere temperature superiori al suo punto di ebollizione in condizioni ordinarie (in H2O si lavora di
solito tra 120 e 220 °C58). In queste condizioni cambiano le proprietà del solvente (quali
viscosità, diffusione e solubilità degli ioni in esso disciolti) dalle quali dipende in ultima analisi il controllo della forma e dimensione delle particelle. Si può variare il solvente o la temperatura.
A seconda delle diverse condizioni si ha un diverso controllo cinetico dei processi di dissoluzione e ricristallizzazione dei grani in sospensione ottenendo dimensioni diverse59.
Anche il pH in soluzione acquosa è un fattore importante: la crescita delle particelle è più lenta in condizioni basiche e neutre rispetto a quelle acide60.
Per preparare nanoparticelle di ceria generalmente viene usata una soluzione acquosa di NaOH61. Alternative possibili sono soluzioni di fosfati62, urea63, ammoniaca64 oppure
perossido di idrogeno65.
Il metodo solvo/idrotermale è una delle vie più efficienti ed economiche in quanto è caratterizzata da un singolo stadio, condotto a temperatura relativamente bassa e in soluzione. Inoltre si ha un controllo più accurato sulla forma e dimensione delle particelle rispetto alla sintesi per precipitazione.
Col metodo sol-gel66 si ottiene una soluzione omogenea disciogliendo in acqua deionizzata
precursori quali nitrati o acetati di cerio(III) insieme a soluzione in etanolo di ammine come surfattanti (dimetiloctilammina, monoetanolammina, idrossido di tetraetilammonio) in modo da ottenere particelle di dimensioni nanometriche. La soluzione viene evaporata a 90 °C fino ad ottenere un gel, il quale viene recuperato ed essiccato a 100 °C per 12 ore. La polvere così ottenuta viene infine calcinata a 500 °C per 4 ore.
Applicando questo metodo in uno studio recente67 è stata ottenuta ceria mesoporosa (pori 3 –
5 nm) con particelle di 5-20 nm.
É descritta anche una sintesi per combustione/decomposizione termica68, con numerose
varianti69. Viene comunque ottenuto un gel da soluzione acquosa del precursore (ad esempio
nitrato) per mezzo di un agente precipitante (es glicina o ossalato) eventualmente evaporando la soluzione. Il gel ottenuto viene poi bruciato all'aria e le polveri ottenute calcinate (400 °C, 20 h).
Si ottengono cristalliti di dimensione medie nell'ordine delle decine di nm70 ma per prolungato
trattamento termico le dimensioni delle particelle sono più grandi per aggregazione dei cristalliti.
Nel nostro lavoro sperimentale la ceria è stata preparata per trattamento termico all'aria a 200°C dei carbonati di Ce(III) ottenuti per idrolisi. I campioni di cerio ottenuti sono di colore giallo acceso, a differenza dei prodotti commerciali: l'ossido cerico Carlo Erba è incolore, mentre un ossido Sigma Aldrich (diametro particelle 50 nm) di colore arancione.
In effetti in letteratura è riportato che il colore della ceria può variare dal biancastro al marrone chiaro fino al giallo (a seconda della purezza e della dimensione delle particelle71).
Per una possibile applicazione in catalisi apparivano desiderabili particelle di piccole dimensioni che offrono una maggiore area superficiale. Per questo appariva importante un controllo della temperatura del processo di decarbonatazione. Temperature troppo elevate favoriscono la sinterizzazione delle particelle con diminuzione dell'area superficiale. Temperature troppo basse, al contrario, non permettono di portare a termine il processo di decomposizione termica del carbonato, se non in tempi troppo lunghi. Sono state quindi condotte prove di decomposizione termica a varie temperature, interrompendo il riscaldamento una volta raggiunto il peso costante. In questo modo è stato possibile verificare che per riscaldamento a 130°C per 12 ore i carbonati perdevano solo le molecole di acqua di cristallizzazione. Ad esempio per la lantanite Ce2(CO3)3∙7H2O è stata misurata una perdita di
peso del 21,7 %, corrispondente alla perdita delle 7 molecole di acqua di cristallizzazione (perdita teorica 21,5 %), mentre per la calchinsite Ce2(CO3)3∙4H2O una perdita del 13,8% di
peso, corrispondente alla perdita delle 4 molecole di acqua di cristallizzazione (perdita teorica 13.5%).
Nell'arco di ore la temperatura di 130°C è sufficiente ad allontanare l'acqua di cristallizzazione, mentre per tempi lunghi (settimane) si ha anche decarbonatazione anche se
incompleta. Ad esempio Ce2(CO3)3∙4H2O in 21 giorni perde il 33,7 % di peso contro un
teorico di 35,3 %, mentre Ce2(CO3)3∙7H2O perde il 38,1 % in 14 giorni (perdita teorica 41,3
%). A 170°C il processo di decarbonatazione è più veloce, ma anche a questa temperatura la conversione a ceria richiede tempi troppo lunghi per arrivare a completezza.
Una conversione completa a ceria si ottiene invece per riscaldamento di 12 ore a 200 °C (per i risultati vedi la tabella 6).
Tabella 6: perdite di peso percentuali sperimentali e teoriche (tra parentesi) ottenute per trattamento termico dei composti Ce2(CO3)3∙7H2O, Ce2(CO3)3∙4H2O e Ce2(CO3)3∙4H2O.
130 °C (- H2O di cristallizzazione) 200 °C (decarbonatazione) Ce2(CO3)3∙7H2O -21.7 % (21.5 %) -40.6 % (41.3 %) Ce2(CO3)3∙4H2O -13.8 % (13.5 %) -35.1 % (35.3 %) Ce(OH)(CO3) –––––––––––– -19.0 % (20.7 %)
La ceria è un ossido basico. Per esposizione all'aria la ceria ottenuta ha la tendenza a carbonatarsi e deve essere conservata sotto atmosfera inerte. È ragionevole pensare che la velocità e il grado di carbonatazione dipendano dalla dimensione delle particelle: prodotti a granulometria più fine ed a più alta area superficiale si possono carbonatare più velocemente. In figura 20 è riportato lo spettro IR della ceria commerciale (a) che, anche dopo essere stato esposto a lungo all'aria, non mostra avere bande significative attribuibili al carbonato, mentre la ceria preparata da noi si carbonata molto rapidamente.
500 1000 1500 2000 2500 3000 3500 0,8 0,825 0,85 0,875 0,9 0,925 0,95 0,975 1 1,025 1,05 1,075 Wavenumbers [1/cm] Transmittance
Figura 20: a)CeO2 commerciale conservata all'aria. b)CeO2 ottenuta in questo lavoro
lasciata all'aria per 10 minuti. c) Specie del punto b lasciata a lungo in atmosfera di CO2.
La composizione e la purezza dei prodotti sono state confermate tramite analisi per diffrazione di raggi X su polveri. Viene individuata un'unica fase, compatibile con l'ossido di cerio(IV) avente la struttura del minerale cerianite, caratterizzata da una struttura tipo fluorite,CaF2, descrivibile come un reticolo cubico a facce centrate di ioni Ce4+ (numero di
coordinazione 8) e tutte le cavità tetraedriche occupate dagli anioni ossido.
Qui sotto è riportato a titolo di esempio lo spettro di diffrazione della ceria ottenuta per trattamento termico a 200 °C di Ce2(CO3)3∙4H2O (figura 21).
5 10 15 20 25 30 35 40 45 50 55 60 161565 80783 0 PowderCell 2.2 CeO2 2 0 0 2 2 0 3 1 1 2 2 2 CER_O_MG.raw
Figura 21: spettro di diffrazione RX su polveri del campione di ceria ottenuto per calcinazione a 200°C di Ce2(CO3)3∙4H2O (nero), che coincide con la fase cerianite (rosso).
Il diffrattogramma sottoriportato (figura 22) confronta i dati per la ceria ottenuta a 200 °C con quella ottenuta a 850 °C. La larghezza di banda notevolmente superiore nel primo caso rispetto al secondo dimostra come la possibilità di poter ottenere ossidi di cerio per
a b
trattamento termico a temperature relativamente basse permette di minimizzare i processi di sinterizzazione.
Figura 22: confronto dei diffrattogrammi RX su polveri per i campioni di ceria ottenuti per calcinazione di Ce2(CO3)3∙4H2O a 200 °C (blu) e 850 °C (rosso).
La diffrazione di raggi X su polveri può dare infatti informazioni qualitative sulle dimensioni delle particelle.
Bande larghe nello spettro di diffrazione RX sono associate in genere alla presenza di cristalliti di piccole dimensioni. L'equazione di Debey-Scherrer72 (17) correla la larghezza del
picco nello spettro di diffrazione alla dimensione media dei cristalliti mostrando proporzionalità inversa. Calcoli in cui viene applicata la formula agli spettri di diffrazione di polveri dei campioni di ceria da noi ottenuti mostrano un diametro medio dei cristalliti intorno a 4-8 nm.
D = K∙λ / W∙cosθ (17) dove:
W = largezza di picco
D = dimensione media dei cristalliti
K = costante di proporzionalità di Scherrer (nella maggior parte delle applicazioni vale 0.94). λ = lunghezza d'onda dei raggi X
θ = angolo di Bragg (espresso in radianti)
In realtà la formula dà solo una buona approssimazione, in quanto la larghezza di banda tende ad essere aumentata anche da fattori strumentali, scarsa cristallinità del campione ed imperfezioni nel reticolo cristallino. Qualitativamente però la larghezza dei picchi è un fattore che può essere tenuto di conto per valutare la dimensione delle particelle di una sostanza. Ad esempio nel grafico seguente (figura 23) vengono confrontati gli spettri di diffrazione dei vari campioni di ceria ottenuti per calcinazione dei carbonati a bassa temperatura con lo spettro di un campione di ceria ottenuta per calcinazione a 850°C. Come si vede la posizione dei picchi dei vari prodotti coincide, indicando che si tratta della stessa fase (cerianite). La larghezza delle bande è invece differente e presumibilmente indicativa della dimensione delle particelle.
Figura 23: confronto spettri RX su polveri per i vari campioni di ceria. E) CeO2 ottenuta
per trattamento termico ad alta temperatura (850 °C). D) CeO2 ottenuta per trattamento
termico a 200 °C del carbammato preparato durante un'idrolisi RT in presenza di dibutilammina come surfattante. C) CeO2 ottenuta per trattamento termico a 200 °C di
Ce(OH)(CO3) (ancylite). B) CeO2 ottenuta per trattamento termico a 200 °C di
Ce2(CO3)3∙4H2O (calchinsite). A) CeO2 ottenuta per trattamento termico a 200 °C di
Ce2(CO3)3∙7H2O (lantanite).
Per avere conferma diretta delle osservazioni ottenute dalla larghezza di segnale nei diffrattogrammi RX su polveri abbiamo cercato di valutare le dimensioni delle particelle per mezzo della microscopia elettronica.
Le immagini dei campioni non permettono di poter distinguere le dimensioni delle particelle, perchè queste si trovano tutte ammassate ed intrecciate. Per cercare di isolare singole particelle, in modo tale da poterne misurare le dimensioni, alcune particelle sono state sospese in un solvente che poi è stato fatto evaporare sul portacampioni. Sono state fatte prove sia con acetone che con tetraclorometano (figura 24), molto più denso, ma in nessun caso è stato possibile isolare singole particelle in modo da valutarne le dimensioni. Dalle figure è comunque possibile notare la struttura aciculare delle particelleb.
b Le analisi elementari EDS, che sfruttano la fluorescenza di raggi X indotta dalla collisione di un fascio di elettroni ad alta energia indicano la composizione del campione compatibile con la formula della ceria CeO2..
E D C B A
Figura 24: immagine SEM del campione di ceria previamente sospesa in CCl4,
sottoposta a sonificazione e poi evaporata.
Per ottenere ulteriori informazioni siamo passati alle analisi TEM e FESEM, che permettono migliori risoluzioni.
È stato possibile confrontare la forma e la dimensione dei due campioni di ceria da noi preparati secondo procedure diverse. I campioni sono stati ottenuti per trattamento termico (200 °C) di Ce2(CO3)3∙7H2O, ottenuta in un caso per idrolisi a temperatura ambiente (LF 412)
e nell'altro per idrolisi a temperatura ambiente in presenza di un surfattante (dibutilammina) (LF 516).
Con lo scopo di ottenere particelle separate e più facilmente osservabili, prima di effettuare le analisi TEM, i campioni sono stati sospesi in acqua all'interno di una provetta e sono stati soggetti a sonicazione (circa 10-15 minuti). In questo modo le particelle andavano in sospensione e la soluzione rimaneva torbida per un tempo indefinito poiché le loro piccole dimensioni rendevano molto lento il processo di decantazione. Il campione è stato raccolto dalla sospensione con una retina di rame dotata di fori di dimensioni intorno ai 100 μm rivestita di un velo di pochi atomi di carbonio depositato sopra, in modo tale da sostenere ma non oscurare il campione (la grafite è un conduttore elettrico utilizzato per evitare la polarizzazione del campione).
Dopo questo trattamento le particelle risultano sufficientemente separate e non sovrapposte l'una con l'altra. L'immagine in figura 25 A ci mostra l'aspetto delle particelle disperse di LF 516. Solo le regioni periferiche sono trasparenti agli elettroni e permettono l'esame dei dettagli strutturali. Dall'immagine si vede bene come la particella nel suo complesso sia costituita da cristalliti aggregati di dimensioni nanometriche, chiaramente distinguibili l'uno dall'altro per le variazioni locali di contrasto nelle immagini.
Un'immagine a più alta risoluzione (figura 25 B) ci permette di valutare con precisione le dimensioni dei cristalliti aggregati, che risultano essere al di sotto dei 5 nm.
Figura 25: immagini TEM a diversa risoluzione delle particelle di LF 516.
Come è possibile osservare dalle immagini TEM (figura 26 B) anche le particelle di LF 412 sono composte di cristalliti nanometrici aggregati, ma in questo caso il contrasto non è sufficientemente definito da permettere una valutazione precisa delle loro dimensioni.
Quello che invece le immagini (figura 26 A) mostrano chiaramente è la particolare forma piatta ed allungata delle particelle nel loro complesso.
Questa tipica forma a bacchetta è confermata anche dall'acquisizione di immagini FESEM (figura 27). Le dimensioni delle particelle sono varie, ma in linea di massima la larghezza si trova comunque intorno alle centinaia di nm, mentre la lunghezza è nell'ordine dei micron. Coerentemente con quanto osservato al TEM, dalle immagini FESEM a risoluzione maggiore (350000 X) è possibile notare che le particelle sono composte da numerosi cristalliti nanometrici aggregati (figura 27 B).
Figura 27: immagini FESEM a diversi ingrandimenti delle particelle di LF 412. A) 25000 X e B) 350000 X.
Per quanto riguarda il campione LF 516 le particelle osservabili nelle immagini FESEM (figura 28 A) appaiono di forma diversa, vagamente sferica, anche se molto irregolare. Le particelle non appaiono infatti piatte e allungate, come nel caso di LF 412, ma sono più o meno ugualmente sviluppate in tutte e tre le dimensioni spaziali. Le immagini a risoluzione maggiore (figura 28 B) mostrano una morfologia delle superfici esterne molto irregolare, con la presenza di microcavità, dovute probabilmente all'allontanamento durante il trattamento termico della dibutilammina, usata in eccesso durante il processo di idrolisi e rimasta inglobata all'interno delle particelle.
Anche in questo caso comunque le particelle hanno dimensioni molto variabili, che vanno da un diametro medio di centinaia di nanometri fino ai micrometri ed è possibile notare nelle immagini ad alta risoluzione i cristalliti nanometrici che le compongono.
Figura 28: immagini FESEM a diversi ingrandimenti delle particelle di LF 516. A) 25000 X e B) 350000 X