INTRODUZIONE
Nel genoma umano i geni occupano il 25% delle sequenze, ma solo l’1% di queste rappresenta le parti effettivamente codificanti per proteine. Le sequenze ripetute, invece, rappresentano più del 50% del genoma umano e non sono equamente distribuite in quanto ci sono regioni genomiche più ricche di sequenze ripetute rispetto ad altre.
Le sequenze ripetute, si dividono in 4 classi principali:
• I Trasposoni sono la grande maggioranza delle sequenze ripetute rappresentando il 45% del genoma.
• Gli pseudogeni occupano lo 0,1% del DNA totale.
• Le sequenze semplici ripetute occupano il 3% circa del DNA totale. • Le duplicazioni segmentali che ne occupano il 5%
Le duplicazioni segmentali
Il sequenziamento del genoma umano ha permesso di identificare i vari tipi di sequenze che lo compongono. Le duplicazioni segmentali, conosciute anche come LCRs (Low Copy Repeats), fanno parte delle sequenze ripetute ed occupano il 5% del nostro genoma. Esse sono blocchi di DNA che variano da 1 a 400 kb, che risultano duplicati in una o più regioni del genoma, mantenendo un’identità elevata che varia dal 97% al 99%. Questi blocchi contengono tipi diversi di sequenze quali geni, frammenti di geni, pseudogeni, sequenze retrovirali endogene; essi possono essere organizzati come ripetizioni in tandem o come ripetizioni intersperse.
Le duplicazioni segmentali possono essere divise in due grandi categorie: duplicazioni intercromosomiche e intracromosomiche. Sono definite duplicazioni intercromosomiche, i segmenti di DNA che risultano duplicati in cromosomi non omologhi; queste tendono a localizzarsi in regioni pericentromeriche o subtelomeriche, cioè adiacenti alle regioni eterocromatiche. Le duplicazioni intracromosomiche, invece, si trovano sullo stesso cromosoma e tendono a presentarsi anche nelle regioni eucromatiche. Ad esempio nel cromosoma 17 ci sono 3 copie di circa 200 Kb separate ciascuna da 5 Mb e due copie di circa 24 Kb separate da 1,5 Mb. Queste copie sono molto simili tra loro (99% di identità) e spesso sono bersaglio di crossing-over ineguali e conseguenti riarrangiamenti (Strachan and Read 2004).
Nel regione 7q11.23 presa in esame in questo lavoro sperimentale, troviamo entrambi i tipi di duplicazioni. I meccanismi molecolari che sono all’origine di queste duplicazioni segmentali possono essere diversi. La ricombinazione omologa non allelica (NAHR) sembra avere svolto un
ruolo fondamentale nell’evoluzione cromosomica che ha prodotto le duplicazioni portando alla loro presenza così consistente. Non meno importante è il meccanismo di riparazione delle rotture a doppio filamento del DNA conosciuto come Non-Homologous End-Joining (NHEJ), che è fondamentale soprattutto nell’origine e nella propagazione delle duplicazioni segmentali nelle regioni subtelomeriche (Bailey et al. 2003 Jurka et al. 2004).
Uno studio realizzato sulle duplicazioni presenti nella regione 7q11.23 attraverso una analisi comparativa tra uomo ed altri primati, tra cui il macaco, ha permesso di ricostruire la successione di riarrangiamenti che ha portato alla organizzazione cromosomica attuale, mettendone in evidenza la complessità (Fig. 1). Un’osservazione basilare è che le duplicazioni segmentali sono frequentemente fiancheggiate da altre sequenze ripetute, le sequenze Alu. Si ritiene che la presenza di tali elementi abbia predisposto la regione 7q11.23 alla formazione delle ampie duplicazioni segmentali da cui è caratterizzata. Partendo da un ipotetico cromosoma ancestrale che presenta due soli Blocchi B e C fiancheggianti una regione di geni a singola copia, una inversione delle regioni 7q11 e 7q22 e una duplicazione della 7q22 stessa avrebbero portato alla formazione del Blocco A seguita da una duplicazione del Blocco C (Fig. 1 A B C). Successivamente un unico riarrangiamento intracromosomico mediato dalle sequenze Alu presenti ai bordi dei Blocchi avrebbe portato alla formazione di un cromosoma intermedio con un Blocco A e un Blocco B aggiuntivi, che successivamente avrebbe subito un’inversione paracentrica mediata dai Blocchi C con orientamento invertito (Fig. 1D). Un successivo crossing-over ineguale tra il cromosoma con l’inversione e il cromosoma senza inversione, avrebbe portato alla formazione dell’attuale cromosoma umano che ha una duplicazione dei Blocchi C, A, e B in prossimità della regione centromerica (Fig. 1E) (Schubert 2008).
Le duplicazioni segmentali per le loro caratteristiche strutturali sono tuttora soggette a riarrangiamenti; esse rappresentano quindi una importante fonte di instabilità genetica che porta a formazione di anomalie cromosomiche associate a disordini genomici. Le anomalie cromosomiche sono causate soprattutto da ricombinazioni aberranti (ovvero dalla NAHR) che portano a duplicazioni, delezioni, traslocazioni e inversioni.
Bailey e collaboratori hanno valutato e illustrato la caratterizzazione molecolare e i pattern specifici delle duplicazioni segmentali intercromosomiche e intracromosomiche del genoma umano (Fig. 3); sono state identificate 169 regioni di duplicazione (circa 289Mb di sequenza), di cui 24 sono state associate a disordini genomici relativamente frequenti (Fig. 2 e Tab. 1) (Bailey et al 2002).
Fig.1 Successione dei
riarrangiamenti che hanno portato alla organizzazione cromosomica attuale delle duplicazioni
segmentale nella regione 7q11.23 (Schubert 2008).
Tab.1 Loci genomici in cui sono presenti duplicazioni segmentali con lunghezza >15 kb ed identità >95%, associate con patologie (Bailey et al 2002)
Fig.2 Duplicazioni segmentali intercromosomiche (linee blu) e intracromosomiche (linee rosse) identificate nel genoma umano. Le regioni contrassegnate dalle lettere dall A alla X rappresentano regioni legate a disordini genetici noti (Bailey et al. 2002).
La regione cromosomica 7q11.23 e la sindrome di
William-Beuren
La sindrome di Williams deve il suo nome al medico neozelandese che per primo la descrisse nel 1961. Questa sindrome è anche conosciuta come sindrome di Williams-Beuren, WBS, in quanto nel 1962, in Germania, Beuren, Apitz e Harmjanz descrissero parallelamente a Williams un quadro clinico simile, rivelatosi poi essere la sindrome stessa.
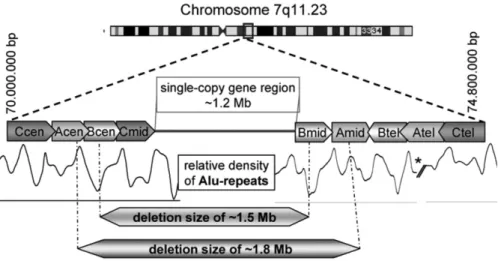
Tale sindrome è causata dalla presenza di CNV (copy number variants) a livello della regione cromosomica 7q11.23. Il riarrangiamento più comune a cui è soggetta tale regione è una delezione che va da 1,5 Mb a 1,8 Mb, che coinvolge circa 28 geni, per alcuni dei quali è stata stabilita una relazione con le caratteristiche fenotipiche della sindrome. Tra questi geni è stato individuato il gene ELN che codifica per l’elastina importante componente dei tessuti connettivi e soprattutto delle arterie.
I primi dati sulle basi genetiche di questa sindrome sono stati ottenuti grazie alla scoperta nel 1993 di una traslocazione tra i cromosomi 6 e 7 (traslocazione t(6;7)(p21.1;q11.23) in famiglie affette da SVAS (stenosi aortica sopravalvolare). La traslocazione interessa il gene dell’elastina (ELN) a livello dell’esone 28, modificandone struttura e funzione (Duba el al. 2002). Basandosi sulla similarità del fenotipo tra i pazienti affetti da SVAS e quelli portatori della WBS, è stato ipotizzato che l’aploinsufficienza del gene ELN fosse la causa anche della WBS. Studi successivi hanno confermato che l’emizigosi del gene per l’elastina è una delle maggiori cause della Sindrome di William-Beuren.
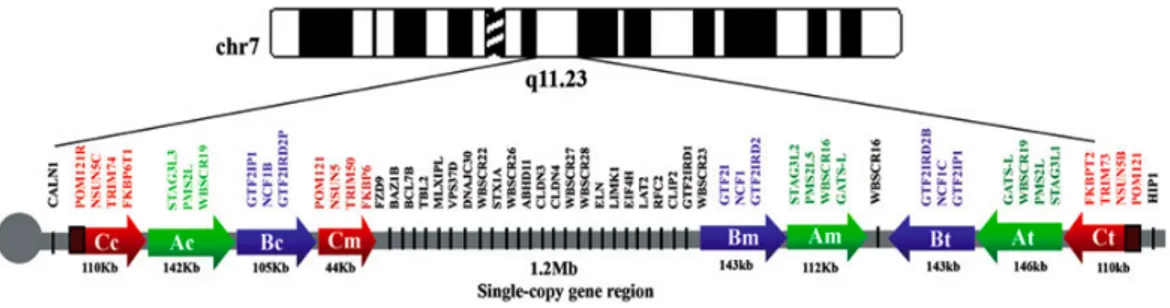
L’architettura della regione cromosomica 7q11.23 è complessa; comprende una regione di 1,2 Mb con geni a singola copia, fiancheggiata da tre Blocchi di sequenze ripetute LCR (Low Copy Repeats) chiamati A B e C di circa 320 Kb ciascuno, ove ciascun Blocco è a sua volta presente in tre complessi: centromenrico (Acen Bcen Ccen) telomerico (Atel Btel Ctel) e mediano (Amed Bmed Cmed), localizzati nelle omonime posizioni all’interno della regione 7q11.23 (Fig. 3).
Lateralmente rispetto ai Blocchi LCR sono presenti regioni ricche in GC ed elementi ripetuti, in particolare sequenze Alu. L’ordine con cui i Blocchi si dispongono all’interno della regione 7q11.23 è: C > A > B per il complesso centromerico e telomerico, C > B > A per il complesso mediano. La direzione di trascrizione dei geni ripetuti interni ai Blocchi, invece, è uguale per il complesso centromerico e mediano (filamento +), inversa per il complesso telomerico (filamento -) (Fig. 4). La mutazione cromosomica trovata con più frequenza in pazienti affetti da WBS è la delezione, ma recentemente sono stati descritti casi di pazienti con WBS portatori di duplicazioni o traslocazioni e in casi ancor più rari di inversioni. Normalmente la WBS insorge in maniera sporadica ma sono stati riportati casi di trasmissione da parte di un genitore con un pattern di WBS lieve o oligosintomatico (Schubert 2008).
Delezione
Il tipo di delezione più comune, riscontrata nel 95% dei pazienti affetti da WBS, interessa una sequenza di 1,5 Mb con Breakpoint all’interno dei Blocchi LCR Bmed e Bcen. Il restante 5% dei pazienti WBS mostra invece una delezione di 1,8 Mb con Breakpoint all’interno dei blocchi LCR Acen e Amid (Fig. 4).
In ambedue i casi la delezione comporta la perdita di 28 geni contigui (Schubert 2008). Il gene di cui si hanno maggiori informazioni riguardo al coinvolgimento nel fenotipo proprio della patologia è il gene ELN, che codifica per l’elastina, componente essenziale del tessuto connettivo.
Le delezioni sono prodotte da una ricombinazione omologa non allelica, rispettivamente tra il blocco Bcen e Bmed e tra il blocco Acen e Amed.
Si ritiene che due fattori importanti giustifichino la maggiore frequenza di ricombinazione tra i Blocchi Bcen e Bmed rispetto ai blocchi Acen ed Amed. Uno riguarda il grado di omologia, infatti l’ identità che c’è tra i due Blocchi B ( 99,6%) è maggiore dell’identità che c’è tra i due Blocchi A (98,2%). L’altro riguarda la distanza che intercorre tra i due blocchi, la lunghezza dell’intervallo genomico tra i Blocchi Bcen e Bmed (circa 1,5 Mb) è minore di quella tra i Blocchi Acen ed Amed (circa 1,84 Mb).
La ricombinazione omologa non allelica può avvenire in maniera intercromosomica, intercromatidica o intracromatidica con la conseguente formazione, nei primi due casi, di un cromosoma con delezione e di uno con la reciproca duplicazione della regione compresa tra le LCR coinvolte nell’appaimento (Fig. 5A), nel terzo caso di un cromosoma deleto e un cromosoma acentrico (Fig. 5B).
Fig.5 A:Schema di
ricombinazione omologa non allelica intercromosomica o intercromatidica tra i blocchi LCR Bmed e Bcen, con conseguente formazione di una delezione e della reciproca duplicazione. B: Ricombinazione omologa non allelica intracromatidica, con formazione di un cromosoma deleto e un cromosoma acentrico.
L’analisi degli aplotipi indica che due terzi delle delezioni derivano da crossing over ineguale tra cromosomi omologhi durante la meiosi mentre in un terzo dei casi le delezione è dovuta a riarrangiamenti intracromosomici tra cromatidi fratelli.
Gli effetti fenotipici in pazienti portatori della delezione sono stati ampiamente identificati e studiati da diversi autori. Nonostante i tratti del viso siano una caratteristica distintiva della sindrome, questi non sono evidenti nei bambini fino circa ai 18-24 mesi di vita; tali tratti si accentuano con l’età e questo è uno dei motivi per i quali la diagnosi può essere tardiva (intorno ai 4-5 anni).
Le principali caratteristiche facciali tipiche di questa sindrome sono: fronte larga, sopracciglia rade, pienezza dei tessuti periorbitali, rime palpebrali corte, epicanto, iridi azzurre o verdi con caratteristica forma stellata, radice del naso infossata, guance prominenti e cadenti, bocca larga con labbra grosse e varie anomalie dentarie (ritardata eruzione, denti piccoli, radici dentarie piccole e sottili, malposizione e malocclusione, eccessivo spazio interdentale e frequente sviluppo di carie). Durante i primi anni di vita i pazienti sono caratterizzati da un’eccessiva irritabilità probabilmente associata all’immaturità del sistema nervoso centrale, ai dolori provocati dall’esofagite o alla ipercalcemia che provoca nei piccoli pazienti vomito e stipsi.
Con l’avvento della pubertà, molto precoce in entrambi i sessi ma con uno scarso sviluppo fisico, questi sintomi di irritabilità diminuiscono o svaniscono del tutto. L’inadeguato sviluppo fisico può derivare da due patologie contingenti alla WBS quali l‘ipotiroidismo e la celiachia. Molti ricercatori hanno riscontrato, infatti, con una prevalenza del 2-40% la presenza di ipotiroidismo, riduzione del volume e forme anomale della ghiandola tiroidea stessa. La delezione del gene ELN per l’elastina, importante componente dei tessuti connettivi, in particolare delle arterie, è responsabile nei pazienti delle malformazioni vascolari e connettivali.
I difetti cordiovascolari rappresentano la maggiore causa di morte per i pazienti WBS e la principale fonte di diagnosi precoce; tra questi difetti cardiovascolari vi sono: la stenosi aortica opraalvolare (SVAS), la stenosi dell’arteria polmonare e l’ipoplasia dell’arco aortico, difetti che insorgono con una percentuale del 50-80%.
Oltre all’apparato cardiovascolare, le anomalie colpiscono anche altri apparati alla base dei quali vi è il tessuto connettivo, come l’apparato gastrointestinale, urogenitale la pelle e l’apparato muscolo-scheletrico. Tra le caratteristiche fenotipicamente visibili si riscontra: l’invecchiamento precoce della cute e la fragilità della stessa, la facilità alle ernie (ombelicali e inguinali), l’iperlassità articolare, la caratteristica voce roca e profonda.
Inoltre anomalie uditive caratterizzano più del 95% degli individui con sindrome di Williams che generalmente manifestano iperacusia o ipersensibilità al suono. L’iperacusia può manifestarsi come un incremento della reazione di trasalimento e di fastidio (ad esempio coprirsi le orecchie)
dell’individuo di fronte ad alcuni suoni, così come una tendenza a sentire le cose, meglio e prima degli altri. Gli individui affetti da WBS hanno, infine, un profilo cognitivo e comportamentale caratteristico, numerosi studi hanno valutato il profilo cognitivo e linguistico associato alla sindrome. Questi studi si sono evoluti da una primitiva generale osservazione basata sui test per la valutazione del QI ad analisi dettagliate degli specifici profili cognitivi e linguistici. Nell’ambito di questi studi la sindrome di Williams è risultata di particolare interesse per il fatto che sembra mostrare un profilo neuropsicologico particolare e definito: cioè prestazioni verbali particolarmente brillanti a fronte di scarse capacità di elaborazione in prove visuo-spaziali, un carattere particolarmente affabile e socievole.La maggior parte degli individui con sindrome di Williams ha un’atipica padronanza del linguaggio: la comprensione è molto più limitata dell’espressione, la quale tende ad essere corretta dal punto di vista grammaticale, complessa e fluente a livello superficiale. Tali risultati rafforzano una concezione modulare dell’intelligenza, cioè l’idea che intelligenza e linguaggio possano essere fra loro abbastanza indipendenti. In media gli individui con sindrome di Williams rientrano nella categoria del ritardo mentale lieve o moderato. In vari studi i punteggi medi di QI degli individui con sindrome di Williams sono risultati compresi tra 50 e 60.
Duplicazione
Le duplicazioni nella regione 7q11.23 sono il risultato dello stesso meccanismo di ricombinazione omologa non allelica descritto per le delezioni; i Breakpoint, anche nel caso delle duplicazioni, si ritrovano all’interno del Blocco LCR Bcen e Bmed. Per un terzo dei casi la duplicazione viene ereditata dai genitori, nei restanti due terzi insorge in maniera sporadica.
La duplicazione della regione compresa tra il Bcen e il Bmed provoca un considerevole aumento dell’espressione dei geni a singola copia della regione di 1,2 Mb compresa tra i Blocchi.
I tratti fenotipici dei pazienti portatori sono stati studiati solo recentemente e il quadro clinico non è completamente definito; tuttavia risulta evidente che le caratteristiche patologiche, così come le caratteristiche facciali sono più miti rispetto alla sindrome di William – Beuren dovuta a delezione; per questo motivo si ritiene possibile che casi di duplicazioni possano rimanere non diagnosticati. Il tratto distintivo dei pazienti con duplicazione è il linguaggio, il ritardo nell’espressione e il mal utilizzo dell’espressione orale, elementi che si riscontrano in tutti i pazienti.
Recentemente l’identificazione del primo caso di triplicazione della regione WBS in pazienti con gravi disfunzioni linguistiche, ha confermato l’ipotesi secondo la quale, il sovradosaggio di alcuni geni coinvolti nella duplicazione provochi l’insorgere di gravi danni alle capacità espressive e al linguaggio degli individui affetti.
autismo, in linea con le caratteristiche fenotipiche di altre sindromi da duplicazione.
In questo tipo di anomalia infine non sono stati riscontrati difetti nella crescita o una bassa statura, e nemmeno problemi legati all’apparato cardiovascolare e al tessuto connettivo , solo in alcuni casi si evidenzia la presenza di SVAS e stenosi dell’arteria polmonare.
Fig.6 schema di un cromosoma con duplicazione. (Merla et al. 2010)
Effetti fenotipici
Delezione 7q11.23
Duplicazione 7q11.23
Caratteristiche facciali Fronte ampia Bassa radice nasale Labbra carnose
Fronte ampia Alta radice nasale Labbra sottili Difetti dello sviluppo e del
sistema endocrino
Ritardo di crescita Ipercalcemia
Normale sviluppo nella crescita
Normocalcemia Anomalie Cardiovascolari SVAS
Ipertensione Difetti cardiaci congeniti
Anomalie del tessuto connettivo
Connettivo e articolazioni lasse
Connettivo e articolazioni lasse
Disfunzioni neurologiche Ipotonia Ipotonia
Difetti a livello cognitivo Ritardo mentale, difetti visuospaziali, particolari potenzialità a livello espressivo,difetti nello sviluppo
Ritardo mentale in pazienti con genitori trasmettenti asintomatici, difetti gravi a livello espressivo, nessun difetto visuospaziale, difetti nello sviluppo.
Difetti comportamentali Eccessiva socievolezza Carattere aggressivo, mancata interazione sociale, autismo.
Tab 2. Differenze nelle caratteristiche cliniche della sindrome di William-Beuren tra portatori di delezione e portatori di duplicazione.
Traslocazioni
Oltre alla traslocazione già citata, t(6;7) )(p21.1;q11.23) , che ha fatto da apri pista allo studio della sindrome di William, Duba e collaboratori hanno caratterizzato una traslocazione ricorrente familiare, la t(7;16) (q11.23;q13), associata ad una espressione molto variabile del fenotipo. Questa si riscontra infatti sia in soggetti essenzialmente asintomatici, che in pazienti con sintomi caratteristici come SVAS ma privi di ritardo cognitivo e dismorfismo facciale o in pazienti che invece presentano anche queste caratteristiche (Fig. 7).
Fig.7 Cariotipo parziale rappresentativo della traslocazione t(7;16)(q11.23;q13)
Il punto di rottura e fusione nei due cromosomi derivati (der7 e der16) è stato clonato e sequenziato; all’interno del cromosoma 7 esso mappa in corrispondenza dell’introne 5 del gene dell’elastina ELN, mentre a livello del cromosoma 16, mappa in corrispondenza del primo introne del gene TM7XN1 (Fig. 8). Dall’espressione del gene di fusione prodotto dalla traslocazione su uno dei due cromosomi derivati si ha una proteina di fusione la cui influenza sul fenotipo della sindrome è ancora in discussione, dal momento che la funzione stessa della proteina TM7XN1 che compone una parte del peptide di fusione non è ancora chiara.
Fig.8 Traslocazione (7;16) (q11.23;q13) posizione Breakpoints
La spiegazione più probabile riguardo alla variabilità del fenotipo nei pazienti portatori di questa traslocazione, si ritiene sia la presenza di un effetto di posizione, che farebbe variare in maniera differenziale l’espressione dei geni contigui a ELN. La differenza tra i fenotipi nelle due traslocazioni suggerisce che Breakpoints diversi a livello del gene dell’elastina possono variare l’effetto fenotipico della sindrome, e dunque è difficile definire una correlazione genotipo-fenotipo che sia univoca (Duba et al. 2002).
Inversione
Dati recenti dimostrano l’esistenza di un’inversione paracentrica nella regione in esame, presente in circa un terzo dei genitori trasmettenti il cromosoma deleto. Si pensa che i soggetti portatori di tale inversione abbiano un rischio maggiore del 25% nel trasmettere il cromosoma deleto alla progenie, perché la probabilità che si verifichi un appaiamento errato durante la meiosi è maggiore a causa della struttura diversa dei due omologhi. L’inversione presente in questi soggetti è causata dall’orientamento invertito delle duplicazioni telomeriche (Atel, Btel e Ctel) rispetto a quelle centromeriche (Acen, Bcen, Ccen). Questa struttura genomica favorirebbe un loop della sequenza tra le due regioni duplicate che, seguito da un evento di ricombinazione, ha come risultato un’inversione (Fig. 9). I geni presenti nella regione invertita non subiscono alterazioni, per cui l’inversione non comporta conseguenze associabili a fenotipi patologici. La lunghezza del frammento invertito varia da 1,79 Mb (se a ricombinare sono i Blocchi Bcen e Bmed) a 2,56 Mb (se a ricombinare sono i Blocchi Ccen e Ctel). I cromosomi risultanti dalle diverse ricombinazioni sono 3 e hanno un’organizzazione genomica diversa a seconda dei Blocchi che hanno ricombinato.
Fig.9 Meccanismo di formazione delle inversioni nella regione 7q11.23.
Contenuto genico della regione 7q11.23:
Modelli animali e trial clinici condotti su diverse famiglie in cui è presente la sindrome di William-Beuren hanno cercato di mettere in luce il rapporto tra il contenuto genico della regione coinvolta e il fenotipo della malattia. L’aploinsufficienza del gene ELN, dovuta alla delezione caratteristica della sindrome, provoca nei pazienti, seri danni al sistema cardiovascolare e al tessuto connettivo in generale. Severe arteropatie e ipertensione sono comunemente osservate nei portatori di WBS. Uno studio ha mostrato che topi transgenici Eln-/- muoiono immediatamente dopo la nascita e presentano iperplasia delle cellule del tessuto muscolare liscio dei vasi sanguigni, mentre topi
Eln+/- che produco il 50% di proteina funzionante, sopravvivono e presentano le caratteristiche
cardiovascolari dei pazienti affetti da WBS anche se con differenze (Li et al. 1998). Queste linee transgeniche sono state create inserendo una mutazione nulla nel gene ELN tramite la ricombinazione omologa in staminali embrionali.
Oltre al gene dell’ ELN, che resta comunque quello di cui si hanno maggiori informazioni, altri due geni, presenti nella regione di geni a singola copia, possono essere considerati importanti nella determinazione del fenotipo della sindrome. Uno è BAZ1B che codifica per una componente del complesso di rimodellamento della cromatina WSTF coinvolto in molte funzione quali la trascrizione la replicazione e la riparazione del DNA, si ritiene essere importante, in quanto è coinvolto nello sviluppo cardiaco, topi Baz1b -/+ e Baz1b -/- mostrano addirittura un parziale sviluppo cardiaco.
L’altro gene è STX1A, il quale codifica per una proteina della membrana plasmatica espressa abbondantemente nei neuroni, è per questo implicata nel mantenimento della plasticità neurale, per questo motivo, sembrerebbe che l’aploinsufficienza di questo gene causata dalla delezione, cointribuisca a determinare gli effetti fenotipici a livello neurologico della WBS.
Nonostante il contributo degli altri geni coinvolti nella delezione non sia ancora del tutto chiaro per alcuni di essi la funzione è conosciuta o presunta. In particolare è chiaro il contenuto genico dei Blocchi A B e C (Fig. 11).
Il Blocco A è composto da quattro differenti pseudogeni: STAG3, PMS2, GATS e WBSCR19, i quali derivano da duplicazioni di altre regioni del cromosoma 7. Il gene attivamente trascritto di cui PMS2 è una duplicazione, ha una funzione di mismatch repair, mentre le funzioni di tutti gli altri geni non sono note.
Il Blocco B è composto da 3 geni GTF2I, GTF2IRD2, NCF1. Le rotture identificate clusterizzano nelle posizioni in cui c’è maggiore identità tra i due Blocchi, ovvero al centro del blocco Bmed, dove mappano i geni GTF2I e NCF1 e al centro del blocco Bcen, dove mappano gli pseudogeni GTF2IP1 e NCF1P1. Il gene GTF2I, codifica per un fattore di trascrizione della famiglia TFII-I;
le sue due copie sui Blocchi Bcen e Btel codificano per una proteina tronca. Studi effettuati sull’espressione del gene GTF2I hanno offerto l’opportunità di capire i meccanismi genetici ed epigenetici a carico di questo gene, hanno dimostrato l’origine parentale del gene stesso, ciò supporta l’idea di un possibile controllo epigenetico del locus e l’ipotesi che possa essere soggetto ad imprinting. Questo aspetto risulta interessante in questo lavoro sperimentale in quanto i geni soggetti ad imprinting sono soggetti ad asincronismo, caratteristica presa in considerazione in questo lavoro. Il gene NCF1 codifica per una componente della NADPH-ossidasi che gioca un importante ruolo nella difesa immunitaria; anche in questo caso le due copie sui blocchi Bcen e Btel codificano per una proteina tronca. Recenti studi hanno dimostrato che pazienti con il Breakpoint all’interno del gene NCF1 presentano una significativa ipertensione rispetto ai pazienti con delezione all’interno del gene GTF2I.
L’analisi genomica del Blocco C ha mostrato la presenza di quattro geni POM121, NSUN5, TRIM50, FKBP6. Il gene POM121, presente sia nel Blocco Ccen che nel Blocco Ctel, è espresso in entrambi i loci e codifica per una componente intermembrana del complesso del poro nucleare. Il gene ancestrale NSUN5 è localizzato nella parte mediana del Blocco C e codifica per una proteina con possibile funzione di metiltrasferasi all’interno del nucleo; le sue due copie che si trovano nella parte telomerica e nella parte centromerica del Blocco C, codificano per una proteina tronca che, così come quella ancestrale, è ubiquitariamente espressa.
Il Tempo di Replicazione
Il ciclo cellulare è la serie di eventi che avvengono in una cellula eucariote tra una divisione cellulare e quella successiva. Il ciclo cellulare è un processo geneticamente controllato, costituito da una serie di eventi coordinati e dipendenti tra loro, dai quali dipende la corretta proliferazione delle cellule eucariotiche. Gli eventi molecolari che controllano il ciclo cellulare sono ordinati e direzionali: ogni processo è la diretta conseguenza dell'evento precedente ed è la causa di quello successivo. Le fasi che costituiscono il ciclo cellulare sono:
• G1 (gap) : nella quale la cellula monitora le sue dimensioni e l’ambiente esterno e cresce sintetizzando RNA e proteine;
• S (sintesi) : durante la quale avviene la replicazione del materiale genetico;
• G2: rappresenta una fase in cui avviene il controllo dell’avvenuta replicazione di tutti i cromosomi e una crescita secondaria prima della mitosi;
• M (mitosis): si compone di profase, metafase, anafase, telofase e citochinesi.
Durante la fase S i segmenti cromosomici si replicano tramite un innesco sincrono di clusters di origini di replicazione. Questi segmenti, detti domini di replicazione, si replicano in un ordine temporale ben definito, relativo ai diversi tipi cellulari (Watenabe and Maekawa 2010).
Inizialmente tali domini di replicazione sono stati studiati tramite la marcatura con bromodeossiuridina (BrdU), analogo della timidina, che somministrata alle cellule a diversi intervalli di tempo durante la fase S, per brevi periodi, detti pulse, mostra un pattern d’incorporazione sui cromosomi in metafase e un caratteristico bandeggio corrispondente al bandeggio G ottenuto attraverso la colorazione con Giemsa. Il bandeggio G definisce nei cromosomi due tipi di bande, le G (più scure) ricche in AT e le bande R (più chiare) ricche in GC. L’analisi con BrdU dà una prima dimostrazione della corrispondenza tra domini a tarda replicazione, identificati nelle bande G, e regioni ricche in AT a basso contenuto genico e la corrispondenza tra domini a replicazione precoce identificati nelle bande R e regioni ricche in GC ad elevato contenuto genico (Mendez 2009; Gilbert et al. 2009).
Lo studio del tempo di replicazione si è servito di metodi molecolari quali la PCR e la FISH. Nel primo metodo il DNA viene isolato a diversi intervalli durante la fase S, purificato e amplificato con primer specifici per i loci d’interesse; comparando poi i vari prodotti di PCR corrispondenti ai vari intervalli della fase S si ottiene una stima del tempo di replicazione delle sequenze scelte. La FISH invece utilizza l’ibridazione in situ con sequenze locus specifiche e permette di distinguere loci non replicati, che all’analisi microscopica presentano un solo segnale di fluorescenza, dai loci replicati che presentano invece due segnali di fluorescenza (Mendez 2009). Comparando la
frequenza dei due tipi di segnali, si può avere una stima del tempo di replicazioni in ciascun locus (Watenabe and Maekawa 2010).
Con la scoperta e l’evoluzione dei microarray, il tempo di replicazione è stato analizzato anche a livello genomico. Questa metodica prevede l’utilizzo di sonde marcate in grado di quantificare il numero di copie delle diverse regioni del genoma. Uno dei vantaggi di questa tecnica è che permette di analizzare allo stesso tempo diverse regioni del genoma e di confrontare i dati ottenuti con caratteristiche genetiche ed epigenetiche (Mendez 2009). Per l’analisi del tempo di replicazione vengono utilizzate cellule non sincronizzate in fase S e in fase G1, il cui DNA viene fatto ibridare con le sonde presenti sull’array che coprono l’intero genoma ad intervalli di 1MB. Ogni regione genomica viene classificata a replicazione precoce (S1) intermedia (S2-S3) o tardiva (S4) in base al rapporto S:G1; la percentuale di nuclei nella fase S in cui una specifica sequenza risulta replicata è proporzionale al tempo in cui questa stessa si replica. Le sequenze che si replicano precocemente hanno un rapporto S:G1 pari a 2:1, perché sono regioni che essendo già replicate hanno un contenuto doppio di DNA in S rispetto a G1; mentre le regioni che sono tardo replicanti hanno un rapporto 1:1, dal momento che in piena fase S ancora non hanno raddoppiato il loro DNA. Sulla base di questi dati è stato definito il tempo di replicazione medio dei cromosomi umani (Tab. 3) (Woodfine et al. 2004).
Tab.3 Tempo di replicazione medio per ciascuno dei 24 cromosomi umani.
Gli studi eseguiti hanno mostrato una stretta correlazione tra il tempo di replicazione e particolari caratteristiche genomiche. In particolare si riscontra una correlazione positiva tra tempo di replicazione precoce, alto contenuto in GC, alta densità genica e presenza rilevante di elementi Alu (Strachan and Read 2006 UTET). Questa analisi è stata realizzata anche a livello cromosomico ad una più elevata risoluzione, in particolare sul cromosoma 22 (Woodfine et al. 2004) e sul cromosoma 6 (Woodfine et al. 2005). I risultati rispecchiano quanto detto:le regioni cromosomiche che hanno più geni si replicano prima di quelle con meno geni, seguendo un pattern di replicazione non uniforme lungo ciascun cromosoma, ma con un alternanza di regioni a replicazione precoce e tardiva, che corrispondono rispettivamente alle bande R e alle bande G. Le bande ricche in GC si replicano nella prima metà della fase S, le regioni povere in GC si replicano tardivamente.
A partire dai questi risultati, è stato poi dimostrata la correlazione dei cambiamenti del tempo di replicazione con l’organizzazione spaziale della cromatina all’interno del nucleo, in cellule diploididi primati, attraverso la FISH in 3D (Grasser et al. 2008). Sonde specifiche per sequenze precedentemente identificate come replicanti precocemente (S:G1 > 1,19) o tardo replicanti (S:G1 <1,5) sono state marcate con fluorocromi differenti. L’analisi è stata condotta sia su tutto il genoma, sia su due cromosomi con valori limite del rapporto S:G1, cioè il cromosoma 5 con replicazione prevalentemente molto tardiva, e il cromosoma 17 con replicazione prevalentemente molto precoce La stima dei risultati quantitativi ottenuti tramite la FISH 3D rivela una distribuzione non casuale all’interno del nucleo delle diverse regioni,quelle replicate precocemente si posizionano nella parte interna del nucleo, mentre le regioni a replicazione tardiva si posizionano nella parte periferica (Fig. 12) (Mendez 2009, Grasser et al. 2008). Questi risultati sono supportati dal ritrovamento delle proteine responsabili della condensazione della cromatina alla periferia del nucleo.
Molti studi effettuati sull’intero genoma umano hanno inoltre messo in luce una stretta relazione tra il tempo di replicazione e l’espressione genica. Infatti si osserva che i geni housekeeping, ossia quelli costitutivamente trascritti e tradotti ad un livello relativamente elevato in tutte le cellule, sono replicati nella fase S precoce. In questa fase sono replicati, esclusivamente nelle cellule in cui sono espressi, anche i geni tessuto specifici; un classico esempio è rappresentato dal gene della β-globina il quale rappresenta, in cellule eritroidi, una regione a replicazione precoce caratterizzata da cromatina acetilata, in cellule non eritroidi una regione tardo replicante caratterizzata da cromatina ipoacetilata (Gilbert 2002). I geni non espressi si replicano nella fase S tardiva e l’eterocromatina, che è completamente inattiva dal punto di vista trascrizionale, è replicata nella fase S molto tardiva (Mendez 2009; Gilbert 2002).E’ interessante notare come i geni coinvolti nella stress cellulare e nella regolazione del processo apoptotico, che quindi non sono espressi durante la normale proliferazione, ma che devono essere rapidamente trascritti in particolari circostanze, rientrano nelle regioni a replicazione precoce (Mendez 2009). In base a quanto già detto, e grazie ad altri studi è stata dimostrato, quindi, un nesso tra tempo di replicazione, struttura della cromatina e l’attività trascrizionale.
La relazione tra il tempo di replicazione e l’attività trascrizionale è spiegata da due teorie diverse ma non alternative (Gilbert et al. 2009). Secondo la prima teoria il DNA replicato precocemente avrebbe un vantaggio competitivo nei confronti del legame con attivatori della trascrizione, rispetto al DNA tardo replicante, per cui tali attivatori, presenti nella cellula in basse concentrazioni, andrebbero a legare solo il DNA replicato precocemente, permettendo l’espressione dei geni lì
Fig.12 Localizzazione nucleare di domini a replicazione precoce e domini tardo-replicanti.
presenti (Fig. 13 a). Il secondo modello prende in considerazione la struttura della cromatina come regolatore della replicazione del DNA; questa teoria prevede che le regioni tardo replicanti abbiano una struttura della cromatina più compatta che impedisce fisicamente l’accesso alle proteine responsabili della replicazione, in questo modo, essendo la struttura della cromatina correlata all’espressione genica, essa regolerebbe sia la trascrizione che la replicazione del DNA (Fig. 13 b).
Fig.13I due modelli che spiegano il legame tra la replicazione e la trascrizione del DNA.
Recentemente Cedar e collaboratori, per dimostrare la relazione tra tempo di replicazione e modificazioni della cromatina, hanno mostrato che il reclutamento dell’enzima istone acetil trasferasi (HATs) a livello del locus della globina, in linee cellulari umane non eritroidi, è sufficiente a cambiare il tempo di replicazione da tardivo a precoce; viceversa, l’azione dell’enzima istone deacetilasi in cellule eritrocitarie è sufficiente a cambiare il tempo di replicazione del locus della globina da precoce a tardivo (Mendez 2009).
la replicazione hanno suggerito che il tempo di replicazione del DNA viene ristabilito per ciascun dominio di replicazione ad ogni ciclo cellulare durante la fase G1 precoce attraverso modificazioni della cromatina (Dimitrova and Gilbert 1999). Questo periodo di tempo discreto in fase G1 all’interno del quale viene ristabilito il programma temporale di replicazione prende il nome di
timing decision point (TDP) e corrisponde al momento in cui, dopo la mitosi, il DNA viene
riposizionato nel nucleo (Mendez 2009; Gilbert 2002; Ryba et al. 2010). Il modello è stato proposto con studi in lievito e poi confermato nell’uomo. Secondo questo modello i regolatori della cromatina si disperdono durante la mitosi per poi concentrarsi in compartimenti subnucleari ben precisi. Una volta raggiunti questi compartimenti, la cromatina viene modificata in trans dalla concentrazione locale dei suddetti regolatori. Quindi in base alle caratteristiche strutturali così assunte, viene ridefinito il tempo di replicazione (Gilbert 2002). Il timing decision point coincide quindi con la creazione dei domini cromosomici di eterocromatina ed eucromatina nei nuclei interfasici (Watenabe and Maekawa 2010).
Fattori epigenetici come la stato della cromatina hanno quindi un profondo effetto sul complesso deputato alla replicazione del DNA; un esperimento in particolare ha dimostrato come la sovraespressione del NoRC (nucleolar chromatin remodelling complex) modifica il tempo di replicazione di geni ribosomiali da precoce a tardivo, confermando l’importanza di tale complesso, e dunque dello stato della cromatina nella determinazione del tempo di replicazione (Watenabe and Maekawa 2010).
Il genoma di tutti i vertebrati è composto in larga scala da strutture compartimentalizzate contenenti lunghi tratti di DNA con una percentuale variabile del contenuto in GC. Tali regioni di circa 300KB con contenuto omogeneo in GC prendono il nome di isocore; le isocore del genoma umano sono di 5 tipi, di cui due (L1 e L2) a basso contenuto in GC e tre (Hi, H2 e H3) ad alto contenuto in GC. Tali isocore caratterizzano diversamente le bande R e G; ad esempio le bande R hanno il maggior presenza di isocore H3. Tali bande sono separate tra loro da regioni di transizione del tempo di replicazione, regioni grandi e prive di origini di replicazione. Nelle zone di transizione la forca replicativa proveniente dalla regione di cromatina attiva a replicazione precoce deve subire un periodo di stallo prolungato prima di congiungersi o fondersi con quella della regione tardo replicante confinante. Queste caratteristiche delle regioni di transizione del tempo di replicazione fanno si che esse siano associate ad un’elevata probabilità di danno. Lo stallo della forca replicativa aperta favorisce rotture a doppio filamento del DNA e conseguenti riarrangiamenti cromosomici (Fig. 14) (Watanabe et al. 2004, Watenabe and Maekawa 2010) .
Fig.14 Organizzazione del cromosoma in bande R/G a replicazione rispettivamente precoce e tardiva. Caratteristiche delle zone di transizione del tempo di replicazione.
Caratteristica cromosomica Banda G/Q Banda R Banda T (sottogruppo banda R)
Tempo di replicazione Tardivo Precoce Molto Precoce
GC% Ricca in AT Medio Ricca in GC
Densità Genica Bassa Elevata Molto Elevata
Struttura della cromatina Compatta Lassa Lassa
Tab.4 Relazione tra tempo di replicazione contenuto in GC densità genica e struttura della cromatina
Il rapporto tra tempo di replicazione e differenziamento rappresenta un altro interessante ramo di questo ambito della ricerca. Studi sui cambiamenti che interessano il tempo e i domini di replicazione durante il differenziamento cellulare hanno consentito di mappare ad elevata risoluzione il tempo di replicazione di cellule staminali embrionali e di cellule indotte al differenziamento neurale (NPCs) (Mendez 2009). I confini dei domini di replicazione hanno posizioni ben definite in linee di cellule staminali diverse, dimostrandosi così una caratteristica distintiva della staminalità, ossia della pluripotenza; per dimostrare ciò, sono stati confrontati i domini di replicazione di cellule staminali embrionali (ESCs) e quelli di cellule riprogrammate (iPS) che non hanno mostrato alcuna differenza. Nelle ESCs i domini di replicazione sono molto più piccoli e non coincidono con le isocore e il loro contenuto in GC. Essendo molto più piccoli, il
DNA presenta molte più zone di transizione del tempo di replicazione; questo rende il DNA delle cellule staminale molto più suscettibile a rotture a doppio filamento in quanto la forca re plicativa lavora su una maggiore distanza e aumenta i suoi periodi di stallo (Gilbert et al. 2009; Ryba et al. 2008).
Numerosi studi hanno dimostrato che oncogeni e geni onco-soppressori sono concentrati nelle zone di transizione del tempo di replicazione; un’analisi condotta su 15 di questi geni a livello delle regioni cromosomiche 11q e 21q ha mostrato che essi si trovano in regioni ricche in GC ma adiacenti a regioni povere in GC, ricoprendo quindi le regioni di transizione (Watanabe et al. 2009).
Allo stesso tempo le regioni di transizione sono soggette ad elevata frequenza di ricombinazione. Watanabe et al, hanno evidenziato anche la presenza di SNPs (single nucleotide polymorphism) a livello delle regioni di transizione del tempo di replicazione. Queste regioni inoltre spesso vanno incontro a riarrangiamenti cromosomici (traslocazioni, delezioni, inserzioni e duplicazioni) che coinvolgono gli oncogeni e gli onco-soppressori. Le regioni di transizione del tempo di replicazione sono anche caratterizzate dalla presenza di duplicazioni segmentali che spesso possono assumere conformazioni non-B della doppia elica del DNA. Watanabe et al, hanno dimostrato una maggiore presenza di strutture non-B del DNA a livello di regioni di transizione rispetto ad altre regioni genomiche, cosa che ciò aumenta l’instabilità genetica (Watenabe and Maekawa 2010).
Quanto detto suggerisce che le regioni di transizione del tempo di replicazione corrispondono a regioni instabili del genoma, associate all’aumento del danno al DNA (Watanabe et al. 2008). In merito a duplicazioni segmentali associate a regioni di transizione del tempo di replicazione, un recente lavoro ha dimostrato l’instabilità della regione 22q11.2, contenente 4 blocchi LCRs esede di delezioni, duplicazioni e traslocazioni, associate a disordini genomici, quali la sindrome di DiGeorge (Puliti et al. 2010). L’approccio utilizzato nel nostro lavoro sperimentale rispecchia quello utilizzato nel lavoro sopraccitato. Per valutare se l’instabilità delle regione fosse legata alla presenza di zone di transizione del tempo di replicazione gli autori hanno analizzato lo stato replicativo dei blocchi LCRs stessi, dimostrando la presenza al loro interno di una regione di transizione del tempo di replicazione. Per questa regione è stato inoltre mostrata una correlazione tra la regione di transizione del tempo di replicazione e la presenza di picchi di flessibilità, asincronismo replicativo e suscettibilità allo stress replicativo indotta da Afidicolina (APC), sostanza che induce fragilità nelle regioni genomiche instabili. Hanno inoltre mostrato, come i punti di rottura dei riarrangiamenti che coinvolgono comunemente questa regione, si trovino all’interno di duplicazioni segmentali ed in corrispondenza dei suddetti picchi di flessibilità.
di una regione cromosomica, può contribuire alla comprensione di meccanismi molecolari che stanno alla base dell’instabilità genomica e alla localizzazione, in un certo senso, delle regioni più suscettibili a mutazioni, potenziali causa di patologie.
L’Asincronismo Replicativo
Ogni gene è presente nel genoma di ciascun individuo con due alleli, che si replicano in maniera sincrona ed hanno, in assenza di mutazioni, un’espressione biallelica. Una classe limitata di geni ha tuttavia un’espressione monoallelica. Di questi geni è trascritto solo uno dei due alleli, in quale si replica precocemente mentre l’allele non espresso mostra un ritardo replicativo. Il fenomeno è noto come asincronismo replicativo. Uno studio condotto su una regione rappresentativa dell’intero genoma mostra che circa il 20% di questo si replica in maniera asincrona. Questo pattern di replicazione è definito pan-S, perché le regioni si replicano in più stadi della fase S. Esempi caratteristici di geni con espressione monoallelica sono l’inattivazione del cromosoma X nelle femmine dei mammiferi e i geni sottoposti ad imprinting.
L’inattivazione del cromosoma X nelle femmine di mammifero è un fenomeno casuale, nel senso che può essere inattivato sia il cromosoma X di origine paterna che quello di origine materna con la stessa probabilità. L’inattivazione avviene in una fase precoce dello sviluppo e viene mantenuta durante tutta la vita dell’individuo. Questo avviene per bilanciare la dose trascrizionale tra le femmine XX e i maschi XY, con il risultato che ogni cellula somatica delle femmine ha attivo un solo cromosoma, o quello paterno o quello materno.
L’imprinting parentale è un classico esempio di geni ad espressione monoallelica, caratterizzati da una modalità di espressione determinata in base all’origine parentale. Per i diversi geni soggetti ad imprinting gli alleli sono espressi o solo se ereditati dal padre (si parla in questo caso di imprinting materno visto che l’allele ereditato dalla madre è inattivato) o se ereditati dalla madre (in questo caso invece si parla di imprinting paterno per analoghi motivi). La conseguenza dell’imprinting parentale è che i geni sono espressi come se si trovassero in una condizione di emizigosi, mentre in realtà ci sono due copie di ciascuno di questi geni autosomici in ogni cellula. Il meccanismo esatto con cui avviene l’imprinting è complesso. Il processo è regolato epigeneticamente; un allele deve acquisire un marcatore epigenetico differenziale durante la gametogenesi e questo marcatore è poi mantenuto in maniera autonoma durante lo sviluppo regolando lo stato di espressione/silenziamento genico. Tra i processi che hanno d un ruolo importante nella regolazione del silenziamento, vi è la metilazione del DNA; questo processo agisce in cis sulle isole CpG modulando l’interazione tra DNA e le proteine del complesso di trascrizione. La metilazione del DNA ha una funzione cruciale nel regolare l’espressione monoallelica dei geni; nel cromosoma X, ha un ruolo sia nell’inattivazione sia nel silenziamento dei geni presenti sul cromosoma inattivato. Nell’imprinting parentale, la metilazione rappresenta un marker, grazie al quale è riconosciuta l’origine paterna o materna dell’allele silenziato.
espressione monoallelica citati; le regioni inattive infatti presentano una cromatina molto compattata e dunque inaccessibile alle proteine dell’apparato trascrizionale.
L’asincronismo replicativo caratterizza gran parte dei geni ad espressione monoallelica. Come la metilazione del DNA, esso è un marcatore epigenetico dei geni soggetti ad imprinting. La replicazione asincrona dei due alleli, quindi la replicazione precoce del gene espresso e quella tardiva del gene soggetto ad imprinting, è visibile non appena l’imprinting è stato definito. Questo vuol dire che l’asincronismo replicativo è un marker primario per questi geni. Replicazione asincrona è stata osservata anche per i geni del cromosoma X; è stato suggerito che essa abbia un ruolo nel mantenere inattivo il cromosoma (Watanabe et al. 2000).
E’ stato riscontrato un legame tra asincronismo replicativo e la presenza di aneuploidie, numero alterato dei cromosomi di una cellula; per esempio nelle trisomie umane, la presenza di un cromosoma in più, forza i geni biallelici a cambiare il loro normale modello di replicazione sincrono verso un allele specifico (Korenstein-Ilan et al. 2002). L’asincronismo è stato inoltre mostrato in presenza di patologie tumorali; in particolare, studi su cellule del midollo osseo e del sangue periferico di pazienti con tumori ematologici, su cellule del carcinoma della cervice e anche su linfociti derivati da pazienti con carcinoma renale, hanno dimostrato associazione tra lo stato tumorale e l’asincronismo replicativo di oncogeni, quali c-myc e geni soppressori di tumori, quali RB1.
L’asincronismo replicativo caratterizza anche le duplicazioni segmentali. Uno studio condotto sulla regione 22q11.2 (Fig. 15), evidenzia che tale regione si replica in maniera asincronasia in soggetti sani che malati; di particolare interesse risulta il fatto che l’omologo paterno si replica prima di quello materno in tutti i casi studiati, suggerendo che il fenomeno non avviene in maniera casuale. Questa replicazione asincrona, non casuale, potrebbe rappresentare un fattore di rischio per la formazione di delezioni aumentando la probabilità di un iniziale mal appaiamento tra le duplicazioni segmentali presenti in tale regione.
Fig.15 Rappresentazione schematica di appaiamenti errati in omologhi caratterizzati da asincronismo replicativo. (A) Struttura delle LCRI e LCRII sul cromosoma 22q11.2 responsabili dell’ appaiamento errato. (B) In una situazione in cui c’è replicazione sincrona o asincronismo random le LCR sono libere di appaiarsi. (C) Nel caso in cui l’asincronismo sia legato ad un solo omologo l’appaiamento tra l’LCRII paterna e LCRI materna è possibile che avvenga quando la LCRII materna ancora non è replicata e questo quindi favorisce la ricombinazione omologa non allelica (Baumer et al. 2004).
Da quanto detto si può dedurre che l’asincronismo di tale regione è una caratteristica non associata agli stati patologici, ma è legata alla struttura della regione 22q11.2 e in particolare alla presenza delle duplicazioni segmentali.
La flessibilità del DNA
La molecola di DNA ha una struttura di base molto flessibile perché il suo scheletro non presenta doppi legami che non possono ruotare. Tale struttura quindi sarebbe libera di ruotare nello spazio; in realtà però non è così perché alcuni atomi del DNA vengono stabilizzati da legami con gruppi laterali o dalle interazioni con le proteine. Ci sono dunque, alcune regioni del DNA meno flessibili perché costrette da queste interazioni ad assumere una determinata posizione nello spazio, e regioni più flessibili perché sono libere da queste costrizioni. Le due costrizioni più importanti sono legate agli angoli torsionali all’interno dell’anello dello zucchero e l’angolo torsionale del legame β-glicosidico.
Fig.16 Unità nucleotidica inserita nello scheletro del DNA. Sono indicate tutti i potenziali legami intorno a cui gli atomi possono ruotare e modificare così la geometria del DNA.
Lo zucchero, a causa di forze attrattive e repulsive, non è planare e in particolare può assumere due conformazioni la Envelope (4 dei 5 atomi dell’anello sono complanari) e la Twist (3 dei 5 atomi sono complanari). Così anche il residuo legato al legame β-glicosidico può assumere due conformazioni la Syn e la Anti le quali si differenziano, perché la Anti ruota di 180° intorno al legame. Queste diverse conformazioni ovviamente possono cambiare la struttura del DNA (Sarai et al 1989).
E’ stato riportato che regioni di DNA altamente instabili sono associate a valori di flessibilità abbastanza elevati; cluster di picchi di flessibilità sono stati identificati, ad esempio, a livello dei siti fragili comuni più espressi, il FRA3B e il FRA16D (Mishmar et al. 1998); similmente la regione 22q11.21 è risultata caratterizzata da evidenti picchi di flessibilità in corrispondenza delle regioni di duplicazione segmentale (Puliti et al. 2010). Per questo, anche nel caso della regione presa in esame
in questo lavoro sperimentale può essere utile caratterizzare la posizione dei picchi di flessibilità e confrontarla con le altre caratteristiche genomiche sopra citate.
Fig.17 Coordinate spaziali usate per effettuare le misure relative alle rotazioni delle basi azotate nel DNA.
Perpendicolare al piano dell’elica