Capitolo 4
La fisiologia del metabolismo miocardico
e la sua alterazione nella Cardiomiopatia Dilatativa
Abbiamo finora affrontato il problema del significato della disfunzione del microcircolo coronarico in varie condizioni patologiche. Ci siamo poi soffermati con particolare attenzione sul ruolo che essa può avere nell’ambito della Cardiomiopatia Dilatativa e, più estesamente, nell’insufficienza cardiaca, partendo dalla premessa che la CMD rappresenta un modello di studio della disfunzione contrattile stessa.
L’argomento di questo capitolo sarà il metabolismo del cuore, che al microcircolo coronarico è intimamente collegato per i motivi che di seguito vedremo. Il concetto risulta comunque ovvio di per sé, se consideriamo che tutto ciò di cui necessita il cuore per svolgere la sua funzione altro non è che ossigeno e substrati energetici da ossidare e che solamente una reciproca modulazione tra flusso e metabolismo a livello microcircolatorio può garantire la persistenza di un’adeguata funzione contrattile.
Affronteremo dapprima, così come abbiamo fatto per il microcircolo coronarico, il metabolismo miocardico in termini fisiologici, descrivendo cioè le sue caratteristiche nel cuore normale. Successivamente vedremo come esso risulti cronicamente alterato nella Cardiomiopatia Dilatativa e, più in generale, nell’insufficienza cardiaca.
Metabolismo miocardico in condizioni fisiologiche
L’energia necessaria per il lavoro della cellula cardiaca deriva dall’idrolisi dell’ATP. Grazie alla rottura dei legami fosfato ad alto contenuto energetico sono rese termodinamicamente possibili le tre funzioni chiave del cardiomiocita:
• il lavoro contrattile (interazione actina-miosina e accorciamento delle miofibrille);
• il passaggio del Ca++ nel reticolo sarcoplasmatico, evento indispensabile per
il rilasciamento diastolico;
• il mantenimento dell’equilibrio ionico (principalmente l’attività della pompa Na+ /K+ ATPasi).
In normali condizioni di lavoro circa 2/3 di ATP idrolizzato dal cuore è utilizzato per il lavoro contrattile, il 1/3 restante per le pompe ioniche (figura 4.1).
Interazione actina-miosina Glicolisi anaerobia ATP Captazione di Ca++ Omeostasi ionica 70% 30% Fosforilazione ossidativa NADH/FADH2 Catena di trasporto degli elettroni ATPasi Glucosio Piruvato 2% 98% Interazione actina-miosina Glicolisi anaerobia ATP Captazione di Ca++ Omeostasi ionica 70% 30% Fosforilazione ossidativa NADH/FADH2 Catena di trasporto degli elettroni ATPasi Glucosio Piruvato 2% 98%
Figra 4.1 Produzione di ATP e suo ruolo. La quasi totalità della produzione di ATP deriva dalla fosforilazione ossidativa mitocondriale (98%) e in minima parte dalla glicolisi anaerobia (2%). L’ATP prodotto fornisce energia sia per il lavoro contrattile (70%) sia per l’omeostasi di calcio e ioni (30%).
Così come in ogni cellula del nostro corpo, le principali forme di energia per il cardiomiocita sono costituite da acidi grassi e carboidrati (in particolare il glucosio), con un minor contributo da parte di lattato e corpi chetonici. In normali condizioni di apporto di ossigeno, la maggior parte (>98%) di ATP proviene dalla fosforilazione ossidativa dei mitocondri, la “centrale energetica” delle nostre cellule, mentre solo una piccola frazione (<2%) deriva dalla glicolisi anaerobia (Ingwall, 2001) (figura 4.1). Nella fosforilazione ossidativa, l’energia chimica delle molecole ridotte di NADH e FADH2, prodotte nella β-ossidazione degli acidi grassi,
nell’ossidazione del piruvato e nel ciclo di Krebs, è sfruttata per costituire un gradiente elettrochimico di H+ a livello della membrana mitocondriale interna. Tale
gradiente elettrochimico rappresenta, dal punto di vista termodinamico, la forma di energia potenziale necessaria affinché l’enzima ATPasi sintetizzi la “moneta energetica” delle nostre cellule, l’ATP, a partire da ADP e fosfato inorganico (Pi). Le velocità del consumo di ATP, della fosforilazione ossidativa e del trasferimento di elettroni dagli equivalenti ridotti FADH2 e NADH alla loro catena
di trasporto sulla membrana mitocondriale interna sono regolate dalle esigenze energetiche della cellula. Nel cuore normale la velocità di idrolisi dell’ATP è esattamente equivalente a quella di sintesi, tant’è che il contenuto tissutale di ATP è estremamente costante persino quando il cardiomiocita lavora al massimo della sua capacità. L’importanza della sua continua rigenerazione risulta immediata se si considera che il ridotto contenuto tissutale di ATP (5 μmol/g di tessuto); se esso non venisse continuamente riprodotto, in un tempo di appena 10 secondi si esaurirebbe completamente l’energia delle cellule e dell’intero tessuto miocardio (Ingwall, 2001; Opie, 1991).
L’entità dell’attivazione delle varie vie metaboliche e la velocità di metabolizzazione dei loro substrati sono controllate sia dall’espressione delle proteine coinvolte nel metabolismo cellulare (enzimi e trasportatori) sia da una complessa rete di regolazione che agisce attraverso la modulazione allosterica dell’attività enzimatica ed è influenzata dalle concentrazioni intracellulari di substrati e prodotti. In condizioni di massimo lavoro cardiaco la macchina metabolica cellulare consuma ossigeno all’80-90% della capacità mitocondriale (Mootha et al, 1997), mentre in condizioni di riposo il cuore lavora al 15-25% della sua massima capacità ossidativa. Durante i rapidi incrementi di richieste energetiche la produzione di ATP non viene a mancare proprio grazie al fatto che le vie metaboliche cellulari possono esser rapidamente attivate o disattivate dalla modulazione allosterica degli enzimi regolatori, dalle variazioni di concentrazione di metaboliti stimolatori o inibitori o dalla traslocazione di proteine ad azione metabolica al loro sito d’azione (Fell et al, 1998). Questi meccanismi consentono il rapido adattamento di fronte a stress acuti quali l’esercizio, l’ischemia o il digiuno.
Vie metaboliche del cardiomiocita
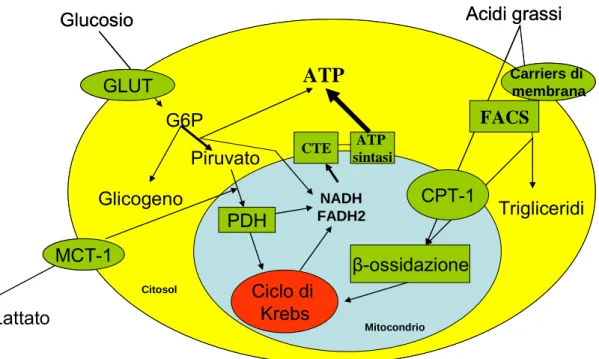
Le principali vie metaboliche dei cardiomiociti sono rappresentate da quella glucidica (che riguarda glucosio e lattato), quella degli acidi grassi e quella dei corpi che tonici.
Il glucosio penetra nel cardiomiocita attraverso un processo di diffusione facilitata mediato da specifici trasportatori di membrana, in particolare il GLUT4. Una volta nel citoplasma, esso viene fosforilato in posizione 6 (glucosio-6-fosfato) ad opera della glucochinasi, il che ne impedisce la retrodiffusione verso l’ambiente extracellulare. Una minima quantità si trasforma in glicogeno, mentre la maggior parte entra nel processo glicolitico, dove glucosio-6-fosfato e NAD+ vengono
trasformati, rispettivamente, in piruvato e NADH con contemporanea produzione di 2 molecole di ATP per molecola di piruvato prodotto. Quest’ultimo, migrato per diffusione semplice nel mitocondrio, viene prima trasformato ad acetil-CoA dalla piruvato deidrogenasi (PDH) per poi entrare nel ciclo di Krebs (o ciclo degli acidi tricarbossilici o ciclo dell’acido citrico) dove viene definitivamente ossidato a CO2
con produzione di NADH e FADH2 (figura 4.2).
Il trasporto del lattato attraverso il sarcolemma cardiaco è facilitato dal trasportatore degli acidi monocarbossilici-1 (monocarboxilic acid transporter-1, MCT-1). Il miocardio non ischemico è un suo consumatore persino in condizioni di lavoro cardiaco sottomassimale ed è noto che in condizioni normali viene estratto dalla circolazione arteriosa ma allo stesso tempo in parte liberato nel torrente venoso (Kaijser et al, 1992). La sua liberazione avviene in quantità maggiore rispetto alla captazione solamente quando la via glicolitica è accelerata e l’ossidazione del piruvato rallentata, condizione che si verifica nell’ischemia (Depre et al, 1998). Gli acidi grassi, liberati dal plasma nell’interstizio cellulare, oltrepassano la membrana in parte secondo un gradiente di concentrazione, in parte mediante trasportatori di membrana, come il FAT/CD36 (acronimo di fatty acids translocase) o FABPmp (plasma membrane fatty acids binding protein) (Schaffer et al, 2002). Nel citoplasma essi si legano ad una molecola di coenzima A grazie all’acil-CoA sintasi, enzima prevalentemente legato al dominio citoplasmatico dei trasportatori di membrana. L’equivalente molecola di acil-CoA per il 10-30% si accumula sotto forma di trigliceridi, mentre il restante 70-90% viene trasportato dalla carnitina palmitoil-transferasi-1 (CPT-1) all’interno del mitocondrio, dove, nel processo della β-ossidazione, viene trasformata in acetil-CoA ed entra così nel ciclo di Krebs (Schaffer et al, 2002) (figura 4.2).
Il cuore estrae ed ossida corpi chetonici (β-idrossi butirrato e acetoacetato) in modo dipendente dalla concentrazione plasmatica (Chen et al, 1984). Dal momento che essa è solitamente molto bassa, i corpi chetonici rappresentano un substrato di minor importanza per il miocardio. In condizione di digiuno, invece, la loro concentrazione plasmatica cresce, a causa dei livelli bassi di insulina ed elevati di acidi grassi, e diventano perciò un substrato miocardico importante (Avogaro et al, 1990).
Glucosio Acidi grassi
Glicogeno β-ossidazione Piruvato Trigliceridi G6P GLUT Ciclo di Krebs Carriers di membrana CPT-1 FACS PDH NADH FADH2 CTE ATP sintasi ATP Mitocondrio Citosol MCT-1 Lattato
Glucosio Acidi grassi
Glicogeno β-ossidazione Piruvato Trigliceridi G6P GLUT Ciclo di Krebs Carriers di membrana CPT-1 FACS PDH NADH FADH2 CTE ATP sintasi ATP Mitocondrio Citosol MCT-1 Lattato
Figura 4.2. Principali vie metaboliche del metabolismo miocardio. (GLUT=trasportatori di glucosio; G6P= glucosio-6-fosfato; PDH=piruvato deidrogenasi; CTE= catena di trasporto degli elettroni; CPT-1= carnicina-palmitoil-transferasi-1; FACS= fatty acids transporter; MCT-1=monocarboxylic acid transporter-1)
Regolazione del metabolismo miocardio
Le vie metaboliche sopra descritte non agiscono indipendentemente l’una dall’altra, ma si influenzano vicendevolmente in senso tendenzialmente inibitorio. Gli esperimenti di Randle di oltre 40 anni orsono misero in luce come l’ossidazione di acidi grassi sopprima quella glucidica principalmente attraverso l’inibizione del complesso della piruvato deidrogenasi (PDH), il principale punto di controllo tra glicolisi e ciclo degli acidi tricarbossilici nel mitocondrio(Randle, 1970) Difatti quando la velocità della β-ossidazione è sostenuta, il che avviene se sono elevate le concentrazioni di FFA circolanti, l’ossidazione di glucosio e piruvato e l’attività della PDH sono ridotte, così come la diminuzione dei livelli di FFA o l’inibizione diretta della loro ossidazione favoriscono l’ossidazione di piruvato (Clarke et al, 1996; Higgins et al, 1981). Il meccanismo di questa inibizione risiede nell’azione della PDH chinasi (PDK), enzima che inibisce la PDH mediante la sua fosforilazione. Difatti elevati rapporti tra acetil-CoA e coenzima A libero e tra NADH e NAD+, risultato di una β-ossidazione normalmente funzionante, si
associano ad un’aumentata attività della PDK (Kerbey et al, 1976). L’ossidazione degli acidi grassi, determinando l’incremento delle concentrazioni mitocondriali, e quindi citosoliche, di citrato, esercita un effetto di rallentamento anche sulla glicolisi per inibizione di un suo enzima, la fosfofruttochinasi (Hue et al, 1987).
Negli anni ’90 Taegtmeyer et al (1994) hanno dimostrato che, in condizioni fisiologiche, anche l’incremento dell’ossidazione di glucosio inibisce in modo parziale quella degli acidi grassi. La molecola segnale di questa via regolatoria è il malonil-CoA, un derivato dalla carbossilazione extramitocondriale dell’acetil-CoA da parte dell’acetil-CoA carbossilasi e le sue concentrazioni intracellulari aumentano in presenza di elevata attività del ciclo di Krebs (Saddik et al, 1993). Il meccanismo d’azione prevede il legame alla CPT-1 dal suo lato citosolico e l’inibizione del trasporto di acil-CoA dal citoplasma al mitocondrio.
Dal momento tuttavia che ciò avviene anche quando la β-ossidazione lavora ad alti regime, l’aumentata produzione di malonil CoA costituisce anche un meccanismo di autoregolazione della via metabolica degli acidi grassi.
L’ossidazione dei corpi chetonici, che risulta elevata nel diabete, inibisce la β-ossidazione, motivo per cui il cuore diabetico presenta una ridotta captazione e metabolismo di acidi grassi. Il meccanismo di questa inibizione non è ben noto, e non sembra correlato alle concentrazioni di malonil CoA o al rapporto tra acetil CoA e coenzima A libero (Stanley et al, 2003).
Possiamo quindi concludere che a livello miocardico, in condizioni fisiologiche, il metabolismo cellulare è fondamentalmente regolato dalla velocità della β-ossidazione degli acidi grassi, e quindi dalla disponibilità di FFA. Essa infatti inibisce la via glucidica agendo sulla PDH e direttamente sugli enzimi della glicolisi, ma contemporaneamente è anche capace di autoregolarsi attraverso il malonil CoA quando il ciclo di Krebs è attivato in modo adeguato alle richieste metaboliche della cellula. Questo meccanismo a sua volta è lo stesso messo in atto dalla via di ossidazione glucidica per rallentare la β-ossidazione.
Vediamo ora qual è il razionale di questa fine regolazione delle principali due vie metaboliche, quella lipidica e quella glucidica, a livello miocardico.
Efficienza metabolica ed efficienza ossidativa
Il concetto fondamentale per capire la dinamica del metabolismo miocardico è che gli acidi grassi rappresentano una risorsa energetica più redditizia in termini di produzione di ATP rispetto al glucosio, mentre quest’ultimo è più efficiente dal punto di vista del consumo di O2.
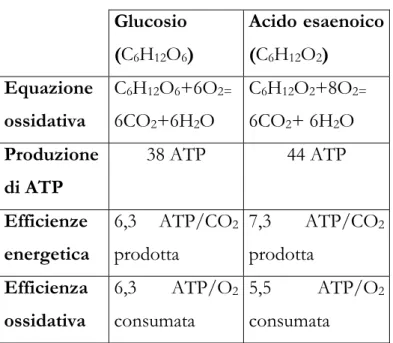
Ciò è esemplificato in tabella 4.1, dove l’ossidazione completa di glucosio a CO2 e H2O è paragonata a quella di un acido grasso a ugual numero di atomi di
carbonio, l’acido esaenoico. I legami chimici della molecola più ridotta, l’acido esaenoico, “contengono” più energia rispetto a quella di glucosio, relativamente più ossidata. Di conseguenza, per ogni mole di CO2 prodotta dalla loro ossidazione,
l’acido esaenoico produce più moli di ATP rispetto al glucosio. Allo stesso tempo, tuttavia, il più ridotto acido esaenoico richiede più ossigeno per completare la sua ossidazione, perciò, per ogni mole do O2 consumata, il glucosio produce una
maggior quantità di ATP.
Glucosio (C6H12O6) Acido esaenoico (C6H12O2) Equazione ossidativa C6H12O6+6O2= 6CO2+6H2O C6H12O2+8O2= 6CO2+ 6H2O Produzione di ATP 38 ATP 44 ATP Efficienze energetica 6,3 ATP/CO2 prodotta 7,3 ATP/CO2 prodotta Efficienza ossidativa 6,3 ATP/O2 consumata 5,5 ATP/O2 consumata
Tabella 4.1. Confronto tra l’ossidazione di glucosio e il corrispondente acido a ugual numero di atomi di carbonio, l’acido esaenoico, in termini di produzione di ATP, efficienza energetica (numero di moli di ATP prodotta per mole di CO2 liberata) ed efficienza
ossidativa (numero di moli di ATP prodotte per mole di O2 consumato).
L’efficienza ossidativa aumenta lievemente se calcolata per acidi grassi a catena di carbonio di maggior lunghezza, senza comunque mai avvicinare quella del glucosio.
Ad esempio, l’ossidazione completa di una mole di palmitato (C16H32O2) richiede 23
moli di O2 producendone 129 di ATP, per un’efficienza ossidativa pari a 5,6 moli di
ATP sintetizzate per mole di O2 consumata.
La maggior capacità del glucosio di produrre energia con minor consumo di ossigeno rispetto quella degli acidi grassi deriva in parte dal processo iniziale del metabolismo glucidico, la glicolisi, dove la trasformazione di una molecola di glucosio, in assenza di consumo di O2, dà luogo a due molecole di piruvato con la
contemporanea produzione di 2 molecole di ATP. In aggiunta, l’ossidazione di acidi grassi risulta meno efficiente dal momento che l’acil-CoA deidrogenasi, un enzima della ossidazione degli acidi grassi, usa come intermedio da ridurre il FAD (a FADH2) anziché il NAD+ (a NADH), il che richiede il consumo di una maggior
quantità di ossigeno (Ingwall, 2001).
Le differenze tra metabolismo glucidico e metabolismo lipidico in termini di “efficienza energetica” ed “efficienza ossidativa” suggeriscono come la preferenza del substrato da ossidare dipenda in gran parte dall’apporto di O2 al tessuto
miocardico. In condizioni di normossia, l’ossidazione di acidi grassi risulta preferibile considerando le elevate richieste energetiche di un organo come il cuore, sottoposto a un lavoro incessante e dispendioso. E’ per questo motivo che, in un cuore a riposo, l’ATP prodotto deriva in maggior parte (60-90%) dall’ossidazione di FFA, mentre solo un 10-40% dal metabolismo glucidico.
Il metabolismo glucidico assume un ruolo centrale per la produzione di energia nel cuore ischemico, dove la carenza di ossigeno determina uno shift verso un metabolismo anaerobio con una rapida stimolazione dell’ingresso di glucosio nella cellula, della glicogenolisi e del flusso glicolitico (Bing et al, 1955; Morgan et al, 1961). L’estrazione di glucosio in particolare è inversamente correlata al flusso coronarico, fino a che l’ischemia non diventa così severa da inibire la glicolisi per l’accumulo dei suoi prodotti (Stanley et al, 1992).
I livelli di acidi grassi aumentano dopo un infarto miocardico acuto o durante procedure chirurgiche, cosicché durante e dopo ischemia il muscolo cardiaco può esser esposto ad alte concentrazione circolanti e il loro effetto deleterio sulle caratteristiche meccaniche ed elettrofisiologiche del cuore è noto da una quarantina
di anni (Oliver et al, 1968; Mueller et al, 1978). Durante la riperfusione miocardica dopo ischemia, l’ossidazione di acidi grassi può rapidamente ristabilirsi e dominare nella fonte della produzione di ATP. Tuttavia, se la stimolazione dell’ossidazione glucidica durante questa fase viene stimolata si verifica un aumento significativo nell’efficienze energetica del cuore, con un parallelo miglioramento della funzione (Broderick et al, 1993, Liu et al, 1996).
Lo “switch” verso la prevalente ossidazione di substrati glucidici, che deriva dall’elevata flessibilità del metabolismo miocardico, è facilmente interpretabile da un punto di vista finalistico. Sappiamo come la cascata ischemica, ovvero la sequenza di eventi che si verificano quando si instaura uno squilibrio tra domanda e offerta di ossigeno, sia caratterizzata, in sequenza, da alterazioni metaboliche e dell’omeostasi ionica, seguite da alterazione della cinesi, da alterazioni elettrocardiografiche ed infine dal dolore anginoso. Così come l’insorgenza del sintomo, momento finale della cascata ischemica, “suggerisce” all’individuo di cessare l’attività fisica per diminuire il lavoro muscolare e conseguentemente le richieste energetiche periferiche, così come l’ipocinesia è finalisticamente diretta a far lavorare di meno il muscolo cardiaco, allo stesso modo risulta comprensibile come sia conveniente al cuore indirizzare il suo metabolismo verso una preferenziale ossidazione di glucosio invece che di acidi grassi.
Gertz et al (1989) hanno inoltre dimostrato che non solo in condizioni di ischemia, ma anche durante un esercizio fisico moderato aumentano sia l’estrazione che l’ossidazione di substrati glucidici da parte del cuore, con una parallela riduzione del metabolism lipidico. Simili osservazioni sono state fatte da Camici et al (1989 e 1991) e Bagger (2000). A differenza del dolore anginoso e della riduzione della cinesi, che insorgono solo quando si è già verificato lo sbilanciamento tra richiesta e apporto di O2 al miocardio, il fenomeno dello “switch” metabolico precede
l’ischemia, è finalizzato anzi, fino a che ciò è possibile, a prevenirla. Esso rappresenta quindi un meccanismo protettivo, che il cuore mette in atto “acutamente”, in modo da usare il substrato più economico dal punto di vista del consumo di ossigeno a spese di un altro, più redditizio in termini di produzione di ATP, ma meno efficiente in funzione della spesa.
Abbiamo finora visto che il metabolismo cardiaco è finemente regolato, e probabilmente con ulteriori meccanismi rispetto a quelli finora noti. Tale sistema consente al cuore di rispondere ai cambiamenti di disponibilità di substrati, ormoni circolanti, (come ad esempio insulina e catecolamine), variazioni del flusso coronarico e del lavoro cardiaco attraverso la selezione dell’appropriato substrato al momento opportuno. In pratica, il cuore non va mai a corto di energia finchè il flusso coronarico rimane adeguato.
Vediamo ora quali sono le alterazioni metaboliche finora note nella Cardiomiopatia Dilatativa e nelle fasi avanzate dello scompenso cardiaco.
Alterazioni del metabolismo miocardico
nella Cardiomiopatia Dilatativa: lo switch metabolico
Fin dagli anni ’50 è stato osservato che il cuore in avanzato stato di scompenso cardiaco modula la scelta dei substrati da ossidare scegliendo la via preferenziale dell’ossidazione di substrati glucidici. Analogo fenomeno si verifica nella Cardiomiopatia Dilatativa, dove il metabolismo del cuore viene a trovarsi in uno stato cronico di “switch” metabolico, caratterizzato da un’aumentata captazione di substrati glucidici, da cui proviene la maggior parte dell’ATP prodotto, con un contributo alla sua produzione da parte della ossidazione lipidica significativamente ridotto. I dati che forniscono la prova di questo fenomeno derivano sia da studi animali che su cuori umani.
In modelli animali di scompenso cardiaco indotto da sovraccarico di volume è stato scoperto che nella progressione dall’ipertrofia alla disfunzione ventricolare si verifica una riduzione dell’espressione genica degli enzimi mitocondriali della ossidazione degli acidi grassi. Il metabolismo miocardico, in altre parole, nel modello animale scompensati ricorda quello fetale, con il glucosio come principale substrato metabolico al posto degli acidi grassi (Kantor et al, 1993; Buttrick et al, 1994)..
Sack et al (1996) hanno trovato un dato simile sul cuore umano di pazienti affetti da CMD, con la riduzione dell’espressione tissutale degli enzimi della β-ossidazione, a sostegno dell’ipotesi che un programma regolatorio genico sia coinvolto nella scelta del substrato da ossidare (Sack et al, 1996)
Nel modello di scompenso cardiaco indotto da quattro settimane di pacing nel cane è stata documentata una riduzione del 70% di captazione di FFA associata ad un rapido incremento di substrati glucidici con rapidi aumenti dei valori di quoziente respiratorio, indicanti che all’aumentata captazione corrisponde anche l’ossidazione di glucosio e lattato (Recchia et al, 1998). Contrariamente alle attese, tuttavia, lo stesso gruppo ha dimostrato che la capacità ossidativa glucidica non è aumentata bensì ridotta, in presenza di un marcato aumento del flusso ossidativo glucidico.In particolare l’espressione miocardica dei trasportatori GLUT1 e GLUT4 e la traslocazione del GLUT4 alla membrana plasmatica non differiscono significativamente rispetto a cuori di controllo (Lei et al, 2004). Questi dati indicherebbero quindi, nel modello animale, che negli stati avanzati di insufficienza cardiaca l’aumento dell’ossidazione glucidica è principalmente dovuto alla riduzione dell’ossidazione di FFA ma che la capacità miocardica di trarre energia da substrati risulta globalmente depressa.
Nell’uomo l’unica tecnica non invasiva capace di studiare il metabolismo miocardico è la tomografia ad emissione di positroni (PET). Tecnica di imaging unica per le sue caratteristiche, la PET consente, oltre che di studiare quantitativamente la perfusione miocardica, di visualizzare anche la captazione di traccianti metabolici marcati. Ciò rende possibile, in modo non invasivo, la valutazione dell’entità dei due tipi di metabolismo, quello glucidico e quello lipidico.
A favore di una ridotto utilizzo di FFA, Yazaki et al (1999) hanno trovato una riduzione della captazione di un analogo degli acidi grassi radiomercato (l’acido
123I-β-metil-iodofenilpentadecanoico) in regioni circoscritte di pazienti affetti da
forme avanzate di Cardiomiopatia Dilatativa. Davila-Roman et al (1996) hanno poi dimostrato che anche negli stadi precoci della malattia in condizioni di riposo è presente sia una ridotta captazione sia una ridotta ossidazione (misurata come prodotto tra captazione miocardica del tracciante per la sua concentrazione plasmatica) di FFA, associata per contro ad un’elevata captazione ventricolare di
11C-glucosio rispetto ad una popolazione di soggetti di controllo (Davila-Roman et
al, 2002).
Van den Heuvel et al hanno studiato una popolazione di pazienti affetti da CMD in condizioni di riposo e durante la somministrazione di dipiridamolo per valutare le variazioni della perfusione (con 13N ammoniaca) e della captazione di 18fluorodesossiglucosio (18FDG). Rispetto ai controlli, i pazienti mostravano una
riserva di flusso globale significativamente inferiore che correlava con un aumentato stress parietale sistolico del ventricolo sinistro. Più del 75% (16 su 22) inoltre presentava un’aumentata captazione di 18FDG, particolarmente accentuata nelle
regioni con riserva di flusso basale maggiormente compromessa (van den Heuvel, 2000)
Un tale quadro di mismatch regionale tra flusso e metabolismo glucidico, caratterizzato cioè da zone di tessuto con un flusso ridotto sia in condizioni basali sia in condizioni di stress (farmacologico o fisico), associato ad un’aumentata captazione di glucosio, è tipico dei territori ischemici, in cui la presenza di una stenosi su una coronaria epicardica riduce il flusso di sangue nel tessuto da essa irrorato.
A B Figura 4.3. Mismatch regionale nella Cardiomiopatia Dilatativa in un’immagine di tomografia ad emissione di positroni (PET). In A si evidenzia una riduzione del flusso basale misurato con 13N-ammoniaca a livello della parete laterale del ventricolo sinistro in confronto alla
regione settale. In B la captazione di 18FDG risulta accentuata nella zona della parete laterale e
dell’apice a fronte di un setto emettente un segnale di minor intensità (immagini gentilmente concesse da Neglia D, IFC-CNR, PISA).
Nella CMD è frequente il riscontro di un blocco di branca sinistro. Tale anomalia di conduzione dell’impulso elettrico si associa ad una asincronia di attivazione e, conseguentemente, di contrazione, tra il setto, precocemente attivato, e la parete laterale del ventricolo sinistro, che si contrae in ritardo, dal momento che la propagazione dell’impulso verso di essa avviene attraverso il miocardio di lavoro e non attraverso quello specifico (di conduzione). La conseguenza emodinamica è che la contrazione della parete laterale avviene contro una maggior resistenza, dovuta alla massa ematica che in fase protosistolica si sposta dal setto verso di essa. In quest’area di aumentato stress parietale il mismatch tra perfusione e captazione di
18FDG risulta particolarmente accentuato (Lindner et al, 2005).
A tal proposito grande interesse ha assunto negli ultimi anni l’osservazione del beneficio emodinamico, clinico e prognostico della terapia di resincronizzazione ventricolare in pazienti con insufficienza cardiaca e asicronia di contrazione intraventricolare. Nowak et al (2003) hanno dimostrato come, a distanza di 14 giorni dall’esecuzione della resincronizzazione, la captazione di 18FDG a livello della parete
laterale diminuiva significativamente a dimostrazione della riduzione dello stress parietali sulla parete laterale.
La cronica attivazione del metabolismo glucidico a scapito di quello lipidico non è presente solo nella CMD, ma si presenta anche nelle fasi avanzate dello scompenso cardiaco (Stanley et al, 2002) e la sua terapia farmacologica può avere anche un effetto indiretto anche sul metabolismo miocardico. I β-bloccanti, ad esempio, esercitano un ruolo fondamentale in tale terapia antagonizzando l’ipertono adrenergico responsabile del peggioramento a lungo termine della funzione contrattile e migliorando il consumo di ossigeno. Wallhaus et al (2001) hanno dimostrato che dopo tre mesi di terapia con carvedilolo, uno dei β-bloccanti maggiormente prescritti nel mondo, si modificava la captazione di acidi grassi. Difatti l’uptake dell’acido 14(R,S)- 18Fluoro-6-tia eptadecaenoico (18FTHA), un acido
grasso a catena lunga marcato, si riduceva del 57% a fronte di un’immodificata captazione di 18FDG.
Finalità e conseguenze dello switch metabolico
Abbiamo finora mostrato che il cronico “switch” metabolico verso un preferenziale uso di glucosio come principale substrato per la produzione di energia al posto degli acidi grassi sembra verificarsi precocemente nella Cardiomiopatia Dilatativa, al contrario di quanto avverrebbe nello scompenso, dove solo in una fase avanzata si avrebbe un’inversione del metabolismo.
Cerchiamo di ipotizzare ora a qual fine ciò avvenga e quali conseguenze possa avere.
Per quanto riguarda il primo punto, ciò che pare più probabile è che il cuore “in difficoltà” metta in atto “cronicamente” il meccanismo di riserva a cui normalmente fa appello solamente “acutamente”, in condizioni di aumentato lavoro cardiaco, così da risparmiare ossigeno ancor prima che esso venga a mancare. Questo
comportamento è compatibile certamente con l’ipotesi di un’ischemia cronica su case microcircolatoria, ma lo è anche se si considera semplicemente che un cuore dilatato e con una massa aumentata, come nelle fasi avanzate dell’insufficienza e nella CMD, compie ad ogni battito un maggior lavoro rispetto alla normalità: da ciò l’esigenza di un più efficiente consumo di ossigeno e, quindi, di un metabolismo prevalentemente glucidico.
E quali le conseguenze del cronico “switch” metabolico? Con l’aumento delle richieste energetiche, come in caso di uno sforzo fisico, ad esempio, il miocardio potrebbe andare a corto di energia, essendo l’ossidazione di glucosio molto meno produttiva in termini di ATP rispetto a quella di acidi grassi. Tale possibilità è suffragata dall’osservazione che in fase tardiva dello scompenso si va a ridurre il rapporto tra fosfocreatina, proteina capace di “ricaricare” rapidamente l’ATP, e ATP stesso (rapporto PCr/ATP), un indice di riserva energetica del cuore. cioè (Neubauer et al, 1997)
Il problema di un cuore non più capace di produrre energia sufficiente al suo fabbisogno potrebbe dipendere non solo da un problema di substrato da ossidare, ma anche da un difetto intrinseco nella produzione di energia per un’alterazione della funzione dei mitocondri. Già nel 1990 Buchwald et al avevano notato come in cuori espiantati da pazienti affetti da CMD il contenuto mitocondriale di citocromo C fosse ridotto significativamente e di come a sua volta risultasse diminuita l’attività dei complessi III e IV della catena di trasporto degli elettroni, di cui il citocromo C è un componente, a differenza dei complessi II e V, che ne sono privi, normalmente funzionanti.
Di solito in medicina, a fini esemplificativi, si è soliti distinguere tra situazioni “acute” e situazioni “croniche”. Questo approccio, sebbene ben si adatti al comune categorizzare i concetti, non può non esser accusato di un eccessivo schematismo mentale, spesso difforme dalla realtà naturale delle cose. Abbiamo parlato all’inizio di come nel cuore sano lo “switch” metabolico acuto eserciti un ruolo protettivo ottimizzando le risorse di ossigeno qualora la sua disponibilità si riduca, o in senso assoluto o in relazione ad un aumentato lavoro cardiaco. Abbiamo poi ipotizzato che anche in una situazione cronica, lo switch metabolico abbia una finalità
protettiva. Ma la natura insegna che per cronico si debba in realtà intendere una sequenza più o meno lunga di situazioni acute che si succedono nel tempo. E’ necessario chiedersi, dunque, come sia capace di reagire il cuore affetto da CMD o in scompenso avanzato quando, a “riserva metabolica” già esaurita, si verifichi un ulteriore aumento delle richieste energetiche. In modo ancora più semplice, cosa avviene, in un cuore già al limite delle sue possibilità, nelle comuni vicissitudini quotidiane a cui ognuno è sottoposto, un’emozione, un momentaneo, seppur modesto, incremento pressorio, la semplice salita delle scale di casa? In queste situazioni il miocardio potrebbe non essere in grado di aumentare ulteriormente il suo metabolismo glucidico, tanto più in presenza di disfunzione mitocondriale, e quindi andar incontro al fenomeno dello “stunning”.
E ancora: l’accumulo di acidi grassi, incapaci di penetrare nel mitocondrio per l’inibizione della carnitina palmitoil-transferasi (CPT-1), e di intermedi della β-ossidazione, non più catabolizzati ad acetil-CoA per la ridotta espressione enzimatica, potrebbe portare, in un ambiente pro-ossidante come quello di un miocardio ischemico, alla formazione di lipoperossidi e ad una ulteriore propagazione del danno.
Possibili meccanismi dello switch metabolico
Analizziamo ora i possibili meccanismi potenzialmente responsabili del cronico “switch” metabolico nella CMD e nella fasi avanzate dello scompenso cardiaco.
Riattivazione di un programma genico fetale
Abbiamo prima accennato a come nella CMD e nel cuore che progredisce verso l’insufficienza contrattile si attivi, o meglio, si riattivi, un programma genico fetale.
E’ ben noto come la vita fetale sia caratterizzata dalla necessità da parte dei tessuti dell’organismo in via di accrescimento di usufruire al massimo delle risorse energetiche derivanti dal sangue materno, considerando in particolar modo l’ambiente relativamente ipossico in cui si trova a lavorare il cuore fetale. E’ per tale motivo che l’espressione genica delle proteine deputate al trasporto e all’ossidazione dei substrati è orientata verso un metabolismo cardiaco prevalentemente glucidico. Razeghi et al (2001) hanno studiato il comportamento dei “geni metabolici” confrontando la loro espressione in cuori umani fetali, adulti e scompensati
Per quanto riguarda il metabolismo glucidico, i cuori dei soggetti sani adulti mostravano, rispetto a quelli fetali e scompensati, un’aumentata espressione dei geni dei trasportatori del glucosio GLUT1 e GLUT4, di due isoforme di piruvato deidrogenasi chinasi (PDK2 e PDK4), enzima chiave nella regolazione dell’ossidazione di glucosio e lattato attraverso la fosforilazione inibitoria del complesso della piruvato deidrogenasi e dell’isoforma muscolare della glicogeno sintasi (mGS).
Per quanto riguarda i geni coinvolti nel metabolismo lipidico, rispetto ai cuori fetali e scompensati, i dati sul tessuto miocardico adulto evidenziavano un’aumentata espressione dell’isoforma muscolare della carnitina palmitoil-transferasi (mCPT1) ma non dell’isoforma epatica (lCPT1), enzima che regola la velocità dell’ossidazione di acidi grassi in quanto controllore del loro passaggio dal citoplasma all’interno del mitocondrio; di due enzimi della β-ossidazione, la LCAD (long chain acyl-CoA deidrogenasi) e la MCAD (medium-chain acyl-CoA deidrogenasi).
Infine, i cuori scompensati, al pari di quelli fetali, mostravano ridotti livelli di trascritti di citrato sintasi, enzima del ciclo degli acidi tricarbossilici e un’attenuata espressione dell’isoforma 2 della proteina disaccoppiante (UCP2).
L’espressione genica appena descritta è coerente con quanto finora detto a proposito del cronico “switch” verso un preferenziale utilizzo di glucosio come primo substrato energetico. Infatti, in un cuore scompensato, il metabolismo degli acidi grassi è rallentato sia dalla riduzione del trasportatore degli acidi grassi nel
mitocondrio (mCPT1) sia dalla ridotta espressione degli enzimi stessi della β-ossidazione (LCAD e MCAD). Nell’ambito del metabolismo glucidico, la riduzione della PDK, rimuovendo l’inibizione della piruvato deidrogenasi, potrebbe giocare un ruolo chiave nell’aumentare il flusso di intermedi glucidici verso il ciclo di Krebs. Di importanza nettamente inferiore sembra invece la riduzione dei trasportatori di membrana del glucosio, in particolare del GLUT4, fatto che anzi appare in contraddizione con l’aumentata captazione di glucosio che chiaramente evidenziano gli studi PET (Sambuceti et al, 1997).
Nel cuore umano i geni per il controllo metabolico esistono in forme costitutive ed inducibili, piuttosto che come isoforme fetali ed adulte Nello scompenso cardiaco perciò il ritorno ad un profilo metabolico fetale avviene principalmente reprimendo geni normalmente attivi piuttosto che esprimendone di nuovi.
Un possibile meccanismo nel controllo del metabolismo cellulare in vari tessuti, compreso il miocardio, chiama in causa il ruolo dei PPAR (acronimo inglese per peroxisome proliferator-activated receptors) e/o dei RXR (retinoid X receptors).
I PPAR rappresentano una superfamiglia di recettori nucleari che, una volta attivati, dimerizzano con i RXR. Il dimero da essi costituito si lega successivamente a specifiche sequenze di DNA, i PPRE (PPAR responding elements) dove promuove la sintesi di prodotti genici coinvolti nel metabolismo lipidico.
Esistono tre isoforme di PPAR: PPARα, PPAR β/δ (anche definiti δ) e i PPARγ, tra cui le principali forme studiate sono le prime due in quanto i PPARγ sono poco espressi a livello miocardico.
Cheng et al (2004)hanno studiato nel topo gli effetti sul metabolismo lipidico della delezione selettiva a livello cardiomiocitario della sottofamiglia dei PPAR-δ. I cuori murini riducevano l’espressione di enzimi chiave della ossidazione degli acidi grassi e quindi la velocità della ossidazione basale. Ne risultava una disfunzione contrattile, progressivo accumulo miocardico di intermedi lipidici, ipertrofia e insufficienza cardiaca congestizia con una riduzione della sopravvivenza. Anche studi condotti su topi knockout per il PPARα, sebbene raramente sopravvivano fuori dall’utero,
hanno evidenziato una ridotta espressione degli enzimi della β-ossidazione (Watanabe et al, 2000)
Osorio et al (2002), nel modello canino di scompenso cardiaco indotto da stimolazione ad alta frequenza, hanno dimostrato la presenza delle tipiche alterazioni metaboliche (captazione elevata di glucosio, ridotta di FFA). Su campioni tissutali hanno poi dimostrato che la riduzione dell’espressione e dell’attività della MCAD, enzima della β-ossidazione, era associata ad una ridotta espressione dell’RXRα.
Questa promettente area di ricerca necessita comunque di conferme su tessuti umani per verificare l’eventuale ruolo della ridotta espressione dei PPAR nella modificazione del metabolismo cardiaco.
Modulazione del metabolismo miocardico mediata dall’ossido nitrico
L’ossido nitrico (NO), a 20 anni dalla sua scoperta, non sembra aver esaurito di riservare sorprese a chi in questo quinto di secolo ne ha seguito il crescente ruolo da primattore nella fisiopatologia dell’endotelio. Molecola estremamente semplice dal punto di vista chimico (un atomo di azoto e uno di ossigeno) e fisico (è un gas), esso è coinvolto nella regolazione di meccanismi fisiopatologici come il tono vascolare, l’attività piastrinica, e addirittura nel più ancestrale strumento di difesa degli esseri viventi, l’infiammazione. Per tali ragioni non solo è ipotizzabile, almeno in via teorica, un suo ruolo nella regolazione di una funzione basale della cellula, cioè il nutrimento, ma, da un punto di vista finalistico, sembra quasi improbabile che la natura lo abbia escluso da tale funzione.
E’ noto come l’insulina, ancor prima di agire a livello cellulare, dove facilita l’esposizione dei trasportatori del glucosio sulla membrana citoplasmatica, “si apre la strada” stimolando la produzione endoteliale di NO in modo da aumentare l’afflusso sanguigno, e quindi di nutrienti glucidici, in un tessuto bersaglio come il muscolo scheletrico. E’ perciò coerente ipotizzare anche un ruolo dell’NO nella
modulazione diretta del metabolismo tissutale, nel senso cioè di un aumento della captazione di glucosio. Ciò che sembra avvenire a livello miocardico, a differenza del muscolo scheletrico, è invece è il contrario.
Depre et al (1998), su cuori isolati perfusi, hanno scoperto dimostrato che, attraverso la via del cGMP, ovvero la via di trasduzione principale dell’ossido nitrico, esso inibisce l’esposizione di GLUT4, riducendo così la captazione di glucosio da parte delle cellule miocardiche. Ciò sarebbe coerente con l’effetto cardioprotettivo del blocco dell’attività della ossido nitrico sintasi durante ischemia, riducendo la produzione di NO da parte dell’isoforma inducibile (iNOS). Lo stesso gruppo ha poi sostenuto che attraverso la stessa via di traduzione, quella del cGMP, l’NO aumenterebbe anche l’attività dell’acetil-CoA carbossilasi, stimolando la produzione di malonil-CoA a partire dall’acetil-CoA; in tal modo si inibisce la carnitina acil transferasi-1 (CPT-1), e quindi tutta la via ossidativa degli acidi grassi. Importanti conferme sul ruolo dell’NO nella regolazione della captazione di glucosio giungono da Recchia et al (2002), che hanno studiato il comportamento del metabolismo e della produzione di ossido nitrico nel modello canino di scompenso cardiaco indotto dalla stimolazione ad alta frequenza. A fronte di un quadro tipico di switch cronico (metabolismo prevalentemente glucidico con bassa captazione di acidi grassi), nel sangue venoso refluo dal miocardio i livelli di nitriti e nitrati, metaboliti stabili dell’NO, erano molto più bassi del normale. Associazione casuale o reale rapporto causa-effetto? Proprio in risposta a tale quesito, lo stesso gruppo ha evidenziato che l’inibizione farmacologica della produzione di ossido nitrico determinava, in cuori di cani normali, un’inversione dell’uso di substrati, come nel quadro di scompenso indotto da pacing, che veniva riequilibrata dalla somministrazione di donatori di NO (Recchia et al, 2002)
L’ipotesi che l’NO inibisca l’ingresso di glucosio nel cardiomiocita e che, al contrario, la sua ridotta produzione lo favorisca, è compatibile con quanto avviene in una situazione acuta come l’ischemia miocardica e in una cronica come lo scompenso. In entrambe le condizioni è presente una riduzione della produzione endoteliale di ossido nitrico. Nell’ischemia ciò è conseguenza, almeno in parte, della riduzione dello shear stress sull’endotelio; nello scompenso è invece parte del
quadro di disfunzione endoteliale cronica, come dimostrato dalla riduzione dell’entità della dilatazione arteriosa flusso-indotta nei pazienti con insufficienza cardiaca.
Cardiomiopatia Dilatativa e ibernazione miocardica
Due sono gli aspetti più suggestivi che emergono dagli studi PET.
Il primo è che sia nella CMD che nello scompenso in fase avanzata si ha un quadro metabolico disomogeneo, lo “switch” metabolico non pare cioè globale, omogeneo tutte le zone cardiache, ma anzi sembra assumere connotati regionali. In presenza di blocco di branca sinistro, ad esempio, la regione settale non mostra un metabolismo alterato al contrario della parete libera del ventricolo sinistro, più sofferente perché cronicamente sottoposta a maggior stress emodinamico (van den Heuvel et al, 2000) Il secondo aspetto di rilievo è che la CMD mostra un quadro caratterizzato da zone di “mismatch” tra flusso e metabolismo, ovvero le zone con maggior compromissione della perfusione mostrano aumentata captazione di 18FDG, indice
di un’aumentata captazione di glucosio. Tale quadro è esattamente identico a quello che si ottiene nei territori perfusi da una coronaria parzialmente occlusa da una placca emodinamicamente significativa, ovvero nei territori cronicamente ischemici (Sambuceti et al, 1997). Il pattern di “mismatch” inoltre si accentua durante uno stress farmacologico con dipiridamolo: si verifica cioè il meccanismo del “furto”, cioè la deviazione del flusso verso distretti attigui con conservata (almeno parzialmente) riserva microcircolatoria, a scapito di quelli in cui le resistenze sono ridotte al minimo già in condizioni basali (van den Heuvel et al, 2000).
Queste osservazioni pongono il sospetto che almeno una parte delle forme di Cardiomiopatia Dilatativa attualmente classificate come “idiopatiche” siano caratterizzate da un quadro molto simile a quello dell’ibernazione miocardica, tipico di condizioni come la cardiopatia ischemica e lo scompenso cardiaco cronico in fase avanzata (Camici, 2004).
Modulazione farmacologia del metabolismo miocardico:
teoria e pratica
Gli interventi terapeutici diretti a un controllo farmacologico del metabolismo cardiaco si basano sul presupposto che aumentando il metabolismo glucidico a scapito di quello degli acidi grassi si riesca a potenziare il fisiologico meccanismo protettivo attuato dal cuore.
Gli inibitori parziali del ossidazione di acidi grassi, ranolazina e trimetazidina, agiscono riducendo direttamente la velocità del metabolismo lipidico e rimuovendo l’inibizione della PDH nel mitocondrio, in modo da aumentare il flusso glucidico. In un modello animale di insufficienza cardiaca caratterizzato da bassa attività della PDH, il trattamento cronico con trimetazidina è stato dimostrato aumentare significativamente la sopravvivenza a breve termine. Nell’uomo, trial clinici con un basso numero di pazienti sembrano per ora affermare che il trattamento con trimetazidina determini un aumento della frazione di eiezione del ventricolo sinistro, anche in questo caso almeno a breve termine (Beardinelli et al, 2001)
Un altro farmaco teoricamente capace di rallentare il metabolismo degli acidi grassi è l’oxfenicina, che inibisce la CPT-1 e quindi il flusso degli acidi grassi all’interno del mitocondrio. Lionetti et al (2005)dimostrato che il trattamento con questo farmaco, nel modello canino di scompenso indotto da pacing, rallenta la progressione dell’insufficienza cardiaca e ritarda la sua insorgenza ben oltre le solite quattro settimane.
Ulteriori approcci sarebbero quelli di agire sul metabolismo glucidico, inibendo l’attività della piruvato deidrogenasi chinasi (PDK), o attivando il trasporto del glucosio o la glicolisi stessa, ma purtroppo farmaci capaci di svolgere tale azione non esistono ancora.
Il razionale di aumentare la via ossidativa del glucosio e di inibire quella degli acidi grassi sembra al momento la prospettiva più promettente. In una patologia
come la CMD e nello scompenso cardiaco in fase avanzata, disporre di un nuovo bersaglio potrebbe ampliare e completare le strategie terapeutiche.
Uno dei principali argomenti a sfavore che tuttavia deve esser tenuto ben a mente è rappresentato dalle possibili conseguenze a lungo termine del potenziamento della risposta metabolica messa in atto dal cuore. Concetto generale della medicina è che non sempre un meccanismo di difesa messo in atto dall’organismo in una situazione critica esita in un reale beneficio, soprattutto a lunga distanza. Un esempio di ciò è il razionale dell’uso di β-bloccanti nell’insufficienza cardiaca cronica. L’ipertono adrenergico tipico dello scompenso costituisce un meccanismo di risposta attuato dall’intero organismo per aumentare le resistenze periferiche, evitare l’ipotensione, aumentare l’inotropismo cardiaco. E’ però purtroppo altrettanto noto come proprio l’attivazione simpatica e l’eccesso di catecolamine circolanti siano essi stessi coinvolti del peggioramento nel tempo della performance cardiaca e, conseguentemente, della prognosi dell’individuo. E’ perciò da tener presente che una terapia basata sull’aumento del metabolismo glucidico a scapito di quello degli acidi grassi, seppur vantaggioso a breve termine, potrebbe presentare controindicazioni nel medio-lungo periodo. L’eccessiva amplificazione del metabolismo glucidico a scapito del consumo di FFA potrebbe portare nel tempo ad un deficit di ATP, per l’attenzione eccessiva volta a garantire al cuore l’”efficienza ossidativa” a scapito di quella energetica.
Considerazioni finali
La CMD rappresenta un quadro quanto mai sconosciuto dal punto di vista della sua eziopatogenesi. In questa breve analisi ci siamo soffermati non tanto su quale potrebbe essere la noxa patogena iniziale responsabile della dilatazione della camere cardiache e dell’insufficienza di pompa, ma sulle cause dell’alterato metabolismo del cuore in tale patologia e sulle conseguenze che ciò potrebbe avere
nella sua progressione. Essendo il cuore un organo così esigente dal punto di vista energetico è improbabile che alterazioni delle vie di ossidazione dei substrati risultino ininfluenti sulla performance cardiaca. Viste le numerose analogie morfologiche, cliniche e metaboliche che accomunano la CMD allo scompenso cardiaco, soprattutto in fase avanzata, gli stessi meccanismi potrebbero essere coinvolti nella progressione dell’insufficienza cardiaca indipendentemente dalla sua causa.
L’approccio terapeutico con farmaci che mirano all’inibizione del metabolismo lipidico e al potenziamento di quello del glucosio risulta, almeno in via teorica, coerente con l’attuale linea terapeutica dello scompenso (ACE-inibitori, β-bloccanti e diuretici), quello di “nutrire un cavallo stanco e a corto di energia” invece che “spronarlo” con agenti inotropi.