11
2. Metodologie analitiche
Secondo la corrente definizione di minerale adottata dall’International Mineralogical Association (IMA), “a mineral is an element or chemical compound that is normally crystalline and that has been formed as a result of geological processes” (Nickel, 1995). Lo studio delle fasi minerali non può pertanto prescindere da indagini chimiche e diffrattometriche volte alla comprensione della loro composizione chimica e del loro assetto strutturale.
I campioni studiati nel corso di questa tesi di dottorato sono stati forniti da vari collezionisti o ottenuti per mezzo di scambi o di acquisti. Ogni campione è stato attentamente osservato al microscopio binoculare (Zeiss Discovery V-8), fino a 128x; in tal modo è stato possibile osservare la morfologia delle fasi presenti, tappa preliminare ed importante per poter successivamente studiare le specie individuate. La loro identificazione è stata generalmente possibile attraverso la raccolta di diffrattogrammi di polvere con camera Gandolfi da 114.6 mm e radiazione CuKα e mediante analisi chimiche semiquantitative con il microscopio elettronico a scansione (SEM, Scanning Electron Microscope) abbinato ad un dispositivo per la microanalisi in dispersione di energia (EDS, Energy Dispersive System) e quantitative utilizzando la microsonda elettronica, operante con il sistema WDS (Wavelenght Dispersive System).
Quando possibile è stato tentato uno studio con tecniche di cristallo singolo, riuscendo a definire la geometria della cella; tali cristalli sono poi stati utilizzati per raccolte di dati di intensità con diffrattometro automatico a 4 cerchi o, laddove le piccole dimensioni dei cristalli lo impedissero, utilizzando radiazione di sincrotrone con rivelatore areale. La natura disordinata e spesso microcristallina dei minerali studiati ha sovente ostacolato, se non impedito, lo studio dei campioni mediante tecniche di cristallo singolo; si è pertanto fatto ricorso anche all’utilizzo di tecniche di polveri.
A complemento delle indagini diffrattometriche e chimiche sono stati raccolti anche dati spettroscopici che hanno consentito di ottenere importanti informazioni a scala atomica sull’assetto strutturale locale attorno ad alcuni particolari elementi; questi dati sono di particolare utilità laddove manchi un ordinamento a lungo raggio.
Infine una parte importante di questo lavoro di tesi ha previsto lo studio del comportamento termico delle fasi studiate. Sono stati pertanto raccolti dati termo-gravimetrici e studiati diffrattometricamente i prodotti riscaldati, sia effettuando raccolte in situ sia lavorando su campioni precedentemente trattati termicamente.
12
2.1 Diffrattometria di raggi X
Un cristallo ideale è caratterizzato dalla ripetizione, secondo le traslazioni reticolari, di un aggregato di atomi o ioni; la determinazione della sua struttura, quindi, implica la definizione sia del periodo di ripetizione, ossia del reticolo, sia della disposizione spaziale degli atomi o ioni che costituiscono il motivo associato a ciascun punto reticolare. La diffrazione di raggi X è un utilissimo strumento per la definizione dei due aspetti ora ricordati, secondo due passi successivi:
• determinazione delle direzioni di diffrazione. Tali direzioni sono funzione degli aspetti geometrici della cella elementare e pertanto la conoscenza delle direzioni di diffrazione consente di ottenere informazioni sul tipo di cella e sulle sue dimensioni. A tale scopo si raccolgono diffrattogrammi con il metodo delle polveri, il metodo del cristallo oscillante, il metodo Weissenberg e mediante il diffrattometro automatico a 4 cerchi;
• determinazione delle intensità delle onde diffratte. L’intensità dipende dalla distribuzione spaziale degli elettroni (che sono le entità fisiche che diffondono i raggi X) e pertanto la misura delle intensità dei singoli riflessi fornisce informazioni sulla distribuzione degli atomi (o ioni) all’interno della cella elementare.
2.1.1 Tecniche di cristallo singolo
Metodo del cristallo oscillante
L’esecuzione di questa tecnica sperimentale prevede di disporre una direzione razionale del cristallo (ad es. uno degli assi di cella) ortogonalmente al fascio di raggi X incidente e di collocare attorno al cristallo una pellicola fotografica montata all’interno di una camera cilindrica, di raggio pari a 28.65 mm, sul cui asse è posto il cristallo, oscillante attorno alla direzione razionale prescelta. Normalmente al vettore diretto giacciono piani del reticolo reciproco che tagliano la sfera di Ewald secondo circonferenze; la costruzione di Ewald ci assicura che quando un nodo di reticolo reciproco taglia la sfera in un punto P, si ha un raggio diffratto lungo la direzione individuata dal centro della sfera (in cui per costruzione si assume sia collocato il cristallo) ed il punto P. Le direzioni di diffrazione si disporranno secondo superfici coniche (coni di Laue) aventi vertice sul cristallo. Esse tagliano la superficie cilindrica della pellicola fotografica secondo circonferenze sulle quali si disporranno le macchie di diffrazione. Una volta che la pellicola venga svolta, le macchie di diffrazione appariranno disposte su diverse linee parallele (strato-linee) corrispondenti ai diversi coni di Laue.
Tale metodo ci consente di risalire agevolmente alla periodicità lungo l’asse di rotazione; inoltre, attraverso un’oscillazione di 10°-12°, è possibile verificare la presenza di un piano di simmetria
13
normale all’asse di oscillazione nella classe di Laue del cristallo esaminato. Difatti la distribuzione delle macchie di diffrazione sulla pellicola fotografica presenterà simmetria m esclusivamente nel caso in cui, perpendicolarmente all’asse di oscillazione, sia presente un piano di riflessione.
Metodo Weissenberg
Questa tecnica diffrattometrica utilizza la stessa camera e la stessa disposizione sperimentale utilizzate per ottenere fotogrammi di cristallo oscillante. Tuttavia in questo caso si registrano soltanto i riflessi relativi ad un piano di reticolo reciproco, corrispondenti ad una determinata strato-linea nel fotogramma di cristallo oscillante. A tal fine si inserisce fra il cristallo e la camera cilindrica uno schermo che presenta una fessura solo in corrispondenza del cono di diffrazione che ci interessa. Le macchie di diffrazione di questa strato-linea sono sparpagliate sulla pellicola fotografica in virtù del moto traslatorio della camera cilindrica, moto accoppiato ad oscillazioni del cristallo di 180°.
La tecnica Weissenberg consente pertanto di ottenere immagini distorte dei vari piani di reticolo reciproco, immagini distorte dalle quali è agevole ottenere il reticolo reciproco “indistorto”; attraverso l’indicizzazione delle macchie di diffrazione e valutando la loro intensità, si ottengono informazioni sulla geometria del reticolo reciproco (e quindi anche i parametri reciproci, necessari per risalire ai parametri diretti), sulla simmetria, sulle assenze sistematiche e, quindi, sulla simmetria di gruppo spaziale del cristallo esaminato.
Raccolta di dati di intensità con diffrattometro automatico a 4 cerchi
I dati di intensità sono stati raccolti presso il Centro Interdipartimentale di Analisi e Determinazione Strutturale (CIADS) dell’Università di Siena mediante un diffrattometro Oxford Xcalibur S e rivelatore CCD, e presso il Laboratorio Raggi X del Dipartimento di Scienze della Terra dell’Università di Pisa, utilizzando un diffrattometro automatico a 4 cerchi Siemens P4. In ambo i casi la radiazione utilizzata è la radiazione MoKα (λ = 0.71073 Å).
Nel diffrattometro automatico a 4 cerchi, il sistema di cerchi concentrici consente al cristallo, montato su una testina goniometrica senza particolari orientazioni, di assumere qualsivoglia orientazione; il cerchio goniometrico 2θ è analogo a quello di un normale diffrattometro per polveri e sul suo bordo è montato il rivelatore a scintillazione il quale fornisce una maggiore accuratezza e rapidità nella raccolta dei dati di intensità rispetto alle pellicole fotografiche. Il cerchio ω, coassiale al precedente, è il supporto di un sistema di altri due cerchi, il cerchio χ ed il cerchio ϕ; su quest’ultimo è montata la testina goniometrica che sorregge il cristallo.
14
Il complesso di cerchi ha lo scopo di portare ogni vettore di reticolo reciproco, prima nel piano orizzontale definito dalla direzione dei raggi X incidenti e del rivelatore (per mezzo dei cerchi ϕ e χ), poi con la sua estremità sulla superficie della sfera di riflessione (cerchio ω). Il cerchio 2θ porta il contatore in posizione appropriata.
Una normale operazione di raccolta di dati di intensità prevede di fissare dei valori angolari entro i quali sono costretti a ruotare i cerchi; in tal modo il diffrattometro esplora tale regione e ricava i valori angolari dei riflessi e quindi le coordinate di un certo numero di nodi di reticolo reciproco. Una volta che ne siano state ricavate un certo numero, il programma calcola i tre vettori più corti
a*, b* e c* ed i rispettivi angoli α∗, β∗ e γ∗. Dai parametri reciproci il calcolatore risale a quelli
diretti e, partendo da questi dati, è ora possibile determinare quali angoli di rotazione imprimere al cristallo per portarlo in condizioni di diffrazione per ogni singolo piano hkl. I dati raccolti, ossia l’insieme delle intensità delle diffrazioni all’interno dei limiti prefissati, vengono successivamente corretti per l’assorbimento e per il fattore “Lorentz-polarizzazione” ed infine ridotti a |F|2, dove F è il fattore di struttura.
Raccolta di dati di intensità con luce di sincrotrone su cristallo singolo
Utilizzando sorgenti di radiazioni X convenzionali soltanto alcune righe caratteristiche (ad esempio CuKα, MoKα) sono sufficientemente intense e di lunghezza d’onda appropriata per trovare impiego nella risoluzione di problemi di cristallografia strutturale. La radiazione di sincrotrone, prodotta quando elettroni in movimento a velocità prossime a quella della luce sono deflessi da campi magnetici, ha invece uno spettro molto più ampio e consente di scegliere la lunghezza d’onda più opportuna per minimizzare l’assorbimento ed i danni da radiazione; comunque la proprietà più importante di questa radiazione è la sua alta brillanza, intesa come il numero di fotoni al secondo, per unità di area della sorgente, per angolo solido unitario e per larghezza di banda relativa dello 0.1% (ossia, ad esempio, per fotoni di energia pari a 10 keV si considerano quelli aventi energia fra 9995 e 10005 eV).
La raccolta di dati di intensità con luce di sincrotrone è stata eseguita presso il laboratorio Elettra, a Basovizza (Trieste). Gli elettroni sono generati all’interno di un LINAC (Linear Accelerator), lungo 66 metri, prima di essere inseriti all’interno di un anello di accumulazione di terza generazione, di circa 40 metri di raggio. L’energia degli elettroni circolanti all’interno dell’anello può raggiungere i 2.4 GeV. La brillanza della “luce di sincrotrone” ottenuta nel laboratorio Elettra è dell’ordine di 1019 fotoni/s/mm2/mrad2/0.1% bw; si tratta di una radiazione 1012 volte più intensa di quella ottenibile attraverso sorgenti convenzionali. La “linea di luce” XRD1, utilizzata per raccogliere i dati di intensità sui cristalli studiati in questa tesi, è stata disegnata appositamente per studi
15
cristallografici sulle macro-molecole. La stazione sperimentale è equipaggiata con un rivelatore CCD da 165 mm di diametro, oltre ad un sistema di raffreddamento per effettuare studi a bassa temperatura (solitamente 100 K). La lunghezza d’onda può essere variata in funzione delle esigenze sperimentali.
Le peculiari caratteristiche della radiazione di sincrotrone consentono di effettuare raccolte su cristallo singolo in tempi molto brevi (da mezz’ora ad un’ora) anche da cristalli di dimensioni estremamente minute o di scarsa qualità. Inoltre è possibile utilizzare anche le informazioni contenute nei riflessi deboli e debolissimi che non sono misurabili con sufficiente accuratezza con radiazione generata da sorgenti convenzionali. Durante la raccolta dei dati, il cristallo, posto su una testina goniometrica senza particolari orientazioni, viene ruotato di un angolo φ definibile dall’operatore; per ogni valore di ∆φ viene raccolta dal rivelatore areale un’immagine nella quale compaiono i riflessi presenti in quella regione di reticolo reciproco. Raccolto il numero di frames stabilito all’inizio dell’esperimento (generalmente in numero tale da coprire un intervallo angolare di almeno 180°), le immagini vengono processate con opportuni programmi che consentono di ricavare i parametri reciproci (e quindi quelli diretti) della fase studiata, andando così ad indicizzare tutti i riflessi raccolti e ad integrarne le intensità.
2.1.2 Tecniche di polveri
La diffrazione di raggi X da polveri consente di effettuare analisi qualitative e quantitative dei campioni studiati.
Le informazioni tridimensionali di un diffrattogramma di cristallo singolo vengono compresse in una dimensione e pertanto l’estrazione delle informazioni è più complessa rispetto all’altra metodologia. I diffrattogrammi di polvere contengono tre tipi di informazioni: geometriche (posizione angolare dei riflessi, descritta dall’equazione di Bragg), strutturali (intensità integrate) e relative allo stato fisico del campione (profilo del picco, dovuto alla convoluzione di tutti gli effetti che controllano la distribuzione angolare dell’intensità).
L’analisi qualitativa consiste nel riconoscimento delle fasi presenti, attraverso un confronto del diffrattogramma con banche dati esistenti oppure, nel caso in cui non siano mai stati depositati dati di diffrazione da polveri, attraverso il confronto con il diffrattogramma calcolato a partire dai dati strutturali, se noti. Per l’analisi qualitativa dei campioni abbiamo utilizzato sia la camera Gandolfi che il diffrattometro Bragg-Brentano; quest’ultimo strumento è stato utilizzato anche per la raccolta di dati di intensità da polveri.
16
Diffrattogrammi con camera Gandolfi
Come detto sopra, per l’identificazione delle fasi solide la diffrazione di polveri è una tecnica particolarmente utile in quanto costituisce una sorta di “impronta digitale” che può essere ottenuta rapidamente ed in maniera riproducibile senza nessuna conoscenza a priori circa i parametri di cella e la struttura cristallina del materiale (Gregorkiewitz, 1994). Può pertanto essere utile ottenere diffrattogrammi di polvere anche da cristalli singoli; ciò è possibile riorientando continuamente il cristallo attraverso la sua rotazione attorno a due assi che si incontrano nel centro della camera. Questa tecnica, introdotta da Gandolfi (1967), ha una geometria semplice e consente di raccogliere effetti di diffrazione su un intervallo angolare, in 2θ, compreso fra 0° e 180°. L’angolo χ fra i due assi di rotazione è variabile in funzione del tipo di camera utilizzata; nel corso di questo lavoro abbiamo utilizzato una camera Gandolfi con un angolo χ di 45°. La registrazione degli effetti di diffrazione avviene su pellicola; su di essa compaiono archi di circonferenza, simmetrici rispetto ai fori di entrata e di uscita dei raggi X, rappresentanti i coni di diffrazione dei vari piani reticolari. Misurando la distanza, sulla “mezzeria” della pellicola, fra gli archi simmetrici e conoscendo il diametro della camera utilizzata, si risale agevolmente, applicando l’equazione di Bragg, al valore dell’angolo di diffrazione θ e quindi al corrispondente valore dhkl
I diffrattogrammi Gandolfi sono stati raccolti presso il laboratorio Raggi X del Dipartimento di Scienze della Terra dell’Università di Pisa e presso la sezione di mineralogia del Museo di Storia Naturale e del Territorio dell’ateneo pisano; ogni campione è stato esposto per periodi di tempo compresi fra 24 e 48 ore (in funzione delle dimensioni degli esemplari studiati) alla radiazione CuKα (λ = 1.54178 Å) in una camera Gandolfi di diametro pari a 114.6 mm. Ogni pellicola così ottenuta è stata successivamente sottoposta a scansione a 300 dpi (dots per inch; risoluzione pari a circa 0.05 mm) ed elaborata con il programma X-RAY (O’Neill et al., 1993) al fine di ottenere un set di dati “I-2θ”. Attraverso una successiva elaborazione con il programma WINFIT (Krumm, 1997) abbiamo ottenuto un diagramma “I-d
.
hkl” che ci ha consentito, nella maggior parte dei casi, di
identificare la fase esaminata.
Diffrattogrammi con diffrattometro Bragg-Brentano
Il diffrattometro Bragg-Brentano è stato utilizzato per una analisi qualitativa e quantitativa dei campioni studiati. L’impiego più frequente è consistito nella verifica della composizione mineralogica dei campioni di polvere impiegati in altre tecniche analitiche (analisi termo-gravimetriche, spettroscopia 29Si NMR). Durante queste raccolte si è privilegiata la rapidità (tipicamente i diffrattogrammi sono stati raccolti in 1-2 h) e la conservazione del campione di
17
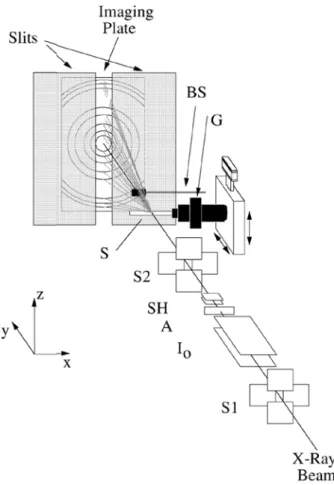
Fig. 2.1 – Apparato sperimentale per la raccolta di dati di
diffrazione durante riscaldamento in situ (da Meneghini et
al., 2001).
polveri il quale era stato preventivamente preparato macinando in un mortaio di agata frammenti del campione in studio. Le condizioni sperimentali prevedevano la raccolta dei diffrattogrammi nell’intervallo angolare 3°<2θ<60°, con un passo di 0.02° in 2θ e un tempo di raccolta di 1 s, impiegando la radiazione CuKα (λ = 1.54178 Å).
Nel caso di alcune fasi studiate, per le quali non erano disponibili campioni adatti a studi di cristallo singolo, si è tentata la raccolta di dati di intensità con diffrattometro Bruker AXS D8 con rivelatore PSD lineare e radiazione CuKα ponendo la polvere, di granulometria inferiore ai 10 µm, all’interno di un
capillare di diametro 0.5-0.7 mm messo in rotazione attorno al proprio asse. Ogni raccolta è stata preceduta da una calibrazione dell’errore di zero utilizzando come standard polvere di corindone (SRM 676), valutando errori di zero di 0.01° in 2θ. La raccolta dei dati di intensità ha avuto luogo con passi di scansione di 0.015° in 2θ, con velocità variabili. Fra 5° e 60° la velocità è stata di 16 s/step; fra 60° e 110° è stata di 32 s/step. I dati di intensità così raccolti sono stati utilizzati per tentare la determinazione ed il raffinamento strutturale di alcuni campioni studiati, scontrandosi tuttavia con i problemi di disordine strutturale manifestati dagli stessi.
Raccolta di dati in situ con luce di
sincrotrone su polveri
La diffrazione di polveri è stata utilizzata per studiare le trasformazioni in situ all’interno di campioni progressivamente riscaldati. Per consentire tale studio è stato impiegato l’apparato sperimentale descritto da Meneghini et al. (2001). Esso è installato presso la linea di diffrazione GILDA (General Italian Line for Diffraction and Absorption) dell’European Synchrotron Radiation Facility (ESRF) di Grenoble (Francia).
Tale apparato consiste di una slitta che consente di selezionare una porzione degli effetti di diffrazione generati da un campione di polvere posto in rotazione attorno ad un asse φ mentre il sistema di raccolta, rappresentato da un image plate, trasla con
moto rettilineo uniforme dietro la slitta. Viene pertanto raccolto in continuo un diffrattogramma di polveri in funzione del tempo e, assumendo un tasso costante di riscaldamento del campione, in
18
funzione della temperatura. La lunghezza d’onda della radiazione utilizzata, la velocità di riscaldamento e la temperatura raggiunta al termine dell’esperimento sono descritte in dettaglio nel seguito del lavoro di tesi.
Tecniche di raffinamento da dati di polveri: il metodo Rietveld ed il metodo Le Bail
I dati di polvere sono stati trattati con il pacchetto di programmi GSAS (Larson & Von Dreele, 1994) attraverso l’interfaccia grafica EXPGUI (Toby, 2001). In funzione della problematica da affrontare, sono stati eseguiti raffinamenti Rietveld e Le Bail.
L’intensità di un riflesso hkl è esprimibile come
Ihkl = SF x Mhkl x LP(θ) x A(θ) x Phkl x Ehkl x |Fhkl|
dove SF è il fattore di scala; M
2
hkl è il fattore di molteplicità; LP(θ) è il fatt ore di Lorentz e
polarizzazione; A(θ) è l’assorbimento dei raggi X incidenti e diffratti; Phkl è la correzione per
l’orientazione preferenziale; Ehkl è il fattore di estinzione; Fhkl
Il raffinamento Rietveld consente di modellare parametri strutturali e strumentali (divergenza assiale, spostamento del campione, errore di zero, fondo), in modo da minimizzare, attraverso un algoritmo di minimi quadrati, la differenza fra spettro di polvere osservato e quello calcolato. Premessa indispensabile è l’utilizzo di un modello strutturale ragionevolmente corretto, tanto più accurato quanto maggiore è la complessità del sistema studiato.
è il fattore di struttura.
La Commission on Powder Diffraction dell’International Union of Crystallography ha stilato un insieme di linee guida generali per il raffinamento strutturale attraverso il metodo Rietveld (McCusker et al., 1999). Per un raffinamento Rietveld è di estrema importanza la fase di raccolta dei dati. Sono infatti da considerare alcuni aspetti: 1) geometria del diffrattometro; 2) qualità dell’allineamento e della calibrazione dello strumento; 3) tipo di radiazione impiegata (raggi X convenzionali, raggi X di sincrotrone) e lunghezza d’onda; 4) appropriata preparazione del campione; 5) spessore del campione; 6) dimensione delle slitte; 7) tempo di raccolta. Si deve cercare di eliminare ogni possibile orientazione preferenziale; nel nostro caso, l’impiego di una configurazione strumentale che vede un capillare montato orizzontalmente ruotare su se stesso potrebbe virtualmente eliminare gli effetti dell’orientazione preferenziale, benché cristalli aciculari possano tendere a disporsi con l’asse lungo parallelamente all’asse del capillare. La dimensione ideale della polvere è compresa fra 1 e 5 μm.
La descrizione delle forme dei picchi è critica per il successo del raffinamento Rietveld; la forma dei picchi è funzione del campione (dimensione dei domini coerenti, stress/strain, difetti) e dello strumento (radiazione, geometria, dimensione delle fenditure) e può variare in funzione del 2θ. In alcuni casi si possono osservare differenze anche in funzione dell’indice hkl dei riflessi (casi di
19
riflessi di famiglia netti ed intensi e riflessi caratteristici deboli e diffusi). La funzione di profilo più utilizzata è la Pseudo-Voigt, una combinazione lineare di componenti lorentziane e gaussiane. Il fondo è stato raffinato con la funzione Shifted-Chebyshev, con un numero di termini dipendente dalla complessità del profilo.
Oltre ai raffinamenti Rietveld, sono stati effettuati anche raffinamenti Le Bail (Le Bail et al., 1988) dei parametri di cella. Questo tipo di raffinamento viene eseguito senza utilizzare alcun modello strutturale, assegnando ai riflessi il fattore di struttura correlato all’intensità osservata ed alla molteplicità dei riflessi, funzione quest’ultima della simmetria della fase studiata. Il raffinamento Le Bail è stato utilizzato per studiare l’andamento dei parametri di cella durante gli esperimenti di riscaldamento in situ. Attraverso di esso è anche possibile estrarre dati di intensità mediante i quali tentare una soluzione strutturale ab initio partendo da dati di polveri.
Per valutare la bontà di un processo di raffinamento il modo migliore è probabilmente rappresentato dalla linea delle differenze; tuttavia la valutazione può essere effettuata anche numericamente mediante una serie di indici di accordo R. Fra di essi i principali sono rappresentati dai seguenti:
• Rwp = {Σwi [yi(obs) – yi(calc)]2 / Σwi [yi(obs)]2}1/2 , con yi(obs) corrispondente all’intensità
osservata all’i-esimo step, yi(calc) l’intensità calcolata e wi
• R
il peso; il valore di questo indice è fortemente influenzato dalla tecnica di raffinamento e/o sottrazione del fondo;
p = Σ[yi(obs) – yi(calc)]/Σyi
• R
(obs);
exp= [(N-P)/Σwiyi(obs)2]1/2
• χ
, con N corrispondente al numero di osservazioni e P al numero di parametri raffinati.
2
= Rwp/Rexp; tale rapporto, noto come goodness-of-fit, dovrebbe idealmente convergere a 1.
Nel caso di dati ridondanti esso può essere significativamente maggiore di 1; viceversa, per raccolte troppo veloci, con un piccolo numero di osservazioni rispetto ai parametri, il χ2
• R
può essere minore di 1.
B = Σ│Ihkl(obs) – Ihkl(calc)│/Σ│Ihkl(obs)│, con Ihkl = mF2hkl (m = molteplicità).
2.2 Analisi chimiche
2.2.1 Analisi chimiche in modalità EDS
La caratterizzazione chimica qualitativa e semiquantitativa dei campioni studiati è stata eseguita attraverso il sistema SEM-EDS del Dipartimento di Scienze della Terra dell’Università di Pisa; questo strumento è formato dall’unione di un microscopio elettronico a scansione (SEM) con un dispositivo per la microanalisi in dispersione di energia (EDS). Gli standard utilizzati per la
20
calibrazione dello strumento sono: olivina (Mg, Fe, Si), albite (Na, Al, Si), ortoclasio (K, Al, Si), diopside (Ca, Mg, Si)
Il campione, preventivamente reso conduttivo mediante grafitizzazione, viene bombardato mediante un fascio elettronico sottoposto ad una differenza di potenziale di 20 kV. L’interazione elettroni incidenti – campione è all’origine di vari fenomeni che possono essere utilizzati per ottenere informazioni sulla chimica del campione e per raccogliere immagini dettagliate della morfologia superficiale dell’oggetto.
Un primo tipo di segnale è costituito dagli elettroni secondari (SE, Secondary Electrons); si tratta di elettroni con energia inferiore ai 50 eV, emessi dalla porzione superficiale del campione. Pertanto essi sono il segnale usato per lo studio morfologico. Un altro tipo di segnale è rappresentato dagli elettroni retrodiffusi (BSE, Back-Scattered Electrons), ossia quella porzione del fascio incidente che viene riflessa dal campione. Essi hanno energie comprese fra 50 eV sino a quella di incidenza. La quantità di BSE emessi dipende, oltre che dalla morfologia della superficie del campione, anche dal numero atomico medio degli atomi costituenti il campione stesso. Questo fatto consente di individuare eventuali disomogeneità composizionali all’interno dell’esemplare studiato. Infine un ulteriore segnale che si origina per l’interazione fra il fascio elettronico e gli atomi del campione è rappresentato da raggi X caratteristici che, raccolti mediante un cristallo di Si drogato con Li, consentono di conoscere la composizione chimica del volume bombardato dagli elettroni. Lo spettrometro opera, come detto sopra, in modalità EDS e riceve simultaneamente tutti i fotoni della radiazione X, separarandoli successivamente in funzione delle loro energie.
L’utilizzo di questo particolare strumento consente dunque di ottenere dettagliate immagini del campione ed analisi chimiche qualitative e semi-quantitative. Per poter ottenere dati chimici sufficientemente accurati è necessario utilizzare un campione piano nel quale il volume incognito di cui vogliamo conoscere la composizione chimica, sia maggiore del volume di interazione del fascio elettronico con il campione.
2.2.2 Analisi chimiche in modalità WDS
La caratterizzazione chimica quantitativa dei silicati idrati di calcio è stata eseguita con microsonda ARL-SEMQ dell’Università degli Studi di Modena e Reggio Emilia. Le condizioni operative sono: 15 kV, 20 nA, diametro del fascio 15 μm. Gli standard utilizzati sono: anortite (Si, Ca), microclino (K, Al), albite (Na), CaF2
I fenomeni di interazione fra il fascio elettronico ed il campione sono i medesimi già descritti per il sistema SEM-EDS. La differenza consiste nel fatto che la microsonda effettua una spettroscopia in dispersione di lunghezza d’onda (sistema WDS, Wavelength Dispersive System) anziché in
21
dispersione di energia. Lo spettroscopio contiene una serie di cristalli analizzatori curvi che si muovono lungo il cerchio di focalizzazione (o cerchio di Rowland) secondo differenti angoli e riflettono i raggi X emessi dal campione secondo la nota equazione di Bragg, focalizzandoli in un contatore. In questo modo si ha un’alta risoluzione nella separazione delle varie lunghezze d’onda, consentendo analisi chimiche quantitative accurate. Passo essenziale nella raccolta di dati chimici è la scelta di appropriati standard, quanto più possibile simili al campione da investigare, con i quali il campione stesso possa essere confrontato.
2.3 Analisi spettroscopiche
2.3.1 Spettroscopia
29Gli spettri
Si NMR
29
La spettroscopia NMR (Nuclear Magnetic Resonance) è un potente strumento per lo studio della struttura e del comportamento dinamico delle fasi condensate. Essendo specifica di un determinato elemento, essa consenta di ottenere informazioni addizionali e complementari rispetto a quelle che possono essere raccolte mediante la diffrazione di raggi X e le spettroscopie vibrazionali. L’applicazione della spettroscopia NMR allo studio delle fasi solide si è evoluta in maniera esplosiva a partire dall’inizio degli anni Ottanta, con il lavoro di Lippmaa et al. (1980). Le basi teoriche di questa spettroscopia sono esaurientemente illustrate in Kirkpatrick (1988) e Fechtelkord (2004). Esse si fondano su una proprietà quantizzata di molti nuclei atomici, nota con il termine spin, che può venire visualizzata come il movimento di rotazione dell’atomo attorno al proprio asse. NMR sono stati raccolti presso l’Istituto per i Processi Chimico-Fisici (IPCF) del CNR-Pisa mediante uno spettrometro Bruker AMX300WB.
Soltanto i nuclidi che posseggono un momento magnetico (numero di spin diverso da zero) sono di interesse per la spettroscopia NMR.
Ogni nuclide ha 2I+1 livelli energetici, descritti dal numero quantico m, che, in assenza di campi magnetici esterni, sono degeneri (hanno cioè la stessa energia). L’applicazione di un campo magnetico induce una interazione fra questi livelli energetici ed il campo magnetico (effetto Zeeman), generando così una differenza di energia fra di loro. La differenza di energia ∆E fra questi livelli sarà:
∆E= |γ(h/2π)H|
dove γ è il rapporto giromagnetico, h è la costante di Planck e H il campo magnetico nei pressi del nucleo.
Un nucleo potrà variare la propria energia assorbendo o emettendo un fotone di frequenza ben definita, corrispondente alle differenze energetiche fra i vari livelli. La frequenza ν del fotone sarà:
22 ν = (γ/2π)H
Negli esperimenti di spettroscopia NMR si misura la frequenza ν della radiazione interagente con i nuclei atomici; tale radiazione cade nel dominio delle frequenze radio.
Gli elettroni posti in prossimità di un atomo schermano il nucleo dal campo magnetico applicato. Pertanto il segnale misurato sarà funzione dell’ambiente locale degli atomi, ossia gli angoli di legame, il numero di coordinazione, la simmetria locale e la natura degli atomi presenti all’interno della prima e della seconda sfera di coordinazione. Di conseguenza, il campo magnetico H nei pressi del nucleo sarà:
H = H0
dove H
(1-σ)
0
ν = (γ/2π)H
è il campo magnetico applicato e σ è l’effetto di schermatura degli elettroni posti in prossimità del nucleo. Pertanto la frequenza misurata sarà:
0
Considerando le difficoltà nella misura del valore assoluto di H (1-σ).
0
Uno dei principali problemi da affrontare nell’ottenimento di spettri NMR è quello dell’allargamento dei picchi legato ad una serie di fenomeni magnetici interatomici. Questo allargamento si supera mediante la tecnica chiamata Magic-Angle Spinning (MAS) che consiste nel far ruotare il campione su un asse posto ad un angolo di 54.7° dal campo magnetico applicato H
, è difficile ottenere valori accurati della frequenza NMR. È per tale motivo che le frequenze di risonanza sono espresse come spostamento chimico δ, in ppm (parti per milione), relativamente ad uno standard. Un altro fenomeno che ha luogo nei nuclidi con I > 1/2 è l’interazione quadripolare legata all’asimmetria nella distribuzione delle cariche elettriche, all’origine di un momento quadripolare. Quest’ultimo può interagire con il gradiente del campo elettrico locale, anch’esso caratteristico dell’ambiente locale intorno al nuclide studiato.
0.
Fra i vari tipi di esperimenti eseguibili con la tecnica NMR, alcuni fanno ricorso all’eccitazione contemporanea di due nuclidi. La tecnica cross polarization (CP) è una delle più usate e consiste nel trasferire lo spin di
Altri problemi, difficilmente risolvibili, riguardano la presenza di elementi paramagnetici (Fe, Mn), anch’essi all’origine di un allargamento dei picchi.
1
H allo spin del sistema di interesse; essa consente di aumentare il rapporto segnale/fondo e di ottenere informazioni strutturali addizionali. Infatti il segnale dei nuclidi vicini agli atomi di idrogeno viene esaltato rispetto a quello dei nuclidi posti lontano, fornendo informazioni strutturali aggiuntive ed eliminando dallo spettro il segnale delle fasi anidre.
23
2.3.2 Spettroscopia micro-Raman
Spettri micro-Raman su campioni di alcuni silicati idrati di calcio sono stati raccolti presso il Dipartimento di Fisica dell’Università di Parma utilizzando un apparato Jobin-Yvon Horiba “Labram”, equipaggiato con un microscopio Olympus. La radiazione incidente è rappresentata da un laser a He-Ne, la cui potenza è controllata mediante una serie di filtri di densità. La risoluzione laterale e verticale è dell’ordine di pochi μm. Prima di ogni acquisizione, il sistema è stato calibrato utilizzando la banda a 520.6 cm-1
La spettroscopia micro-Raman è una spettroscopia vibrazionale che implica l’utilizzo di una radiazione luminosa monocromatica per studiare il comportamento vibrazionale dei sistemi molecolari, attraverso un esperimento di diffusione della luce. La maggior parte della radiazione incidente esce dal campione senza modifiche mentre una piccola frazione (intorno a 10
del Si; gli spettri sono stati raccolti con acquisizioni multiple (da 2 a 6), con tempi di conteggio variabili fra 20 e 180 s.
-3
) viene diffusa. Di questa, la massima parte subisce una diffusione elastica (senza cambiamento di energia), nota come diffusione Rayleigh; soltanto una piccola parte (10-6
La spettroscopia micro-Raman consente di eseguire analisi in situ non distruttive, senza una preventiva preparazione del campione, che può quindi essere successivamente utilizzato per altre indagini strumentali.
del fascio incidente) viene diffusa in maniera anelastica, mutando quindi la propria energia. Quest’ultimo processo prende il nome di diffusione Raman. L’energia della luce diffusa è analizzata tramite uno spettrometro ed i picchi Raman appaiono come deboli picchi spostati rispetto alla linea della diffusione Rayleigh. Poiché lo spettro vibrazionale è funzione delle forze interatomiche, esso sarà sensibile alla struttura microscopica ed ai legami presenti nel composto studiato. Pertanto lo spettro Raman è una sorta di impronta digitale in grado di consentire l’identificazione del campione indagato. Inoltre, la presenza di particolari bande può consentire di ipotizzare la presenza di certi gruppi strutturali; mediante una opportuna calibrazione, sarebbe possibile ottenere anche informazioni quantitative.
Una accurata descrizione dei principi teorici e sperimentali della spettroscopia micro-Raman sono in McMillan & Hofmeister (1988).
2.4 Analisi termo-gravimetriche
Le analisi termo-gravimetriche sono state condotte presso il Dipartimento di Scienze della Terra dell’Università di Pisa, utilizzando uno strumento modello Netzsch STA 449C Jupiter accoppiato a uno spettrometro di massa ThermoStarTM GSD 301 T2. Gli esperimenti sono stati condotti su campioni di polvere (quantitativi di circa 25 mg) posti all’interno di crogiuoli di Al2O3, da
24
temperatura ambiente (20°C) sino a 1000°C, con velocità di riscaldamento di 10°C/min, in atmosfera di N2
Le analisi termo-gravimetriche (TG) consentono di osservare le variazioni di peso come funzione della temperatura, permettendo di osservare le eventuali perdite di fasi volatili e stimandone la corrispondente percentuale in peso. Lo spettrometro di massa consente inoltre di verificare quali siano le specie volatili rilasciate durante il processo di riscaldamento. In aggiunta allo studio termo-gravimetrico, è possibile raccogliere anche informazioni calorimetriche (DSC, Differential Scanning Calorimetry), in grado di fornire informazioni sulle eventuali reazioni (esotermiche o endotermiche) che hanno luogo durante l’incremento di temperatura. Tali reazioni possono indicare sia processi di disidratazione sia vere e proprie transizioni di fase.