3)
M
ETODI DI ANALISI
3.1) ESTRAZIONE DEL DNA
Da un’aliquota del campione di sangue intero raccolto per ciascun paziente è stato estratto il DNA, al fine di poter effettuare l’analisi genotipica. L’estrazione del DNA è stata fatta utilizzando il kit QIAamp DNA Blood della Qiagen, seguendo le operazioni del protocollo standard. Il processo di estrazione prevede diverse fasi purificazione del materiale estratto, volte ad eliminare tutte le componenti cellulari e proteiche che possono inquinare il campione. In un primo passaggio, ad un volume standard di 200 µl di sangue vengono aggiunti 20 µl di proteasi K, la cui azione serve a degradare il materiale proteico, e 200 µl del tampone di lisi AL, necessario per la degradazione delle membrane biologiche. Il tutto viene agitato per pochi secondi e poi incubato in bagnetto termostatico a 56° per 10 minuti. La temperatura è importante per l’azione della proteasi K e per favorire la lisi cellulare. Al termine dei 10 minuti, la provetta contenente la miscela di reazione viene centrifugata ad 8000 rpm per 20 secondi. Si aggiungono a questo punto 200 µl di etanolo puro, che servono alla precipitazione del DNA, si agita per pochi secondi ed infine si centrifuga a 8000 rpm per 20 secondi. La soluzione così ottenuta si applica ad una colonnina QIAamp, costituita da una resina silicea che interagisce col DNA, bloccandolo nella colonna. La colonnina viene posta all’interno di uno specifico tubo di raccolta da 2 ml e centrifugata a 8000 rpm per 1 minuto. Al termine della centrifugazione, viene recuperata e inserita in un nuovo tubo di raccolta, mentre il materiale eluito viene eliminato. Cominciano a questo punto una serie di lavaggi successivi volti a purificare il DNA estratto. Alla colonnina vengono applicati 500 µl di tampone di lavaggio AW1, ed il tutto viene sottoposto a nuova centrifugazione a 8000 rpm per 1 minuto. La colonnina viene nuovamente recuperata e posta su un nuovo tubo di raccolta da 2 ml, mentre il materiale eluito viene ancora una volta eliminato. Si applicano alla
colonna 500 µl di un altro tampone di lavaggio, AW2, e si centrifuga a 14000 rpm per 3 minuti. Per il recupero del DNA, la colonnina viene posta su una eppendorf con chiusura di sicurezza da 1,5 ml e su di essa vengono applicati 200 µl di tampone AE, un tampone di eluizione necessario per il recupero del DNA. La colonna viene lasciata a temperatura ambiente per circa 5 minuti e successivamente centrifugata a 8000 rpm per 1 minuto. A questo punto, il DNA estratto si trova in soluzione nella eppendorf da 1,5 ml.
Il DNA viene tenuto alla temperatura di –20°C, men tre una piccola aliquota necessaria alle operazioni di genotipizzazione quotidiane viene conservata a 4°C per non più di un mese.
3.2) QUANTIZZAZIONE E NORMALIZZAZIONE DEL DNA ESTRATTO
Una volta estratto, per poter standardizzare le analisi genotipiche successive, il DNA deve essere normalizzato, ovvero portato alla medesima concentrazione di partenza per tutti i campioni estratti. Per poter procedere alla normalizzazione del DNA, è necessaria prima la sua quantizzazione, ovvero una procedura che permette di valutarne la concentrazione.
Questo protocollo si avvale dell’uso di una sostanza fluorescente, denominata Pico Green, che si coniuga al DNA e che emette a lunghezze d’onda comprese tra 480 nm e 520 nm ed attraverso la quantizzazione della sua luce emessa si risale alla concentrazione di DNA nel campione.
Il kit di reazione utilizzato per la quantizzazione del DNA è riportato nella
tabella 3.1.
Pico Green 1 ml soluzione in DMSO (dimetilsulfossido) 20 X TE 25 ml Tris-HCL 200 mM, EDTA 20 mM, pH 7.5 DNA standard fago Lambda 1 ml 100 µg/ml in TE
La quantizzazione del DNA avviene mediante la costruzione di una curva di taratura, effettuata attraverso l’utilizzo di uno standard di DNA a concentrazione nota. Lo standard, costituito solitamente da DNA del batteriofago Lambda, è fornito ad una concentrazione iniziale di 100 µg/ml, che può essere diluito 50 volte in TE per ottenere una concentrazione di 2
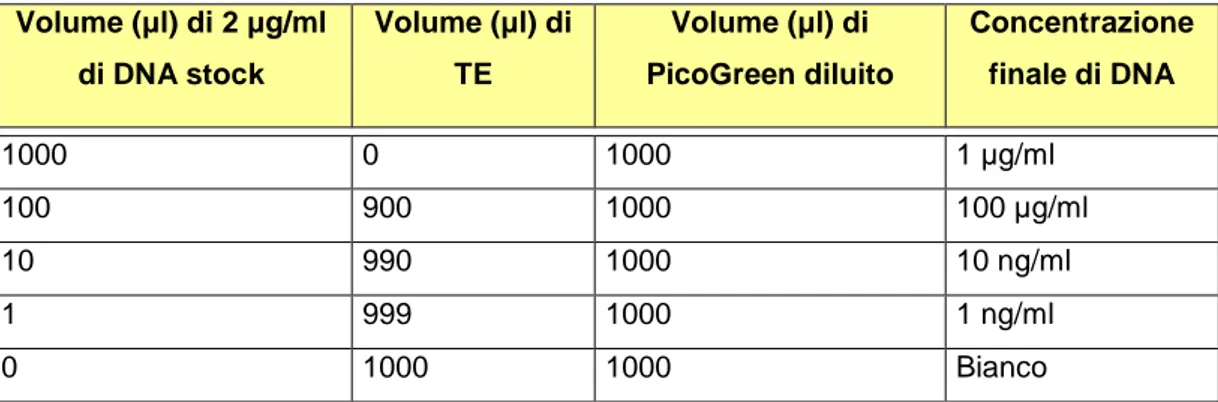
µg/ml di soluzione da lavoro. Una curva di calibrazione può essere disegnata, ad esempio, utilizzando 30 µl di DNA standard, mescolati a 1.47 ml di TE. In tabella 3.2 è riportato il protocollo per una curva standard di DNA. Volume (µl) di 2 µg/ml di DNA stock Volume (µl) di TE Volume (µl) di PicoGreen diluito Concentrazione finale di DNA 1000 0 1000 1 µg/ml 100 900 1000 100 µg/ml 10 990 1000 10 ng/ml 1 999 1000 1 ng/ml 0 1000 1000 Bianco
Tabella 3.2. Protocollo per la preparazione di una curva standard di taratura
I campioni ugualmente diluiti in TE e PicoGreen vengono posti su piastre a 96 pozzetti insieme al DNA standard e al controllo negativo (soluzione priva di DNA). La piastra viene inserita nello spettrofluorimetro, uno strumento in grado di rilevare e quantificare l’emissione del Pico Green, che, con una elaborazione computerizzata, calcola la concentrazione di ogni singolo campione. Infine, i campioni vengono opportunamente diluiti al fine di portarli tutti alla stessa concentrazione finale.
3.3) PCR ED ELETTROFORESI DI CONTROLLO
Prima di procedere alla genotipizzazione, è sempre buona norma sincerarsi che il DNA estratto sia amplificabile (integro e purificato): il DNA viene testato mediante PCR ed il risultato è controllato con elettroforesi su gel di agarosio.
La PCR (Polimerase Chain Reaction) è una tecnica che permette di ottenere copie multiple di un certo segmento di DNA. In questa reazione sono necessari i primers, o inneschi (Fw e Rw), che delimitano la regione da amplificare, i nucleotidi necessari per l’estensione e l’enzima DNA polimerasi per la sintesi dei filamenti di DNA. Questi reagenti vengono addizionati ad una soluzione contente anche MgCl2 ed un tampone ottimale per l’attività
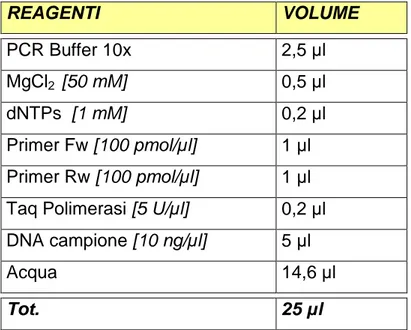
dell’enzima ed, ovviamente, il DNA genomico da amplificare. Il volume finale della miscela di amplificazione è di 25 µl. Nella tabella 3.3 è riportata la composizione della miscela di amplificazione.
REAGENTI VOLUME PCR Buffer 10x 2,5 µl MgCl2 [50 mM] 0,5 µl dNTPs [1 mM] 0,2 µl Primer Fw [100 pmol/µl] 1 µl Primer Rw [100 pmol/µl] 1 µl Taq Polimerasi [5 U/µl] 0,2 µl DNA campione [10 ng/µl] 5 µl Acqua 14,6 µl Tot. 25 µl
Tabella 3.3. Miscela di reazione per PCR di controllo
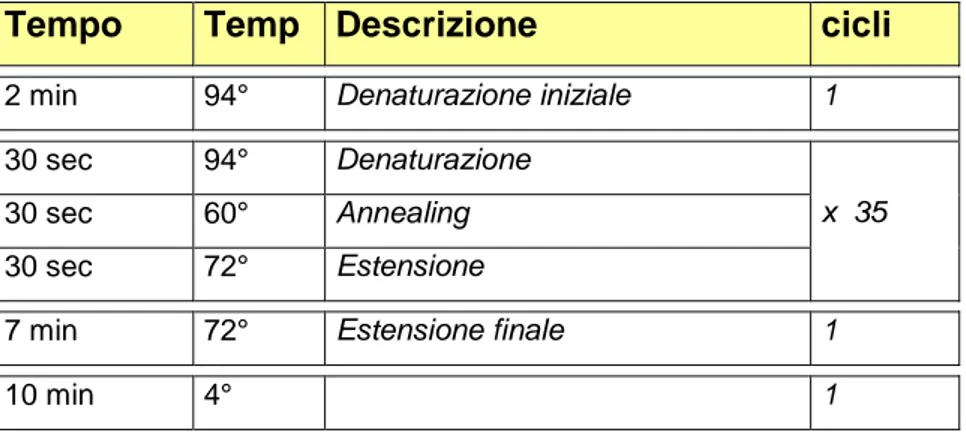
La reazione di PCR viene condotta in termociclizzatori, in grado di variare ciclicamente, secondo un programma prestabilito, la temperatura della reazione. Generalmente, la reazione di PCR prevede una fase di denaturazione, necessaria alla separazione dei due filamenti, una fase di appaiamento, per permettere ai primers di appaiarsi in maniera complementare al DNA stampo, ed una fase di estensione, in cui la DNA polimerasi amplifica la sequenza di interesse. Ciascun ciclo di denaturazione-appaiamento-estensione viene ripetuto più volte, al fine di ottenere un elevato numero di copie. Il protocollo di reazione, effettuato in termociclizzatori Perkin Elmer, viene riportato di seguito (Tab. 3.4).
Il risultato dell’amplificazione viene poi controllato mediante elettroforesi su gel di agarosio al 2%. Il gel viene preparato utilizzando una soluzione di 200 ml di tampone TBE, diluito 1x, al quale vengono aggiunti 4 g di agarosio in polvere. La beuta viene agitata manualmente per favorire l’iniziale dissoluzione dell’agarosio nel tampone e successivamente riscaldata fino a quando la soluzione non assume un aspetto limpido e trasparente e la beuta viene posta a raffreddare.
Tempo Temp Descrizione cicli
2 min 94° Denaturazione iniziale 1
30 sec 94° Denaturazione
30 sec 60° Annealing
30 sec 72° Estensione
x 35
7 min 72° Estensione finale 1
10 min 4° 1
Tabella 3.4. Protocollo per la reazione di PCR di controllo
Da questo momento in poi le operazioni vengono eseguite sotto cappa chimica, in quanto si utilizzano sostanze tossiche come il bromuro d’etidio e gli stessi vapori del gel. Il gel viene fatto raffreddare per circa 15 minuti e solo quando è sufficientemente freddo viene aggiunto il bromuro d’etidio ad una concentrazione di 0,5 µg/ml. Il bromuro d’etidio è un intercalante del DNA, che se illuminato con luce ultravioletta, permette la visualizzazione delle bande sul gel a corsa avvenuta. Il gel viene quindi versato nell’apposita vaschetta, precedentemente preparata con un pettine ad una delle estremità, che verrà tolto quando il gel sarà solidificato e lascerà liberi i pozzetti di caricamento del campione. La vaschetta viene quindi coperta per evitare il decadimento alla luce del bromuro d’etidio ed il gel viene lasciato circa un’ora a solidificare.
In seguito, la vaschetta viene posta nella cella di corsa, riempita con un tampone (TBE) e collegata ad un alimentatore a corrente continua, in grado di applicare una differenza di potenziale alla vasca. Si procede quindi al caricamento dei campioni amplificati per poi eseguirne l’elettroforesi. Solitamente, vengono caricati 10 µl di prodotto di PCR, ai quali viene
addizionato 1 µl di loading buffer necessario alla visualizzazione della corsa. Il loading buffer è costituito da un colorante, spesso il blu di bromofenolo, glicerolo e saccarosio, necessari a favorire la discesa del DNA nel pozzetto di caricamento. Comunemente, il primo pozzetto viene caricato con un ladder, una soluzione di frammenti di DNA a peso molecolare noto, utile per stabilire le dimensioni dei frammenti di DNA visualizzati sul gel. Al termine della procedura di caricamento, che deve comunque avvenire in tempi brevi per evitare che il DNA diffonda attraverso il gel, si applica la corrente alla camera di corsa. I campioni vengono fatti correre a 110 volt per circa 30 minuti. La presenza del colorante, comunque, permette di visualizzare il fronte più avanzato della corsa.
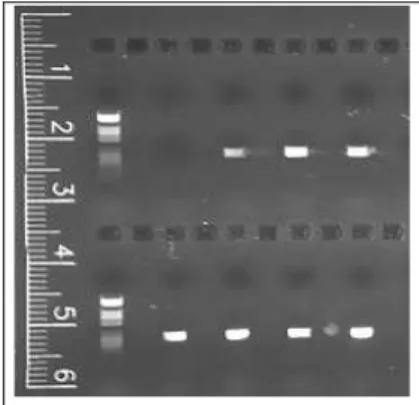
Una volta spento il l’alimentatore, la vaschetta contenete il gel viene rimossa dalla cella di corsa e portata al transilluminatore, uno strumento a luce UV che permette la visualizzazione della bande sul gel. Un esempio di gel di controllo per una PCR è riportato in figura 3.1.
Figura 3.1. Gel di controllo per PCR
3.4) LA GENOTIPIZZAZIONE DEL POLIMORFISMO C3435T:il metodo TaqMan in
Real Time PCR
La genotipizzazione al locus C3435T di MDR1 è stata condotta attraverso l’utilizzo della Real Time PCR con il metodo TaqMan, un sistema di rilevazione in tempo reale che consente di determinare il genotipo in base
all’emissione di fluorofori legati a sonde allele-specifiche. Il sistema di amplificazione e rilevazione utilizzato è il sistema iCiclerTMiQ della Biorad.
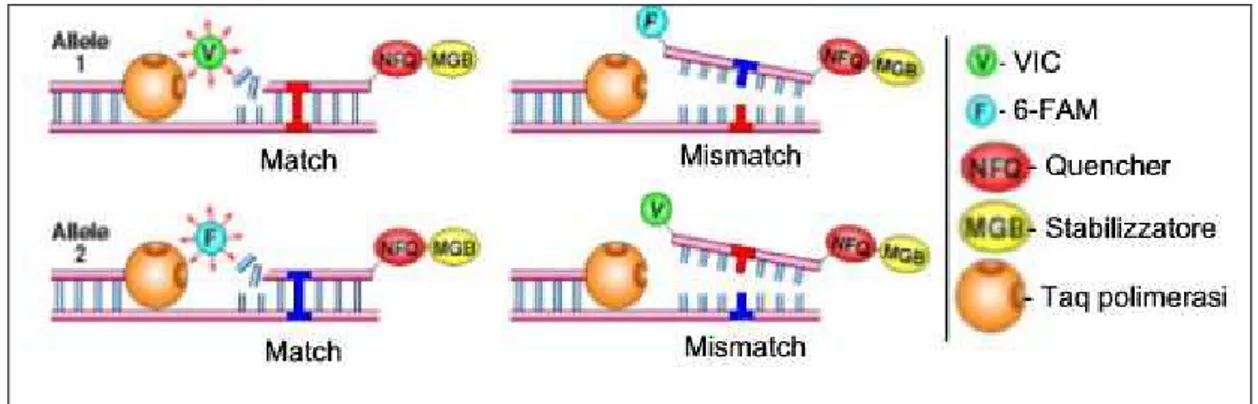
Attraverso l’uso di sonde allele-specifiche per gli alleli 3435C e 3435T, ciascuna delle è quali legata ad un differente fluoroforo, è possibile monitorare la reazione di PCR in real time, grazie all’ausilio di un computer. Il metodo TaqMan in particolare, è stato ideato per la discriminazione allelica di polimorfismi a singolo nucleotide. Nella tabella 3.5 sono riportate le sequenze oligonucleotidiche dei primers utilizzati per la reazione di PCR.
PRIMERS SEQUENZE
MDR1 (forward) 5’-CTGTTTGACTGCAGCATTGCT-3’ MDR1 (reverse) 5’-ATGTATGTTGGCCTCCTTTGCT-3’
Tabella 3.5. Oligonucleotidi per l’amplificazione del gene MDR1
Le sonde oligonucleotidiche utilizzate per la genotipizzazione del gene MDR1 sono marcate all’estremità 5’ con due fluorofori differenti, ed in particolare la sonda specifica per l’allele wild type (3435C) è marcata con il fluoroforo VIC, mentre la sonda specifica per la sequenza mutata (3435T) è marcata con il fluoroforo 6-FAM. Il fluoroforo VIC emette alla lunghezza d’onda di 530 nm, mentre il fluoroforo 6-FAM emette a 490 nm. In tabella
3.6, sono riportate le sonde allele-specifiche utilizzate. Ciascuna sonda, poi,
porta legato all’estremità 3’ un silenziatore (Quencher), che assorbe la luce emessa dal fluoroforo quando si trova nelle sue immediate vicinanze.
FLUOROFORI SONDE TaqMan ALLELE
VIC 5’-VIC-TGAGGCGGGTGGATCACT-Q-3’ 3435C
6-FAM 5’-6-FAM-AGGCTGAGGCAGGTGGAT-Q-3’ 3435T
Tabella 3.6. Sonde allele-specifiche per gli alleli C/T al sito 3435 di MDR1
Dal punto di vista teorico, il principio sul quale si basa il metodo TaqMan è piuttosto semplice. I primers forward e reverse sono disegnati in maniera tale da delimitare una sequenza di DNA all’interno della quale è compreso il sito di mutazione. Durante la fase di annealing, oltre ai primers,
anche le sonde allele-specifiche si appaiano alla loro sequenza complementare. Quando una sonda per un allele si appaia alla sequenza specifica per l’altro allele, si crea un mismatch. La Taq DNA polimerasi utilizzata nella fase di estensione è dotata, oltre che dell’attività polimerasica, anche di un’attività esonucleasica 5’-3’, che permette all’enzima di degradare eventuali frammenti di DNA che incontra legati lungo il filamento stampo. Grazie all’apposito disegno dei primers, durante la fase di estensione la Taq DNA polimerasi incontra le sonde allele-specifiche che si sono ibridate alla loro sequenza complementare sul DNA stampo. Quando l’appaiamento tra sonda e DNA stampo è perfetto, ovvero privo di mismatch, l’attività esonucleasica della Taq DNA polimerasi provvederà alla degradazione della sonda, liberando così il fluoroforo all’estremità 5’ che, allontanandosi dal silenziatore, potrà emettere alla sua lunghezza d’onda specifica. Al contrario, quando l’appaiamento tra DNA stampo e sonda presenta mismatch, la DNA polimerasi non degrada la sonda, bensì provvede semplicemente a scalzarla intatta, senza liberare il fluoroforo dall’effetto silenziatore del Quencher. In questo modo, quindi, la presenza di emissione è indice di un perfetto appaiamento della sonda, e la discriminazione tra i due fluorofori utilizzati permette di stabilire qual è la configurazione genotipica al locus considerato. E’ anche importante sottolineare che la fluorescenza registrata è proporzionale alla quantità di DNA amplificato in ogni momento della reazione, poiché viene liberata una molecola di fluoroforo per ogni copia di DNA duplicata.
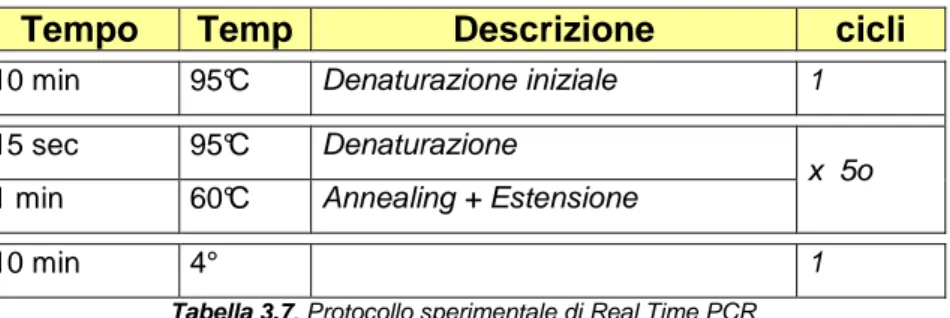
La figura 3.2 riassume gli eventi che si verificano in una amplificazione con metodo TaqMan. Il protocollo sperimentale utilizzato per la reazione di amplificazione in Real Time, come quello di una comune PCR, prevede più cicli di denaturazione, annealing ed estensione (Tab 3.7).
Tempo Temp Descrizione cicli
10 min 95°C Denaturazione iniziale 1
15 sec 95°C Denaturazione
1 min 60°C Annealing + Estensione x 5o
10 min 4° 1
Tabella 3.7. Protocollo sperimentale di Real Time PCR
I valori di fluorescenza emessi vengono rilevati in tempo reale da un opportuno dispositivo fluorimetrico collegato ad un computer che provvede all’elaborazione dei dati. Nelle prime fasi della reazione di PCR, la fluorescenza si mantiene a valori piuttosto bassi.
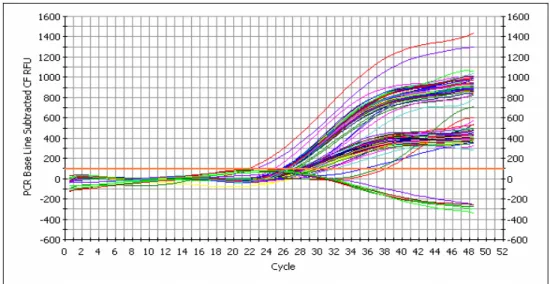
Il primo ciclo di amplificazione in cui viene rilevato un aumento evidente della fluorescenza è definito come “ciclo soglia”. Da questo punto in poi la reazione entra nella fase detta di amplificazione esponenziale, ovvero quella fase in cui tutti i campioni, indipendentemente dalla quantità di DNA iniziale, sono amplificati con la medesima efficienza. Ciascun campione ha un proprio ciclo soglia, anche se l’utilizzo di DNA precedentemente normalizzato fa sì che, più o meno, il ciclo soglia sia lo stesso per tutti i campioni. Ciascun ciclo soglia corrisponde al punto in cui le curve di amplificazione intersecano la linea di base della fluorescenza. Nelle figure
3.3 e 3.4 sono riportate le curve di amplificazione rispettivamente per i
fluorofori VIC e FAM relative alla genotipizzazione di alcuni campioni.
La quantità di campioni analizzabili contemporaneamente dipende dallo strumento: il termociclizzatore da me utilizzato lavora con piastre a 96 pozzetti, per cui possono essere analizzati circa 90 campioni contemporaneamente. I restanti 6 pozzetti sono solitamente riempiti con i controlli negativi (senza DNA) e positivi (DNA a genotipo noto).
Figura 3.3. Spettro di emissione per VIC-530 in Real Time
Figura 3.4. Spettro di emissione per FAM-490 in Real Time
L’allestimento della reazione di PCR prevede la preparazione di due miscele distinte. Si utilizza una miscela 2x, contente buffer, MgCl2, nucleotidi
e Taq polimerasi, ed una miscela 20x contente le sonde ed i primers necessari. Il volume dei pozzetti nel nostro dispositivo può essere tarato a 15
µl o a 25 µl. Solitamente, ogni pozzetto contiene 15 µl di soluzione, costituiti da 4 µl di DNA da genotipizzare e 11 µl di miscela finale.
In tabella 3.8 è riportato un esempio di miscela di reazione per un unico campione. L’identificazione dei campioni analizzati è fornita ad un software, che attribuisce il genotipo a ciascun campione in base alla fluorescenza rilevata, riportata in RFU (Relative Fluorescence Unit).
MISCELA DI REAZIONE Buffer dNTP Taq Mix 2x (7.5 µl) Vic e 6-Fam (sonde)
Primers H2O Mix 20x (3.5 µl) DNA Campione 4 µl Tot. 15 µl
Tabella 3.8. Miscela di reazione per Real -Time PCR
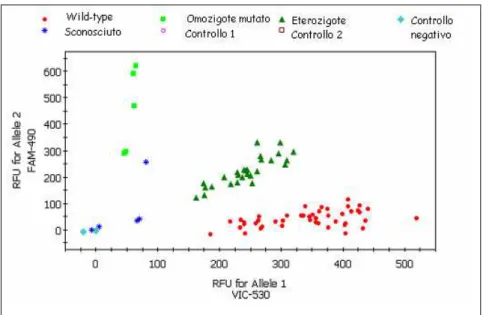
Nella figura 3.5 è visibile l’output dei genotipi per alcuni campioni analizzati.
Figura 3.5. Attribuzione dei genotipi per MDR1 (iCiclerTM Biorad)
In rosso (allele 1) sono riportati gli omozigoti wild-type (3435C), in verde scuro gli eterozigoti (3435C/T) ed, infine, in verde chiaro (allele 2) gli omozigoti mutanti (3435T/T). Nella figura poi compaiono anche alcuni campioni in blu, che corrispondono a genotipi incerti, che necessitano quindi di un ulteriore controllo per essere attribuiti in modo univoco. In celeste sono riportati i controlli negativi. I dati ottenuti in seguito alla genotipizzazione di tutti i campioni a disposizione sono stati poi sottoposti ad un severo controllo per garantire l’univocità del genotipo attribuito.
3.5) LA GENOTIPIZZAZIONE DEL POLIMORFISMO G2677T/A: analisi dei
polimorfismi dei frammenti di restrizione (RFLP)
La genotipizzazione al locus G2677T/A di MDR1 è stata effettuata attraverso l’analisi dei polimorfismi di lunghezza dei frammenti di restrizione (RFLP), una metodica che si basa sulla presenza o assenza di un sito di restrizione per uno specifico enzima determinata dal cambiamento di nucleotide al sito del polimorfismo. Questo tipo di tecnica prevede infatti una digestione enzimatica, operata con un enzima opportunamente scelto, cui segue la visualizzazione del pattern di frammenti originati mediante elettroforesi su gel di agarosio. In questo modo, è possibile discriminare tra alleli differenti e quindi attribuire genotipo certo ad ogni individuo.
Nel caso specifico dello SNP G2677T/A il sistema di discriminazione risulta leggermente complicato a causa natura triallelica del polimorfismo. In questo caso, infatti, si è resa necessaria la messa a punto di un sistema di discriminazione basato su una doppia digestione enzimatica e sul confronto dei relativi pattern di elettroforesi.
In letteratura si ritrovano esempi differenti di protocolli di genotipizzazione mediante RFLP per il polimorfismo G2677T/A tra i quali il più consolidato e utilizzato rimane quello proposto nel 2001 da Cascorbi et al[57]. Tuttavia, a causa della ridotta differenza nelle dimensioni dei frammenti di restrizione, la visualizzazione risulta notevolmente laboriosa. Un’alternativa al protocollo proposto da Cascorbi e colleghi risulta dal lavoro di Tang et al (2002)[67] , i quali utilizzano sia primers che enzimi di restrizione differenti.
Il protocollo messo a punto per questo lavoro di tesi è il frutto della sintesi tra i due lavori suddetti, in quanto così facendo sono stati ottmizzati alcuni aspetti della metodica di genotipizzazione rendendo il protocollo più rapido ed efficace al tempo stesso.
Il DNA di ciascun individuo è stato amplificato mediante PCR con due coppie di primers, costituite da un forward comune e due reverse specifici rispettivamente per ciascuna delle due digestioni enzimatiche. I primers utilizzati sono riportati nella tabella 3.9
.
In linea teorica, sarebbe stato possibile eseguire questo tipo di analisi amplificando il DNA di ciascun soggetto con un’unica coppia di primers ed utilizzare poi due enzimi di restrizione differenti in grado di discriminare due dei tre alleli del polimorfismo. Tuttavia, non sono stati individuati due enzimi di restrizione, il cui sito di taglio comprendesse il sito del polimorfismo, in grado di permettere il disegno di un sistema discriminante per i diversi alleli.
Per questa ragione, il sistema ha assunto un’architettura più complessa, in particolare per l’utilizzo di un primer mismatch (MDR-D), che introduce il sito di taglio per l’enzima BanI nella sequenza amplificata (Fig. 3.6).
C* mismatch
5’- TGAAAGATAAGAAAGAACTAGAAGGTGCTGGGAAGGTGAGTCAAACTAAA……… -3’
………CTTCCACGGCCCTTCCACTCAGTTTGATTT –5’ (MDR-D)
Figura 3.6: introduzione del sito di taglio per l’enzima BanI grazie all’uso del primer mismatch MDR-D.
Per ciascun individuo quindi sono stati generati due amplificati, di dimensioni diverse, sottoposti a due digestioni differenti.
Gli enzimi di restrizione utilizzati sono stati le endonucleasi di restrizione BanI e BseYI. Un primo vantaggio ottenuto nel protocollo proposto è quello che entrambi gli enzimi utilizzati esplicano la loro attività endonucleasica a 37°, anche se in presenza di buffe r differenti. In questo modo è stato possibile eseguire le due digestioni in parallelo.
L’enzima BanI è stato impiegato nella digestione del frammento amplificato con la coppia di primers MDR-A/MDR-D. Il sito di taglio di questo enzima, generato attraverso l’introduzione di un mismatch due basi a valle dello SNP, è presente se al sito del polimorfismo si trova una base purinica (G o A). Il pattern di frammenti generato varia tra le 224 bp del frammento non tagliato (T), alle 198 bp e 26 bp dei frammenti digeriti (A o G) (Fig. 3.7).
PRIMERS SEQUENZA
MDR-A (forward) 5’- TGCAGGCTATAGGTTCCAGG…… –3’ MDR-D (reverse) 5’- TTTAGTTTGACTCACCTTCCG*… -3’ MDR-K (reverse) 5’- AGAGCATAGTAAGCAGTAGG… –3’ Tabella 3.9: primers per l’amplificazione del DNA utilizzati per la genotipizzazione al sito G2677T/A
L’enzima BseYI è stato, invece, impiegato nella digestione del frammento amplificato con la coppia di primer MDR-A/MDR-K. Il sito di taglio per l’enzima è presente quando la base al sito del polimorfismo è una G, per cui il cui pattern di frammenti ottenibili prevede un frammento non tagliato di 300 bp (A o T), e due frammenti di digestione di 201 bp e 99 bp (G) (Fig. 3.7).
Figura 3.7: sito di restrizione e relativa localizzazione sul DNA per gli enzimi BanI e BseYI.
Uno schema dei pattern di frammenti di DNA ottenuti nelle due digestioni è schematizzato nella figura 3.8 sottostante.
Primer: MDR-A + MDR-C Primer: MDR-A + MDR-Kim Enzima:
BanI
Enzima:BseYI
200 150 100 50 224 198 198 26 224 26 200 150 100 50 300 201 99 300 300 201 99
L’analisi dei prodotti delle digestioni è stata effettuata mediante elettroforesi su gel di agarosio. Le ridotte dimensioni dei frammenti generati, soprattutto nella digestione con l’enzima BanI, ha richiesto la messa a punto e l’ottimizzazione di un gel ad alto potere risolutivo.
Per questo motivo è stato utilizzato un agarosio “low melting”, quale il NuSieve, in un rapporto di 3:1 con agarosio standard. Sono stati eseguiti numerosi test a concentrazioni di agarosio differenti per saggiare la soluzione ottimale alla risoluzione delle bande di digestioni. La combinazione finale adottata, che offre il miglior compromesso tra velocità di corsa e risoluzione delle bande, è quella che prevede l’impiego di un gel NuSieve/Agarosio 3:1 al 3%. Dal confronto tra le possibili combinazioni degli eventi di taglio enzimatico è possibile ricostruire uno schema per la genotipizzazione (Fig. 3.9). Genotipo G/G Genotipo G/T Genotipo T/T Genotipo G/A Genotipo T/A Genotipo A/A BseYI Ban I BseYI Ban I BseYI Ban I BseYI Ban I BseYI Ban I BseYI Ban I
+ + + - + - - - + - + - + - - + Figura 3.9: schema dei genotipi ottenubuli dal confronto tra le due digestioni con BanI e BseYI.
In questo modo, sono stati analizzati tutti i campioni ricavandone il genotipo attraverso il confronto tra le digestioni.
A causa delle numerose criticità evidenziate nel metodo, il protocollo è stato sottoposto a verifica mediante il sequenziamento di alcuni campioni. I risultati del sequenziamento hanno evidenziato la correttezza del metodo impostato, essendo in perfetto accordo con quanto i genotipi assegnati.