17
2.
Alterazioni geniche della P-gP
La P-gP è una proteina di membrana codificata dal gene MDR (ABCB); sono stati identificati 2 tipi di geni nel genoma umano, MDR1 e MDR2, e 3 tipi di geni nei roditori mdr1a, mdr1b e mdr2. Il gene MDR1 sembra essere implicato nel meccanismo della Multidrug Resistance [4]. Entrambi i geni sono localizzati sul cromosoma 7 umano; MDR1 è costituito da 28 esoni [5] ed è altamente polimorfico.
2.1.Polimorfismo genetico
Il polimorfismo genetico può essere definito come una variazione genetica che si manifesta nella popolazione con una certa frequenza (in genere maggiore dell’1%). Questi polimorfismi possono trovarsi all’interno di regioni codificanti (esoni) e non codificanti (introni) e sono responsabili di alterazioni nella sequenza nucleotidica di un segmento di DNA. Il più comune è il cambiamento di un singolo nucleotide definito “Single Nucleotide Polimorfism” (SNP). Un SNP può provocare una variazione della sequenza degli amminoacidi nella proteina codificata. Questi cambiamenti costituiscono il 90% delle variazioni genetiche umane. Altri polimorfismi genetici comprendono inserzioni e delezioni di uno o più nucleotidi e duplicazioni [25].
Il polimorfismo genetico può portare ad un aumento o una riduzione dell’espressione genica, dell’attività della proteina codificata da quel gene e della risposta farmacologica delle cellule che esprimono queste proteine. Diversi polimorfismi genetici sono stati evidenziati nei geni che codificano per i trasportatori ABC.
Nel 1989, attraverso studi condotti su cellule tumorali, sono stati individuati da Kioka e collaboratori diversi tipi di SNPs nel gene MDR1 (ABCB1) [26]. Oggi sono conosciuti più di 50 tipi di SNPs: alcuni sono “silenti “ e non provocano cambiamenti nella sequenza amminoacidica della proteina (i.e. C1236T, C3396T e C3435T); altri invece generano cambiamenti degli amminoacidi, in particolare, alcuni SNPs codificano amminoacidi localizzati nel dominio transmembranale della proteina o nel sito di legame per l’ATP, alterando così l’attività della proteina stessa.
Tra gli SNPs, il più studiato è il C3435T localizzato sull’esone 26. Questo è un cambiamento “silente” di una C con una T ed responsabile di alterazioni dell’attività e dell’espressione della P-gP [26].
18 In particolare questo SNP è stato associato ad una riduzione dell’ espressione del gene MDR1, che codifica per la P-gP, nelle cellule epiteliali dell’intestino e ad un aumento della concentrazione della digossina nel plasma a causa di una sua ridotta eliminazione [26]. La P-gP si trova localizzata a livello della membrana apicale delle cellule endoteliali intestinali dove trasferisce substrati direttamente nel lume intestinale, favorendo la loro eliminazione tramite le feci; inoltre, la digossina è un suo substrato. Una diminuzione dell’espressione della P-gP comporta una ridotta eliminazione della digossina e di conseguenza un aumento della sua concentrazione plasmatica.
Diversi studi inoltre hanno dimostrato che il polimorfismo C3435T è implicato anche nella resistenza ai farmaci antiepilettici. Questi esperimenti sono stati condotti su 315 pazienti affetti da epilessia: 200 sono risultati resistenti alla terapia farmacologica, 115 hanno risposto alla terapia [27]. Il ruolo di questo SNP nella comparsa della resistenza ai farmaci antiepilettici è però ancora da chiarire in quanto, un recente studio condotto in Australia su 401 pazienti che non rispondono ai farmaci antiepilettici e su 208 che rispondono positivamente, nega una diretta associazione tra il polimorfismo C3435T e la resistenza a questo tipo di farmaci [28].
Polimorfismi del gene MDR1 sembrano essere implicati nello sviluppo di leucemie e nella patogenesi di disturbi neurodegenerativi come il morbo di Parkinson. Il genotipo (TT), associato ad una riduzione dell’attività e dell’espressione della P-gP, appare con maggiore frequenza nei soggetti che si trovano in uno stadio precoce della patologia, con una frequenza intermedia nei soggetti che si trovano in una fase avanzata della patologia e con frequenza minore nei soggetti sani [26].
Il morbo di Parkinson e il coinvolgimento della P-gP nella patogenesi di questa malattia verranno trattati in modo dettagliato nei capitoli seguenti.
Dai casi discussi sopra risulta che il polimorfismo del gene MDR1 contribuisce in maniera significativa ad una ipoespressione o iperespressione della P-gP con conseguente riduzione o aumento della sua attività e alterazioni della risposta farmacologica delle cellule che esprimono questa proteina.
19 2.2. Regolazione dell’espressione genica
Negli ultimi anni sono stati fatti importanti progressi per comprendere i meccanismi che regolano l’espressione dei geni che codificano per i trasportatori ABC, in particolare del gene per la P-gP. L’espressione di questi geni è controllata da fattori di trascrizione appartenenti al gruppo dei recettori nucleari orfani (orphan nuclear receptors)[29]. Tra questi troviamo il PXR nei roditori (pregnane X receptor), l’SXR nell’uomo (steroid and xenobiotic receptor), il CAR (constitutive androstane receptor) e l’FXR (farnesoid X-activated receptor). Questi recettori regolano l’espressione di diverse proteine implicate nell’eliminazione degli xenobiotici, come ad esempio gli enzimi metabolizzanti di fase I e II ed i trasportatori ABC come la P-gP e le MRPs.
L’espressione genica è influenzata anche da stimoli che evocano uno stress per l’organismo come ad esempio agenti citotossici, shock termico, infiammazione, citochine, ecc. Sono stati fatti esperimenti su cellule endoteliali cerebrali di topo per esaminare l’effetto dello stress ossidativo sulla espressione della P-gP [30]. Da questi studi è emerso un aumento dei livelli di espressione della P-gP. Il fattore responsabile di questa iperespressione non è stato ancora ben identificato; sembra che l’attivazione del fattore di trascrizione NF-kB sia implicata nell’induzione del gene mdr1b che codifica per la P-gP nei roditori [31].
È molto interessante notare che il glutammato, un neurotrasmettitore del SNC di tipo eccitatorio trasportato dalla P-gP a livello della BEE, è in grado di aumentare l’espressione dei geni mdr1a e mdr1b nelle cellule endoteliali cerebrali di topo [30].
2.3.Multidrug Resistance (MDR)
Il fenomeno della Multidrug Resistance (MDR) è un tipo di resistenza tipica di microorganismi e di cellule tumorali verso determinati farmaci chemioterapici strutturalmente non correlati tra loro e con diverso meccanismo d’azione. La resistenza delle cellule tumorali è il principale ostacolo al successo della terapia antineoplastica [32].
La MDR può essere intrinseca, quando le cellule hanno una resistenza innata agli agenti chemioterapici, o acquisita, se compare dopo l’inizio della terapia.
20 La MDR può essere dovuta a diversi meccanismi biochimici [32]:
• alterato trasporto di membrana dovuto ad un ridotto ingresso di farmaco nella cellula o aumentata eliminazione;
• attivazione di particolari enzimi come il glutatione e il citocromo P450 responsabili del metabolismo dei farmaci;
• alterazioni nell’attivazione e degradazione del farmaco;
• aumento della riparazione del DNA danneggiato;
• fallimento dell’apoptosi.
Questi meccanismi possono coesistere oppure manifestarsi singolarmente. Quello più studiato è l’alterazione del trasporto attraverso la membrana plasmatica, dovuto ad una iperespressione di proteine di membrana che funzionano da pompe di efflusso, come la P-gP [9]. L’elevata espressione di queste proteine comporta un aumento della loro attività che consiste nel trasportare farmaci e xenobiotici fuori dalla cellula riducendo così la loro concentrazione intracellulare. Proprio questo ridotto accumulo di farmaco nella cellula è responsabile del fenomeno della resistenza e del fallimento della terapia. La P-gP, essendo localizzata anche a livello delle cellule epiteliali intestinali, trasferisce direttamente farmaci nel lume, favorendone l’eliminazione. Questa attività riduce notevolmente l’assorbimento di farmaci somministrati per via orale.
Goldstein e collaboratori [34] nel 1989 analizzarono più di 400 tipi di tumori e li classificarono in base ai livelli di espressione del gene MDR1 che codifica per la P-gP. I tumori furono divisi in 3 classi: positivi per il gene MDR1 (con resistenza intrinseca, ad esempio tumori del colon, fegato, reni e pancreas), a volte positivi (con resistenza acquisita come neuroblastomi, leucemia linfocitica acuta, feocromocitoma), negativi (melanomi, tumori alla prostata, ecc). L’associazione tra l’elevato livello di espressione del gene MDR1 e la resistenza intrinseca o estrinseca di alcuni tumori, conferma l’importanza clinica del gene MDR1. Gli autori arrivarono alla conclusione che le cellule tumorali che iperesprimono la P-gP rispondono negativamente alla terapia antineoplastica [34].
21 Poiché la P-gP si trova espressa anche a livello della BEE, questa è implicata anche nella resistenza dei tumori cerebrali [35]. La chemioterapia non è molto efficace nel trattamento di questi tumori; questo perché i trasportatori ABC, tra cui anche la P-gP, a livello della BEE, legano diversi farmaci antitumorali e li trasportano direttamente nel sangue. Il ridotto accumulo di questi farmaci nel cervello spiega l’inefficacia della terapia. I principali farmaci antitumorali substrati della P-gP sono: gli alcaloidi della vinca (vincristina e vinblastina), le antracicline (doxorubicina, daunorubicina, epirubicina), i taxani (paclitaxel, taxolo, docetaxel), il metotrexato e il mitoxantrone. I trasportatori ABC, in particolare la P-gP, sono implicati anche nella resistenza del virus HIV. Il cervello è un ottimo sito di replicazione per questo tipo di virus e nello stesso tempo , è anche un importante tessuto bersaglio per i farmaci antiretrovirali. I farmaci inibitori delle proteasi dell’HIV hanno un’azione limitata a livello del SNC perché vengono eliminati dalla P-gP, espressa sulla membrana luminale della BEE [11]. Diversi studi hanno dimostrato un aumento della concentrazione cerebrale di farmaci antiretrovirali tra cui il saquinavir, il nelfinavir e l’indinavir in topi mancanti del gene mdr1a [11]. Questi dati dimostrano che la P-gP limita l’ingresso di questi farmaci nel cervello e di conseguenza anche la loro azione terapeutica è ridotta.
La P-gP è espressa anche nei linfociti, in particolare nelle cellule CD4+, il principale target del virus HIV. Negli individui infetti, una iperespressione della P-gP è associata con un ridotto accumulo intracellulare di farmaci inibitori delle proteasi [11].
2.3.1. Approcci per superare il fenomeno della MDR
La principale strategia farmacologica per cercare di rendere reversibile il fenomeno della MDR è quella di somministrare sostanze capaci di inibire la P-gP e di consentire così l’accumulo di farmaci antitumorali nelle cellule. Studi preclinici hanno dimostrato che la penetrazione cerebrale di farmaci antitumorali come ad esempio il paclitaxel, il docetaxel e l’imatinib, è aumentata utilizzando composti inibitori della P-gP (ciclosporina A, valspodar, elacridar) [32]. Sono stati sperimentati 3 classi di farmaci inibitori della P-gP. Nessuno di questi è stato ancora introdotto in terapia a causa degli effetti collaterali e della mancanza di selettività nei confronti delle cellule tumorali. Associazioni tra farmaci antitumorali e inibitori della P-gP sono tuttora in fase di studio.
22 Gli inibitori della P-gP, la loro classificazione e meccanismo d’azione verranno trattati in modo dettagliato nel capitolo seguente.
Altri approcci sperimentali per superare il fenomeno della MDR sono: l’incapsulazione di farmaci antitumorali in liposomi e nanosfere; l’uso di anticorpi contro la P-gP (UIC2, MRK16); oligonucleotidi antisenso e ribozimi che hanno come bersaglio l’mRNA del gene MDR1; proteggere le cellule ematopoietiche dalla chemioterapia mediante l’utilizzo di antitumorali substrati della P-gP; uso di farmaci antitumorali che non siano substrati per la P-gP (es. seconda generazione di taxani, efotiloni).
• INCAPSULAZIONE DI FARMACI ANTITUMORALI IN LIPOSOMI E NANOSFERE
L’utilizzo di liposomi o altri trasportatori può aumentare l’ingresso di farmaci nelle cellule tumorali o in compartimenti intracellulari. I liposomi vengono captati nelle cellule di tumori solidi e vi rimangono per diversi giorni dopo somministrazione per via endovenosa. I livelli di farmaco intracellulare veicolato dai liposomi sono circa 10 volte maggiori rispetto a quelli di farmaco libero [36]. I primi studi sono stati condotti con liposomi composti da fosfolipidi anionici tra cui la fosfatidilserina e la cardiolipina. Questi non mostrarono in vivo una stabilità e un tempo di circolazione da permettere il loro utilizzo nella pratica clinica [36].
Oggi sono usati fosfolipidi neutri come la fosfatidilcolina e la fosfatidiletanolammina.
Questa tecnica è ancora oggi in fase di studio e richiede ulteriori sperimentazioni. Ad esempio, non ci sono ancora dati certi che riguardano l’accumulo intracellulare e gli effetti collaterali della antraciclina veicolata dai liposomi nelle cellule tumorali resistenti [36].
23
• ANTICORPI CONTRO LA P-gP
L’utilizzo di diversi anticorpi monoclonali (MoAbs) potrebbe essere una tecnica per rendere reversibile in fenomeno della MDR. Il trattamento con questi anticorpi come MRK16 e MRK17 modula il trasporto della vincristina e della actinomicina D e inibisce la crescita delle cellule resistenti [37]. Inoltre, è stato dimostrato che MRK16 aumenta l’attività della ciclosporina A e del suo analogo strutturale valspodar, entrambi inibitori della P-gP [37].
Un aumento dell’accumulo della vincristina e della actinomicina D è stato riscontrato anche utilizzando altri 2 MoAbs: HYB-241 e UIC2. Quest’ultimo è in grado di impedire l’eliminazione di substrati della P-gP, aumentando però la loro citotossicità. Con la tecnologia del DNA ricombinante e tecniche di ingegneria genetica è possibile produrre in laboratorio piccoli e funzionali frammenti di anticorpi, come gli scFvs. Un piccolo anticorpo ricombinante scFv è stato studiato e ha prodotto i primi risultati sulla reversibilità della MDR; questo è in grado di inibire il trasporto mediato dalla P-gP [37].
• OLIGONUCLEOTIDI ANTISENSO E RIBOZIMI
Piccoli oligonucleotidi antisenso sono stati utilizzati per ridurre l’espressione del gene MDR1 e quindi la sintesi della P-gP. Questa tecnica non ha dato però i risultati sperati forse a causa degli elevati livelli di espressione dell’mRNA nelle cellule resistenti [36]. Un altro metodo per ridurre l’espressione dell’mRNA del gene MDR1 è l’utilizzo di ribozimi, piccole molecole di RNA che si ibridizzano con sequenze di RNA complementare. Sono stati sperimentati una grande varietà di ribozimi; i più piccoli sono costituiti da 30-40 nucleotidi. Al momento però non ci sono applicazioni cliniche di queste molecole [36].
24
• FARMACI ANTITUMORALI CHE NON SONO SUBSTRATI PER LA P-gP I farmaci antitumorali che non sono substrati per la P-gP e che non vengono eliminati dalle cellule resistenti vengono definiti “inclusive”. Un esempio di questi composti sono gli efotiloni, una nuova classe di farmaci che agisce sul sistema microtubulare. All’inizio sono stati individuati come metaboliti citotossici del batterio Sorangium cellulosum, ma la loro capacità di indurre la polimerizzazione dei microtubuli cellulari è stata scoperta cercando di ridurre resistenza ai taxani. Queste 2 classi di farmaci hanno lo stesso meccanismo d’azione; sembra che gli efotiloni competano con il paclitaxel per il legame con i microtubuli. Gli studi preclinici effettuati dimostrano che questi farmaci hanno un largo spettro d’azione antitumorale sia in vitro che in vivo [38]. Altri farmaci che non legano la P-gP sono quelli appartenenti alla seconda generazione di taxani, ottenuti modificando chimicamente la struttura del docetaxel. Queste sostanze sono ancora in fase di sperimentazione clinica.
• PROTEZIONE DELLE CELLULE EMATOPOIETICHE DALLA CHEMIOTERAPIA CON ANTITUMORALI SUBSTRATI PER LA P-gP
Il principale effetto collaterale della chemioterapia è la mielosoppressione, un’alterazione del midollo osseo con conseguente riduzione della sintesi delle cellule ematopoietiche. Tecniche di trasferimento genico possono essere utilizzate per trasformare alcune cellule progenitrici del midollo spinale in cellule resistenti, senza il rischio che si instauri, dopo la somministrazione di antitumorali, questa mielosoppressione [38]. Le cellule target ideali sono le PBSCs (CD34+ Peripheralblood Stem Cells).
Queste cellule vengono infettate con retrovirus contenenti il gene per la resistenza farmacologica MDR1, si ha il trasferimento del gene e l’applicazione di una dose elevata di farmaco chemioterapico.
25 Studi preclinici effettuati su topi hanno evidenziato che i topi transgenici che esprimono il gene MDR1 nelle cellule progenitrici del midollo spinale sono protetti dall’azione mielosoppressiva dei farmaci antitumorali [38].
Oltre che nelle infezioni e nei tumori, il fenomeno della MDR si verifica anche in alcune patologie come l’artrite reumatoide, l’epilessia e diversi disturbi psichiatrici. Il 40-50% dei pazienti affetti da depressione, schizofrenia e epilessia, risulta resistente a diversi farmaci utilizzati alle massime dosi tollerate dall’organismo [39].
2.4. Epilessia
L’epilessia è una patologia neurologica di cui è affetto circa l’1-2% della popolazione mondiale, caratterizzata da crisi convulsive accompagnate o meno da perdita di coscienza. Sono stati fatti molti progressi al fine di ricercare nuovi farmaci per il trattamento di questa patologia a causa della ridotta efficacia dei farmaci antiepilettici attualmente in uso. Circa il 40% dei pazienti affetti da epilessia cronica è resistente al trattamento [39].
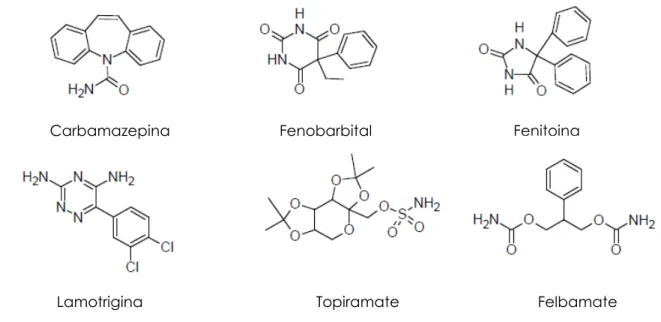
Anche se i farmaci antiepilettici tuttora utilizzati hanno diversi meccanismi d’azione, il fenomeno della resistenza è comune a tutti. La principale causa della resistenza sembra essere il ridotto accesso di questi farmaci al SNC, risultato di un aumento del trasporto attraverso la BEE ad opera di proteine trasportatrici come la P-gP. Tra i farmaci antiepilettici più utilizzati troviamo il fenobarbital, la fenitoina, la carbamazepina, la lamotrigina, il topiramate e il felbamate. Tutti questi farmaci sono substrati della P-gP.(Fig.2.1)
26
Figura 2.1.Principali farmaci antiepilettici
La P-gP si trova iperespressa nel tessuto cerebrale dei pazienti affetti da epilessia cronica che non rispondono alla terapia, in particolare nelle cellule endoteliali dei capillari e negli astrociti che costituiscono la BEE. I livelli di mRNA del gene MDR1 nel cervello di pazienti con epilessia intrattabile sono stati determinati con la tecnica della RT-PCR e paragonati con quelli misurati nel cervello del soggetto che risponde alla terapia [40]. L’iperespressione della P-gP a livello della BEE gioca un ruolo fondamentale nella resistenza ai farmaci antiepilettici , in quanto, limita notevolmente l’accesso di questi farmaci nel tessuto cerebrale e quindi anche la loro efficacia terapeutica. Anche le proteine MRPs si trovano iperespresse nella BEE dei pazienti resistenti alla terapia, mentre i livelli della BCRP sono normali [40]. Non è ancora chiaro se questa iperespressione sia intrinseca o acquisita, e quindi se questa è conseguenza di trattamento cronico con farmaci antiepilettici oppure se è una conseguenza della malattia vera e propria. È stato dimostrato che un lungo trattamento con farmaci antiepilettici, nell’uomo induce l’espressione della P-gP, mentre negli animali da esperimento questo non si verifica [40].
I pazienti resistenti alla terapia mostrano gli stessi effetti collaterali neurotossici dei pazienti che invece rispondono alla terapia. Questo può essere spiegato con il fatto che l’iperespressione dei trasportatori ABC è limitata al focus epilettico, mentre nei tessuti normali adiacenti l’espressione di queste proteine rimane costante.
Carbamazepina Fenobarbital Fenitoina
27 La resistenza ai farmaci antiepilettici non è dovuta solo ad un iperespressione della P-gP nel tessuto cerebrale, ma anche ad un aumento della sua attività nell’intestino, dove la proteina riduce l’assorbimento dei farmaci somministrati per via orale.
Oltre alla resistenza nell’epilessia cronica, i trasportatori ABC sembrano implicati anche nella farmacoresistenza dello stato epilettico. Questo è caratterizzato da continue crisi convulsive con perdita di coscienza dalla durata di circa 30 minuti e richiede quindi un trattamento tempestivo. Il 30% dei pazienti non risponde al primo e secondo trattamento con farmaci anticonvulsivanti. È stata dimostrata una iperespressione della P-gP nei pazienti affetti da stato epilettico e negli animali da esperimento in cui sono state indotte queste crisi [41].
Tutti i farmaci antiepilettici sono substrati per la P-gP; eccezione è il levetiracetam. Questo è il primo farmaco antiepilettico testato in laboratorio che non è un substrato per la P-gP [39]. La sua efficacia terapeutica è stata dimostrata in pazienti che non rispondono ad altri tipi di terapie.
Un approccio terapeutico per risolvere il problema della resistenza ai farmaci antiepilettici è quello di somministrare sostanze in grado di inibire la P-gP, aumentando così l’ingresso di questi farmaci nel cervello e il loro assorbimento dopo somministrazione per via orale.
Associazioni tra inibitori della P-gP e farmaci antiepilettici sono ancora in fase di studio; queste potrebbero aumentare notevolmente la qualità di vita dei pazienti e permettere un maggior controllo delle crisi convulsive [39].
2.5. Depressione
La depressione è una patologia caratterizzata da uno stato generale di ansia, il soggetto tende ad isolarsi, talvolta si possono avere momenti di forte esaltazione alternati a periodi di abbattimento (depressione bipolare). Il 30-50% dei pazienti non risponde ad agenti antidepressivi [39].
La P-gP è implicata nella resistenza ai farmaci antidepressivi: limita l’accumulo di farmaci quali l’amitriptilina, il citalopram e la paroxetina nel tessuto cerebrale, favorendo la loro eliminazione dal SNC. Oltre ad essere substrati per la P-gP, i farmaci antidepressivi sono anche inibitori dei trasportatori per gli ormoni steroidei localizzati nella BEE, tra cui anche la P-gP.
28 Questo fa si che il cortisolo abbia un maggior accesso al SNC e aumenti il meccanismo feed-back negativo sull’asse ipotalamo-ipofisi. La secrezione del cortisolo, infatti, è sotto il controllo dell’ormone ACTH ipofisiario e del fattore di rilascio CRH ipotalamico. Quando la concentrazione del cortisolo aumenta si ha un’inibizione della sintesi di questi 2 ormoni. Un’alterazione di questo meccanismo feed-back negativo sembra implicata nella patogenesi dello stato depressivo [42].
2.6.Patologie neurodegenerative
Disfunzioni della P-gP sembrano essere implicate nella patogenesi di malattie neurodegenerative come il morbo di Parkinson e di Alzheimer. Queste patologie sono dovute all’accumulo di sostanze citotossiche e di proteine nel cervello.
Sono disturbi che colpiscono principalmente gli anziani: esiste una correlazione tra età e ridotta funzionalità della P-gP nella BEE [43].
Una ipoespressione della P-gP è la principale causa della comparsa di patologie neurodegenerative: in particolare, questa sua ridotta attività, è responsabile dell’accumulo di tossine, causa principale del Parkinson, e di una proteina detta β amiloide nell’Alzheimer. In seguito al danno neuronale provocato dall’accumulo di queste sostanze, si ha un aumento dell’espressione della P-gP e della sua attività. Durante il processo infiammatorio neuronale, si verifica un’elevata attivazione delle microglia, cellule di supporto dei neuroni che esprimono la P-gP, e di altri fattori implicati nel processo infiammatorio, tra cui il fattore di necrosi tumorale α (TNF-α), l’interleuchina 6 (IL-6) e l’ossido nitrico, che aumentano l’attività della P-gP [44]. L’aumento del trasporto mediato dalla P-gP durante gli stadi iniziali di queste patologie potrebbe essere un nuovo approccio terapeutico per prevenire l’accumulo di tossine e proteine responsabili dello sviluppo di queste patologie.
L’iperespressione della P-gP è caratteristica quindi di stadi avanzati di disturbi neurodegenerativi ed è responsabile del fallimento della terapia. Questo è dovuto al fatto che l’aumento dell’attività della P-gP diminuisce l’ingresso di farmaci nel SNC, impedendo quindi il raggiungimento del sito bersaglio di queste sostanze. Farmaci utilizzati nel trattamento del Parkinson e dell’Alzheimer sono substrati per la P-gP e di conseguenza la loro permeabilità a livello della BEE è ridotta.
29 2.6.1. Espressione della P-gP nel morbo di Alzheimer
Il morbo di Alzheimer è un disturbo neurodegenerativo che colpisce principalmente gli anziani; in genere si manifesta in persone che hanno più di 60 anni. Negli stadi iniziali della malattia si hanno leggere perdite di memoria, poi compare una progressiva perdita della funzione cognitiva e demenza.
Le principali cause dell’Alzheimer sono: lo sviluppo di placche di β amiloide nel parenchima cerebrale e la formazione di ammassi di neurofilamenti, costituiti da un intreccio di proteine dette Tau che nel soggetto malato sono iperfosforilate. Le placche invece sono costituite da un insieme di peptidi β amiloidi, in particolare Aβ40 e Aβ42 che si accumulano a livello cerebrale.
Questi peptidi vengono prodotti normalmente nelle cellule del sistema nervoso e nei tessuti periferici a partire da un precursore detto APP (Amyloid Precursor Protein). Questo viene scisso da un enzima detto β secretasi, conosciuto anche come BACE, che da origine a peptidi di 40 o 42 amminoacidi (Aβ40 e Aβ42) che poi si accumulano a formare placche insolubili. Sembra che il deposito di queste placche sia la causa principale dell’Alzheimer, mentre la formazione degli ammassi di neurofilamenti è un evento secondario [43]. Le placche si accumulano principalmente nei neuroni colinergici della corteccia e dell’ippocampo, centri deputati all’apprendimento e alla memoria.
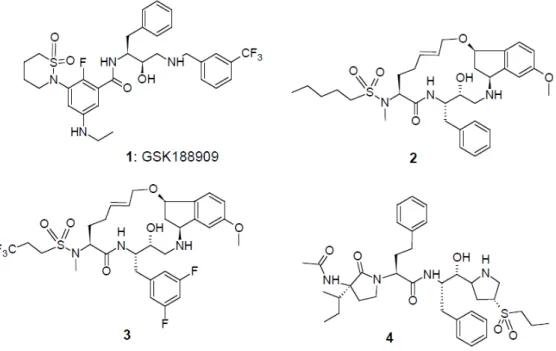
L’enzima BACE è un target terapeutico per il trattamento e la prevenzione del morbo di Alzheimer; la sua inibizione però non ha dato i risultati sperati in quanto, gli inibitori di questo enzima, sono substrati per la P-gP e non raggiungono il SNC.(Fig.2.2)
30
Figura 2.2. Inibitori dell'enzima BACE
Recenti studi hanno dimostrato che il deposito della placche di β amiloide è inversamente proporzionale all’espressione della P-gP a livello della BEE [42]. Gli Aβ peptidi sono substrati per la P-gP; di conseguenza, la diminuzione dell’espressione di questa proteina caratteristica dei primi stadi della patologia , comporta una ridotta eliminazione dei peptidi dal cervello favorendo il loro accumulo sottoforma di placche.
Le placche che si formano a livello cerebrale innescano un processo infiammatorio neuronale: si ha la produzione di citochine, interleuchina 6 e TNF-α che aumentano i livelli di espressione e l’attività della P-gP. Infatti negli stadi avanzati del morbo di Alzheimer la proteina risulta iperespressa. Farmaci in grado di aumentare l’attività della P-gP possono favorire l’eliminazione delle placche di β amiloide dal cervello negli stadi iniziali della malattia. Questo è stato dimostrato mediante sperimentazioni cliniche su pazienti affetti da Alzheimer trattati con rifampicina, un potente induttore della P-gP [45]. Questo antibiotico riduce notevolmente il declino cognitivo in quanto aumenta il trasporto della β amiloide mediato dalla proteina.
31 L’iperespressione della P-gP negli stadi avanzati della patologie è invece un limite per l’efficacia della terapia in quanto farmaci utilizzati nel trattamento dell’Alzheimer, sono substrati per la P-gP. Tra questi troviamo i già citati inibitori dell’enzima BACE. Per risolvere questo problema è stato proposto di utilizzare inibitori della P-gP in associazione con inibitori BACE; in questo modo si otterrebbe una ridotta eliminazione dei farmaci dal SNC e un aumento della loro efficacia terapeutica. Questo però non ha ancora trovato impieghi nella pratica clinica.
Tra gli inibitori BACE troviamo il GSK188909; in vitro riduce la scissione del precursore APP da parte della β secretasi e impedisce il deposito degli Aβ peptidi sottoforma di placche insolubili. Questo composto riduce i livelli di β amiloide anche in vivo e l’inibizione dell’enzima è più efficace se si somministrano contemporaneamente inibitori della P-gP. Ciò dimostra che gli inibitori dell’enzima BACE sono anche substrati per la P-gP e che inibitori di questa proteina influenzano la loro attività cerebrale [46].
Attualmente i farmaci utilizzati per il trattamento dell’Alzheimer non sono farmaci causali, cioè in grado di rimuovere la causa della malattia, ma soltanto “sintomatici”, cioè finalizzati alla riduzione delle manifestazioni cliniche.
Farmaci utilizzati nella fase lieve e moderata della malattia sono gli inibitori dell’acetilcolinesterasi, enzima deputato alla degradazione dell’acetilcolina in acetato e colina. Tra questi il donepezil, lagalantamina e la rivastigmina sono in grado di aumentare i livelli di aceticolina e possono compensare la distruzione dei neuroni colinergici provocata dalla malattia. Possono inoltre migliorare alcuni sintomi cognitivi (memoria e attenzione) e comportamentali (apatia, agitazione); questa proprietà diminuisce però con la progressione della patologia. Il loro impiego clinico è piuttosto ridotto per i numerosi effetti collaterali e per il fatto che molti pazienti non rispondono alla terapia.
Un farmaco utilizzato in fase severa dell’Alzheimer è la memantina, che ha un effetto sull’apprendimento e sulla memoria.
32 Sostanze che intervengono invece sui processi ossidativi che caratterizzano l’invecchiamento sono gli antiossidanti tra cui la selegilina, la vitamina E e il gingko biloba. L’efficacia di questi farmaci nel trattamento dell’Alzheimer richiede ulteriori studi.
2.6.2. Espressione della P-gP nel morbo di Parkinson
Il morbo di Parkinson è un disturbo neurodegenerativo, caratterizzato da lentezza nei movimenti volontari, tremore e rigidità muscolare. È una patologia tipica dell’età senile; la causa principale è una diminuzione della produzione della dopamina a livello di alcune strutture del sistema extrapiramidale tra cui la sostanza nigra e il nucleo striato. Questi centri sono deputati al controllo e all’avvio dei movimenti volontari. I livelli di dopamina risultano diminuiti del 60-70% rispetto ai valori normali.
La diminuzione della dopamina provoca un’alterazione dell’equilibrio con altri neurotrasmettitori extrapiramidali tra cui l’acetilcolina, che diventa prevalente.
L’eziologia del Parkinson non è ancora ben conosciuta: è stato osservato nei soggetti malati la presenza di aggregati proteici detti Lewy Bodies (LBs) [47]. Questi sono costituiti da α-sinucleina, una proteina neuronale che in condizioni patologiche forma aggregati insolubili, che vanno a danneggiare i neuroni dopaminergici.
Non è ancora chiaro se la P-gP è in grado di trasportare questa proteina, e se una sua ipoespressione nelle fasi iniziali della patologia sia responsabile della formazione degli aggregati insolubili. La P-gP partecipa però al trasporto di pesticidi e di neurotossine, implicati nella patogenesi del Parkinson. La scoperta di una particolare tossina, l’MPTP (1-metil-4-fenil-1,2,3,6-tetraidropiridina), composto secondario che si forma durante la sintesi dell’ “eroina sintetica”, ha dato il via alla ricerca di cause tossiche per spiegare la genesi della patologia. Questo composto, infatti, è responsabile di un disturbo motorio reversibile simile al morbo di Parkinson. E’ stato riscontrato inoltre che numerosi pazienti affetti da Parkinson in gioventù avevano fatto uso di sostanze stupefacenti contenenti MPTP [47].
33 Sembra che i soggetti più a rischio di contrarre questa patologia siano persone che si trovano a contatto con idrocarburi, pesticidi, resine, ecc. Una disfunzione della P-gP nelle fasi iniziali della patologia comporta una ridotta eliminazione di composti tossici, che si accumulano a livello cerebrale, provocando la degenerazione dei neuroni che producono dopamina.
Diversi studi hanno dimostrato un’associazione tra esposizione a pesticidi e polimorfismo del gene MDR1, che codifica per la P-gP. In particolare si ha una frequenza maggiore del polimorfismo C3435T, associato ad una ridotta espressione e funzionalità della P-gP, nei pazienti affetti da Parkinson che sono stati precedentemente esposti a pesticidi [26].
Anche in questo caso, come nell’Alzheimer, un’aumento dell’attività della P-gP nella fase iniziale della patologia potrebbe essere un nuovo approccio terapeutico per ridurre l’ accumulo di questi composti tossici e quindi la progressione della malattia.
L’età sicuramente è un importante fattore di rischio; intorno ai 60 anni viene meno la protezione delle cellule contenenti dopamina, quindi le persone anziane sono più predisposte al Parkinson.
Nelle fasi tardive della patologia si ha una iperespressione della P-gP che è responsabile del fallimento della terapia e della resistenza ai farmaci antiparkinson. In seguito al danno neuronale, si ha la produzione di citochine, radicali liberi, attivazione delle microglia, che inducono l’espressione della P-gP [48]. La L-DOPA, farmaco d’eccellenza utilizzato nel trattamento di questa patologia, è un substrato per la P-gP.
Un aumento dell’attività della proteina comporta una ridotta permeabilità di questo farmaco a livello cerebrale, che quindi non riesce a raggiungere il suo sito d’azione. La somministrazione di L-DOPA dopo un periodo prolungato non risulta più efficace perché si instaura il meccanismo della resistenza, e richiede l’utilizzo di dosi sempre crescenti. Questo non è legato solamente all’ iperespressione della P-gP a livello cerebrale ma anche ad un aumento dell’attività della proteina nell’intestino, dove favorisce l’eliminazione dei suoi substrati.
34 La L-DOPA è somministrata principalmente per via orale e la sua biodisponibilità è ridotta dall’azione della P-gP a livello delle cellule epiteliali intestinali, dove trasferisce le sostanze direttamente nel lume favorendo la loro eliminazione tramite le feci.
Per aumentare l’attività di questi composti, sono in fase di studio associazioni tra farmaci antiparkinson e inibitori della P-gP. Queste associazioni, fulcro centrale di questa tesi, verranno trattate nei capitoli successivi.