I
Indice Appendici
Appendice I: Profili geomorfologici sponda Nord e Sud
III.1 Classe A: degrada dolcemente con rottura di pendio
in prossimità dell’acqua. II
I.2 Classe B: gradino in prossimità dell’acqua con bassa battuta di sponda III I.3 Classe C: doppio gradino con bassa battuta di sponda IV
I.4 Classe D: degrada dolcemente con rottura di pendio a circa 3 m
dall’acqua V
I.5 Classe E: gradino in prossimità dell’acqua con alta battuta di sponda VI
I.6 Classe F: doppio gradino con alta battuta di sponda VII
Appendice II: Principali composti disciolti nelle acque e loro
determinazione
VIII
II.1 Anioni e cationi maggiori
VIII
II.2 Cromatografia a scambio ionico X II.2.1 Determinazione degli anioni: cloruri. nitrati, solfati XII
II.3 Spettrofotometria di Assorbimento Atomico XIII
II.3.1 Determinazione dei cationi: sodio, potassio, calcio, magnesio XV
II.4 Spettrofotometria UV-VIS XVI
II.4.1 Determinazione di ammoniaca e COD XVI
Appendice III: Natanti da diporto
XVIII
III.1 Generalità natanti da diporto
XVIII
III.2 La carena planante e dislocante XXI
II
Appendice I: Profili geomorfologici sponda Nord e Sud
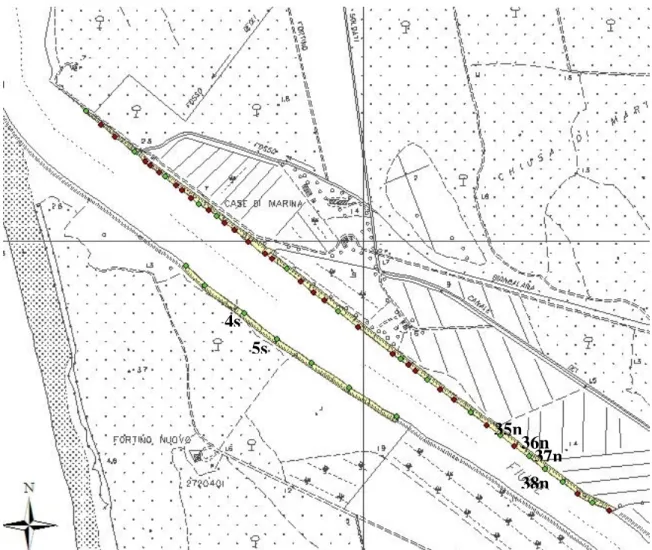
Di seguito sono riportati tutti i profili geomorfologici effettuati nella prima e nella seconda campagna di campionamento come illustrato nel capitolo 5§5.2, visualizzati con Autocad. I profili delle due sponde sono divisi nelle classi che sono state individuate a seguito della prima campagna di campionamento.
I.1 Classe A
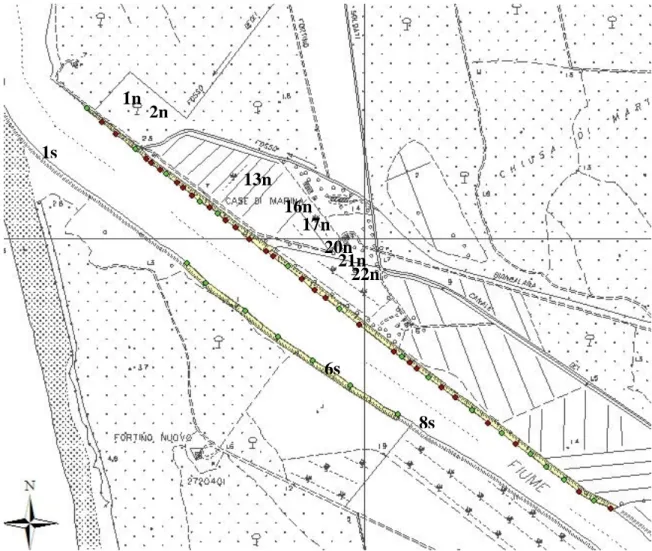
: degrada dolcemente con rottura di pendio in prossimità dell’acqua.Figura I.1: Profili n°: 1, 2, 13, 16, 17, 20, 21, 22 sponda Nord; 1, 6, 8 sponda Sud. 1n 2n 13n 16n 20n 21n 22n 1s 6s 8s 17n
III
I.2
Classe B
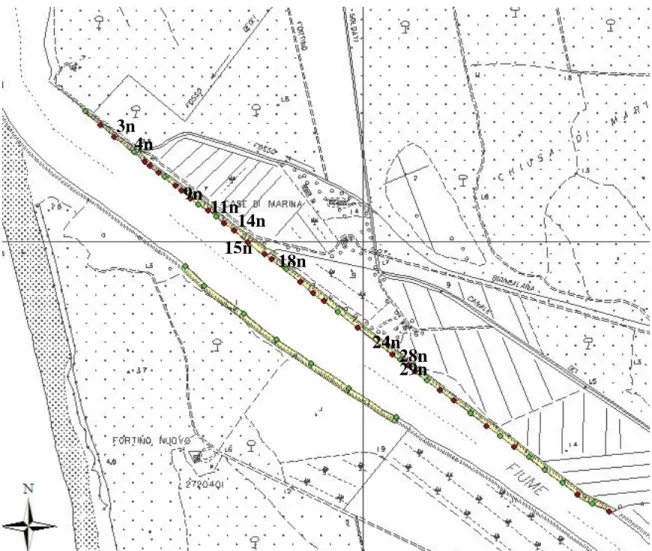
: gradino in prossimità dell’acqua con bassa battuta di spondaFigura I.2: Profili n°: 3, 4, 9, 11, 14, 15, 18, 24, 26, 28, 29 sponda Nord. 3n 4n 9n 11n 14n 15n 18n 24n 28n 29n
IV
I.3
Classe C:
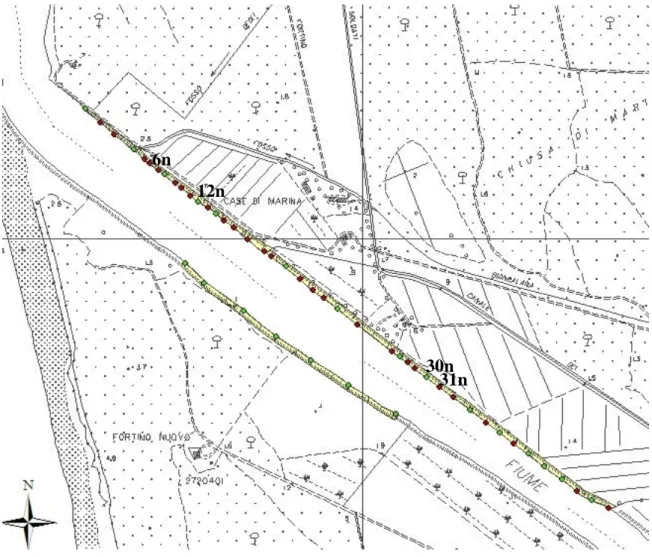
doppio gradino con bassa battuta di spondaFigura I.3:Profili n°: 6, 12, 30, 31 sponda Nord.
6n
12n
30n 31n
V
I.4 Classe D:
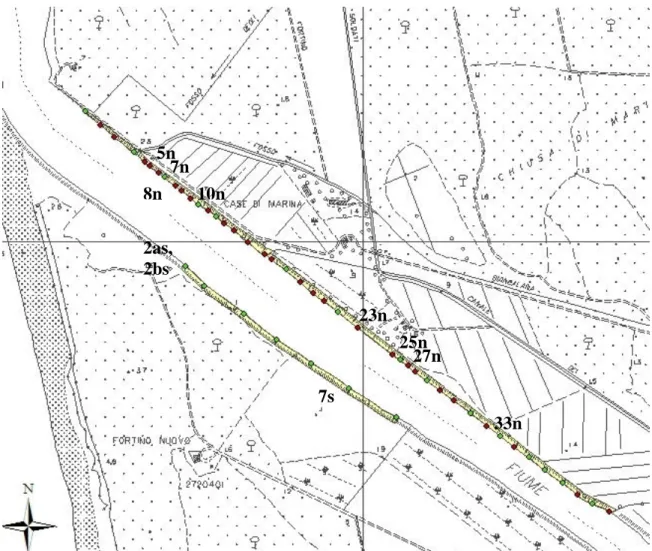
degrada dolcemente con rottura di pendio a circa 3 m dall’acquaFigura I.3: Profili n°: 5, 7, 8, 10, 23, 25, 27, 33 sponda Nord; 2a, 2b, 7 sponda Sud.
5n 7n 8n 10n 23n 25n 27n 33n 2as, 2bs 7s
VI
I.5
Classe E:
gradino in prossimità dell’acqua con alta battuta di spondaFigura I.4: Profili n°: 35, 36, 37, 38 sponda Nord; 4, 5 sponda Sud. 35n 36n 37n 38n 4s 5s
VII
I.6 Classe F:
doppio gradino con alta battuta di spondaFigura I.5:Profili n°: 32, 39, 41 sponda Nord; 3, sponda Sud.
41n 39n 32n
VIII
Appendice II: Principali composti disciolti nelle acque e
loro determinazione
II.1 Anioni e cationi maggiori
La composizione chimica delle acque superficiali deriva dall’interazione di molte variabili, tra cui:
? le caratteristiche dell’acqua piovana che ricara le falde
? la composizione petrologica e mineralogica delle rocce superficiali
? le proprietà idrogeologiche delle rocce, che condizionano l’entità delle interazioni acqua-roccia: velocità elevate del flusso sotterraneo determinano acque relativamente povere in solidi disciolti per i tempi brevi di contatto.
Conoscere il contenuto delle varie specie chimiche è fondamentale per valutare se vi sono incrementi derivanti da fonti antropiche, inclusi gli effetti derivanti dalle diverse strategie di sfruttamento e gestione del suolo. In media le acque superficiali contengono da 30 a 50 cc di gas per litro, di cui i principali sono: anidride carbonica, azoto e ossigeno, mentre tra i sali prevalgono, normalmente, quelli di calcio e di magnesio.
Gli ioni maggiori presenti nelle acque superficiali sono:
? Ione cloruro (Cl-): è uno ione molto mobile la cui concentrazione aumenta con il decorrere dell’interazione con la roccia; forma sali molto solubili ed è lo ione predominante nell’acqua di mare con 19000 mg/l, ma può raggiungere i 22000 mg/l (Custodio e Llamas, 1976). Nelle acque superficiali i contenuti variano da 10 mg/l a 250 mg/l e le fonti principali sono rappresentate da rocce sedimentarie (evaporiti), miscelazione con acque marine nelle zone costiere, precipitazioni influenzate da aerosol marino, scarichi industriali od urbani dove è presente anche in concentrazione di 100 mg/l. Se la concentrazione supera i 250 mg/l le acque non vengono più usate a scopo potabile, in quanto contenuti >300mg/l conferiscono sapore salato alle acque e possono indurre danni fisiologici; con valori superiori a 350 mg/l non ne è consigliabile l’ uso irriguo ed industriale. E’ uno tra i maggiori elementi la cui massa si conserva durante le fasi del ciclo idrologico ed una sua diminuzione è attribuibile essenzialmente a diluizione, per cui può essere usato per seguire fenomeni di miscelazione con acque marine.
IX ? Ione solfato (SO4-): forma sali generalmente molto solubili, eccetto i sali di stronzio
e di bario; rappresenta il secondo ione più abbondante nelle acque superficiali (dopo il bicarbonato) e in quelle marine (dopo i cloruri): nelle acque dolci è presente in quantità limitate, da 2 a 250 mg/l, ma può raggiungere il limite di saturazione di 1500 ppm se sono presenti rocce evaporitiche contenenti gesso ed anidride. Tale limite nelle acque marine è di 7200 mg/l (Custodio & Lamas, 1976). Può derivare oltre che da depositi evaporatici, dall’ossidazione dei solfuri o da scarichi industriali.
? Ione bicarbonato (HCO3-): è il principale responsabile dell’alcalinità dell’acqua. Le
acque del sottosuolo hanno un contenuto in bicarbonato variabile tra 100 e 800 mg/l, mentre l’acqua di mare ha un contenuto di 100 mg/l (Custodio & Lamas, 1976). Le principali fonti degli ioni bicarbonato e carbonato sono rappresentate principalmente dalla dissoluzione delle rocce carbonatiche in presenza di CO2 e di
acidi, secondo la relazione caratteristica degli ambienti carsici: CaCO3 + H2O+ CO2
= Ca++ + 2HCO3-
? Ione Calcio (Ca++): rappresenta lo ione più abbondante nelle acque sotterranee, specialmente in quelle circolanti in litologie sedimentarie. Le concentrazioni variano da 10 a 250 mg/l nelle acque dolci, 600 mg/l nelle acque selenitose e 400 mg/l nelle acque di mare (Custodio & Lamas, 1976). La solubilità di tale ione è condizionata dal pH e dalla presenza di CO2, secondo un complesso equilibrio
descrivibile dalle seguenti equazioni (Stumm & Morgan, 1996): CaCO3(s) ? Ca++ + CO3—
CO3— + H+ ? HCO3- + H+ ? H2CO3
CO2 + H2O ? H2CO3
? Ione magnesio (Mg++): forma composti ad alta solubilità, maggiore di quelli formati dal calcio ma comunque meno abbondanti. Le concentrazioni nelle acque dolci variano da 1 a 100 mg/l, mentre nelle acque di mare si arriva a 1200 mg/l (Custodio & Lamas, 1976). Deriva principalmente da rocce contenenti dolomite, ma anche da rocce evaporitiche, acqua di mare ed inquinamento da miniere.
? Ione sodio(Na+): appartiene al gruppo dei metalli alcalini; normalmente è associato con il cloruro e presenta una solubilità molto elevata. Nelle acque dolci il suo contenuto varia tra 1 e 150 mg/l, ma può arrivare a qualche migliaio (Custodio & Lamas, 1976). L’acqua di mare ne contiene da 10000 mg/l a 100000 mg/l raggiungendo il limite di saturazione di 105000 mg/l (Davis & DeWiest, 1966).
X ? Ione potassio (K+): è caratterizzato da un’elevata solubilità, ma essendo facilmente assorbito dalle argille non si trova in quantità abbondante nelle acque, infatti varia da 0,1 a 10 mg/l nelle acque dolci, e può raggiungere i 400 mg/l nelle acque di mare-.
II.2 Cromatografia a scambio ionico
La cromatografia ionica è un metodo di separazione di natura fisica applicato alla determinazione di soluzioni contenenti specie ioniche in funzione della diversa forza di legame che si instaura tra una fase fissa ed una mobile. Il meccanismo su cui si basa la cromatografia liquida è lo scambio ionico con una soluzione trasportante ed eluente contenente essa stessa ioni.
Poiché il movimento del soluto può avvenire solo nella fase mobile, la velocità media con cui il soluto migra dipende dalla frazione di tempo che esso trascorre in questa fase. Le differenze di velocità risultanti fanno sì che i componenti di una miscela si separino lungo la lunghezza della colonna e escano da essa in tempi diversi. Il fatto che ogni specie esca sempre al solito tempo permette di fare una analisi qualitativa, mentre l’altezza dei picchi o la loro area permettono di fare un’analisi quantitativa La cromatografia ionica (soprattutto la cromatografia di anioni inorganici) consente di eseguire in un tempo ragionevole (massimo 30 minuti) l’analisi contemporanea di molti componenti con minime manipolazione del campione. Nel caso della cromatografia a scambio ionico, gli scambiatori ionici sono costituiti da resine polimeriche dotate di gruppi funzionali in grado di effettuare scambi reversibili di ioni con la soluzione in esame. Si possono distinguere scambiatori di cationi, che possono essere fortemente acidi con i radicali -SO2OH e -CH2SO2OH o debolmente con
il gruppo -COOH, e scambiatori anionici fortemente o debolmente basici. Gli scambiatori anionici devono le loro proprietà ai gruppi amminici sostituiti nella struttura della resina. Le reazioni che intervengono possono essere schematizzate dalle due reazioni seguenti relative ad una colonna anionica:
nRN(CH3)3+OH- + An- [RN(CH3)3+]nAn- + nOH
-Gli analiti ionici sono introdotti in testa ad una colonna impaccata con una adatta resina a scambio ionico. Viene effettuata l’eluizione utilizzando come eluente una soluzione che contenga uno ione in grado di competere con gli analiti ionici per i gruppi carichi sulla
XI superficie della resina. Un cromatografo ionico è uno strumento che permette di determinare le concentrazioni delle specie anioniche presenti nelle acque e può essere schematizzato come in figura II.1
Figura II.1: schema di un cromatografo ionico
La pompa a pistone assicura un flusso costante e continuo dell’eluente nel circuito idraulico, la valvola di iniezione, dispositivo a due posizioni, permette di caricare il loop di campionamento con il campione, in genere tra 20 e 50 µl, poi di iniettare questo volume nel circuito idraulico, prima della colonna cromatografia, che permette la separazione delle specie ioniche presenti in soluzione. Il soppressore serve ad ottimizzare la sensibilità del rivelatore e può essere di tipo elettronico, ossia un circuito elettrico che permette di sottrarre il valore medio della conducibilità dell’eluente alla conducibilità della soluzione nel momento che uno ione passa nella cella conduttimetrica, oppure chimico, che abbassa drasticamente la conducibilità dell’eluente prima che esso giunga nella cella. Nel caso della determinazione degli anioni si fa passare la soluzione, in uscita dalla colonna cromatografica, in uno scambiatore a membrana, all’esterno della quale circola una soluzione contenente H+ che vengono sostituiti con gli ioni Na+ dell’eluente in modo da neutralizzare gli OH- dell’eluente stesso; ciò produce specie poco dissociate come H2CO3,
per eluizione effettuata con una soluzione di Na2CO3 e NaHCO3, ed H2O, per eluizione con
NaOH. In questo modo si abbassa la conducibilità dell’eluente, con un incremento della sensibilità in considerazione di un migliore rapporto tra segnale/rumore di fondo. Il rivelatore conduttimetrico è costituito da una cella di ridottissimo volume (1,5 µl) e da un circuito elettronico di misura.
XII
II.2.1 Determinazione degli anioni: cloruri. nitrati, solfati
E’ stato utilizzato, per la determinazione degli anioni, un cromatografo ionico Dionex, modello 1000, con colonne tipo AG-4a e AG 14-4 della dionex messe in serie, mentre per eluente si usa una soluzione di NaHCO3 e Na2CO3, avente una alcalinità totale di 8 meq/l. Il
cromatografo è collegato a un integratore che fornisce l’area e l’altezza dei picchi. Parametro, quest’ultimo, che è proporzionale alla concentrazione della specie chimica ricercata. Per il buon funzionamento dello strumento l’alcalinità del campione ha effetti sull’altezza dei picchi,, quindi l’alcalinità della fase mobile deve essere circa uguale a quella della soluzione analizzata. Per questo l’eluente con 80 meq/l, è usato per le correzioni di alcalinità sia dei campioni che degli standard. Prima di procedere alle analisi dei campioni vanno preparate le soluzioni standard a concentrazione nota con cui costruire la curva di calibrazione (altezza picco/concentrazione), necessaria poi per risalire alle concentrazioni incognite di cloruri, nitrati e solfati.
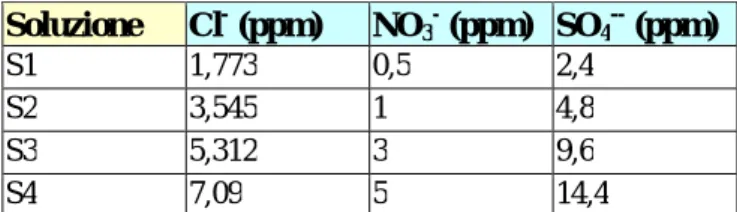
Le scale degli standard utilizzate sono mostrate in tabella II.2.1
Tabella II.2.1: scale degli standard per l’individuazione
di cloruri, solfati e nitrati
Soluzione Cl- (ppm) NO3- (ppm) SO4-- (ppm)
S1 1,773 0,5 2,4
S2 3,545 1 4,8
S3 5,312 3 9,6
S4 7,09 5 14,4
Operativamente si passano prima tutti gli standard, poi 4-5 campioni e poi si passano gli standard che permettono di “riprendere” i picchi dei campioni appena analizzati. Ci si alterna in questo modo fino alla fine delle analisi, in modo da tenere sotto controllo la riproducibilità e la stabilità dello strumento. I campioni devono ricadere nell’intervallo degli standard, altrimenti si cambia la diluizione su cui effettuare l’analisi.
Per correggere l’alcalinità si usa la seguente equazione:
Alc. Campione (meq/l)*Vol.Campione+106*Vol. Eluente = Vol. scelto di diluizione*10,6 Le soluzioni da analizzare sono inserite nella colonna con una siringa da 5 ml e per ogni iniezione è possibile determinare cloruri, nitrati e solfati. I cloruri escono dopo circa 2,4 minuti, i nitrati intorno a 6 minuti e i solfati intorno a 12 minuti.
XIII
II.3 Spettrofotometria di Assorbimento Atomico
La spettrofotometria ad assorbimento atomico consiste nel misurare la concentrazione di un elemento sulla base della capacità di questo di assorbire, allo stato atomico, luce di frequenza caratteristica. L’assorbimento sarà proporzionale alla concentrazione dell’elemento nel campione da analizzare. Gli spettrofotometri di assorbimento atomico sono strumenti capaci di misurare sia l’assorbimento atomico che l’emissione atomica. Ogni elemento ha un numero specifico di elettroni (pari al numero di protoni del nucleo). La configurazione orbitale più stabile di un atomo è conosciuta come stato fondamentale. Se ad un atomo è fornita energia, questa sarà assorbita e un elettrone dell’orbita più esterna passerà ad una configurazione meno stabile (stato eccitato). Poiché questo stato è instabile, l’atomo ritornerà immediatamente allo stato fondamentale rilasciando energia sotto forma di luce. Nell’emissione atomica sono coinvolti due processi: quello di eccitazione e quello di decadimento. Lo spettro di emissione di un elemento non è continuo ma consiste in un numero limitato di lunghezze d’onda chiamate linee di emissione (infatti gli elettroni eccitati hanno comunque ben definite energie). L’intensità di una linea di emissione aumenterà con il numero degli atomi eccitati. L’atomo nello stato fondamentale assorbe energia radiante di una specifica lunghezza d’onda passando allo stato eccitato. La quantità di radiazione assorbita è funzione del numero di atomi allo stato fondamentale presenti sul cammino ottico. L’uso di speciali sorgenti di luce ed una accurata selezione della lunghezza d’onda permette la specifica determinazione di elementi individuali. Quando una soluzione viene inviata sotto forma di minuscole goccioline all’interno di una fiamma, una frazione degli elementi presenti nella soluzione vengono trasformati dallo stato molecolare a quello atomico che è quello capace di assorbire la radiazione caratteristica. Tuttavia, la fiamma ha sufficiente energia per ionizzare una parte consistente di questi atomi. Questo fenomeno è tanto più effettivo quanto più alta è la temperatura della fiamma e quanto più basso è il potenziale di ionizzazione dell’elemento in esame. Pertanto quando si lavora in assorbimento atomico dobbiamo usare una fiamma che garantisca un elevato numero di atomi allo stato fondamentale rispetto a quelli ionizzati. Il contrario è valido quando si lavori in emissione atomica.
Qualsiasi spettrofotometro di Assorbimento Atomico è costituita di 5 componenti basilari: ? La sorgente di luce che emette lo spettro dell’elemento di interesse. Quella più
comunemente impiegata è la lampada a catodo cavo. Il bulbo della lampada è in vetro con una finestra di quarzo trasparente alle radiazioni, l’interno è riempito di
XIV Ne o Ar e quando viene applicata una differenza di potenziale di qualche centinaio di volt, il gas di riempimento si ionizza, gli ioni urtano il catodo e avviene l’espulsione degli atomi superficiali. Questi atomi vaporizzati quando urtano il gas di riempimento emettono energia luminosa. In genere le lampade sono singolo elemento per analizzarne uno alla volta, ma esistono anche lampade multielemento, meno sensibili, che consentono analisi di diversi elementi in successione.
? Una cella di assorbimento in cui vengono prodotti atomi allo stato fondamentale del campione. La più comune cella di assorbimento è la fiamma che può essere di vario tipo: aria-acetilene (2300°C), aria-idrogeno (2045), protossido di azoto-acetilene (2900°C). Il campione viene aspirato attraverso il nebulizzatore, trasformato in aerosol e spruzzato nella camera di premiscelazione. Qui si mescola con il gas combustibile e il comburente; la combustione e l’atomizzazione avvengono poi sulla testata del bruciatore. La soluzione da analizzare viene nebulizzata nella fiamma dove si producono gli atomi allo stato fondamentale
? Un monocromatore, da cui passa il segnale in uscita dalla fiamma, che ha la funzione di selezionare la lunghezza d’onda della radiazione. Gli spettrofotometri ad AA usano generalmente monocromatori a reticolo.
? Un rivelatore (fotomoltiplicatore), che misura l’intensità della luce e amplifica il segnale.
? Un sistema di acquisizione del segnale.
Le misure quantitative in assorbimento atomico sono basate sulla legge di Lambert-Beer. Essa dice che la concentrazione è proporzionale all’assorbanza misurata.
A = log(I0/I) I0 = Intensità radiazione entrante
I = Intensità radiazione uscente
A = x*b*N x = Coefficiente di assorbimento atomico (A = kC) b = Spessore dello strato assorbente
N = Numero totale di atomi liberi
Ad elevate concentrazioni tale legge non è più rispettata per cui bisogna cercare di lavorare nell’intervallo in cui la curva di calibrazione mantiene la sua linearità.
XV
II.3.1 Determinazione dei cationi: sodio, potassio, calcio, magnesio
Lo spettrofotometro di assorbimento atomico “Perkin Elmer” mod.3110 è uno strumento a singolo raggio, dotato di bruciatore a fiamma. Per la determinazione dell’assorbimento atomico venivano usate come sorgenti lampade a catodo cavo:
Perkin Elmer Lumina™ Lamp K+(current operating 12mA, ?=766.5 ? m) Perkin Elmer Intensitron™ Lamp Na+(current operating 8-10mA, ?=589 ? m) Perkin Elmer Intensitron™ Lamp Ca2+(current operating 10-25mA, ?=423 ? m) Perkin Elmer Intensitron™ Lamp Mg2+(current operating 10-25mA, ?=285 ? m) La fiamma usata è quella ottenuta con una miscela aria-acetilene ed è sempre la stessa per i quattro elementi.
? Sodio e potassio: Le scale degli standard utilizzate sono mostrate in tabella II.3.1
Tabella II.3.1: scale degli standard
per sodio e potassio
Soluzione Na+ e K+ (ppm) S1 0,4 S2 0,8 S3 1,2 S4 1,6 S0 0
Per evitare di avere sodio e potassio allo stato eccitato nella fiamma prima dell’arrivo della radiazione, si aggiunge agli standard e ai campioni una sostanza che si ionizza più facilmente degli elementi da analizzare. Questa sostanza è una soluzione di cesio a 10000 ppm che è preparata a partire da polvere di CsCl. Nelle soluzioni da analizzare si aggiunge il cesio affinché la sua concentrazione sia 1000 ppm, cioè ne va aggiunta 1/10 del volume finale. Il cesio, oltre a essere più facile da ionizzare, libera elettroni creando una nube riducente che si oppone ulteriormente alla ionizzazione di sodio e potassio.
? Calcio e magnesio: Le scale degli standard utilizzate sono mostrate in tabella II.3.2
Tabella II.3.2: scale degli standard
per calcio e magnesio
Soluzione Ca++(ppm) Mg++ (ppm) S1 1 0,1 S2 2 0,2 S3 3 0,3 S4 4 0,4 S5 5 0,5
XVI La sostanza da aggiungere per evitare la ionizzazione sulla fiamma di calcio e magnesio è, in questo caso, lo stronzio. Deve essere presente una quantità di 500 ppm e la soluzione di partenza è a 5000 ppm, quindi, anche ora, ne va aggiunto 1/10 rispetto al volume finale.
II.4 Spettrofotometria UV-VIS
I metodi per assorbimento rivelano la struttura molecolare delle sostanze e possono, quindi, essere applicati al riconoscimento ed alla determinazione quantitativa dei composti. Questi metodi si basano sull’assorbimento da parte di una soluzione trasparente di una determinata lunghezza d’onda, secondo la legge di Lambert e Beer: If = I0e-ksc dove k: coefficiente di estinzione; s: spessore della cella; c: concentrazione della specie in soluzione
L’assorbanza, A, è definita come A = logIo/If=ksc
Dato che k è una costante e lo spessore della cella è mantenuto fisso, la relazione tra assorbenza e concentrazione è teoricamente lineare.
L’analisi quantitativa viene eseguita confrontando l’assorbanza delle soluzioni in esame con quella di soluzioni a concentrazione nota. Se la specie chimica assorbe nell’UV non è necessario nessun trattamento particolare della soluzione, se non l’acidificazione; mentre se non assorbe in questa regione dello spettro si può effettuare la misura nel caso in cui formi con opportuni reagenti sostanze che assorbono radiazione visibile.
Le specie chimiche determinate con questa tecnica sono state: NH4+ e COD.
Bisogna sempre considerare le eventuali interferenze che si possono presentare in ogni determinazione, dovute, ad esempio, a: presenza di solidi in sospensione, selettività della reazione, presenza in soluzione di sostanze colorate e loro stabilità.
II.4.1 Determinazione di ammoniaca e COD
La misura di ammoniaca è stata eseguita mediante lo spettrofotometro di assorbimento “Elmer Lambda Spectophotometers” mod.632-0001”, mentre per il COD è stato usato lo spettrofotometro da campo “DR/2000 Spectophotometer HACH”.
? Ammoniaca: L’assorbimento è dovuto alla formazione di un complesso di colore verde contenente ammonio. La lunghezza d’onda necessaria alla determinazione
XVII dell’ammoniaca è 690 nm e la cella da utilizzare è quella di quarzo puro. Il volume totale deve essere 20 cc sia per gli standard che per i campioni e l'acqua da utilizzare per la loro preparazione è l'acqua millipore. La scala degli standard viene preparata a partire da cloruro di ammonio e per lo sviluppo del colore si aggiungono 2,5 cc del reagente 1B (NaOH circa 8N e Tartrato di Sodio) ed una punta di spatola del reagente 2B (ipoclorito), si attendono 15 minuti e si aggiungono 2 gocce del reagente 3B (timolo e nitroprusiato sodico). Si attendono circa 30 minuti dopodiché si esegue la lettura allo strumento. (Standard Methods, 1998).
? COD: La seguente metodologia si basa sulla digestione acida dell'analita. Si prendono aliquote note del campione in esame (2 cc se la concentrazione di cloruri è inferiore a 1000 ppm, altrimenti prima è necessaria una diluizione), e si mettono all’interno di una provetta in vetro che ha la caratteristica di essere una cella spettrofotometrica. All’interno della provetta è presente acido solforico (86%) per mantenere la soluzione a pH acido, solfato di mercurio che permette di precipitare i cloruri a cloruro di mercurio poiché è molto insolubile, e triossido di cromo che opera l’ossidazione della materia organica a CO2. Appena chiusa la provetta va
agitata e poi la si immette in un reattore a 150 °C per 2 ore. La fase finale, cioè la misura, avviene dopo che si sono raffreddati i campioni e dopo aver pulito l’esterno della celletta da possibili interferenze ottiche. La lunghezza d’onda indicata nella metodologia di lavoro è di 420 nm e lo strumento fornisce direttamente la misura di COD in mg/l
XVIII
Appendice III: Natanti da diporto
III.1 Generalità natanti da diporto
Il naviglio da diporto, quello cioè utilizzato per la navigazione effettuata a scopi sportivi o ricreativi dai quali esuli il fine di lucro, viene suddiviso, in relazione alla lunghezza "fuori tutto” (f.t.), in tre grandi categorie:
? navi da diporto, quando la lunghezza f.t. sia superiore ai 24 m;
? imbarcazioni da diporto, quando la lunghezza fuori tutto, inferiore ai 24 m, sia superiore ai 10 m;
? natanti da diporto, quando la lunghezza f.t. sia pari o inferiore ai 10 m.
Con la dizione "lunghezza fuori tutto", s'intende quella indicata nella licenza di navigazione, per le navi e le imbarcazioni; per i natanti si intende quella misurata in linea retta fra il punto estremo anteriore della prora ed il punto estremo posteriore della poppa, escluse tutte le appendici quali delfiniere, bompresso, piattaforme poppiere, falchette e similari.
Quando si parla di barche in genere si è portati a illustrare quelle che sono le loro caratteristiche di stile e design e vengono trascurate le caratteristiche tecniche, che sono basilari per consentire una migliore navigazione. Fondamentale nella progettazione delle barche è la carena, la parte immersa dello scafo che con le sue forme influenza direttamente la resistenza e il comportamento dell’imbarcazione durante il suo moto. La scelta di una carena, che può essere dislocante, semidislocante o planante, è dettata principalmente da due parametri fondamentali: le dimensioni dello scafo e la velocità alla quale si desidera navigare.
Per un'analisi sulla scelta di una determinata forma di carena, bisogna tener presente la resistenza al moto che un'imbarcazione incontra in acqua. La resistenza al moto di una imbarcazione o nave, qualsiasi ne sia la velocità, la grandezza e il tipo, dipende soprattutto da due principali fenomeni:
? l'attrito dell'acqua contro la superficie della carena (resistenza d'attrito)
? la generazione, al passaggio dell'imbarcazione sull'acqua, di onde di superficie (resistenza d'onda)
A questi due principali fattori se ne devono aggiungere altri, meno condizionanti, ma comunque importanti che di seguito enunciamo:
? resistenza di forma ? resistenza dell'aria
XIX ? resistenza delle appendici
? resistenza della carena sporca ? resistenza per mare mosso
La resistenza d'onda e la resistenza di forma sono comunemente chiamate globalmente resistenza residua o d'onda.
La resistenza della carena sporca ha molta influenza sulla resistenza della nave specie con l'andar del tempo. Al fine di prevenire la formazione di sporcizia dovuta a vegetazione marina e a microrganismi che si formano dopo lunghi periodi di mare soprattutto in acque calde, la superficie dell'opera viva è trattata con pitture antivegetative speciali, che periodicamente devono essere rinnovate. Analizziamo, dunque, le prime due resistenze citate, le più importanti: la resistenza d’attrito e la resistenza residua.
La resistenza di attrito rappresenta l'energia da spendere per vincere l'attrito generato dalla
parte immersa dello scafo (opera viva o carena), scorrendo nell'acqua ed è sintetizzata da questa formula:
RF = CF1/2?SV2
Dove il coefficiente CF è funzione del numero di Reynolds
Rn = (VL)/n
? = densità del fluido S = superficie bagnata V = velocità
L = lunghezza al galleggiamento n = viscosità cinematica
La resistenza d’onda rappresenta l’energia da spendere per mettere in movimento le masse
d’acqua costituenti i “treni d’onde” generati dal passaggio dell’imbarcazione.
Il sistema di onde generate dalle imbarcazioni sono di due tipi, quelle divergenti, che si formano lateralmente alla nave e che hanno creste inclinate rispetto al piano di simmetria della barca, e quelle trasversali, che hanno le creste
perpendicolari alla mezzeria. Tale sistema di onde, divergenti e trasversali, è generato sia dalla poppa che dalla prua.
La resistenza d’onda o residua è funzione del numero di Froude
Fn = V/(gL)1/2
dove:
XX V = velocità
g = accelerazione di gravità L = lunghezza al galleggiamento
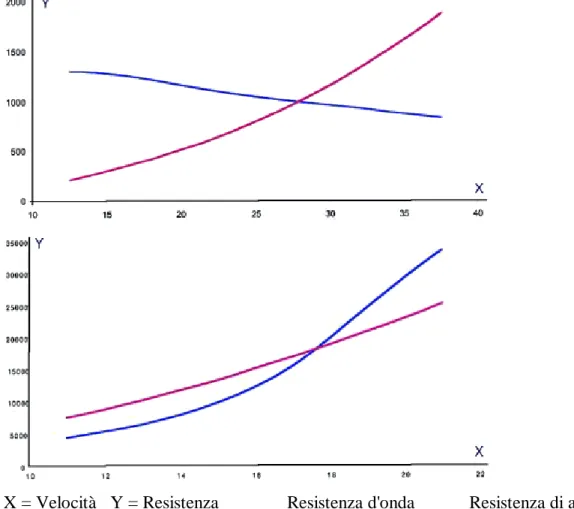
In temini meno tecnici ciò significa che la resistenza di attrito di una carena aumenta rapidamente con l'aumentare della velocità, mentre la resistenza residua diminuisce, a parità di velocità, con l'aumentare della lunghezza al galleggiamento, cioè della lunghezza misurata sulla "linea che traccia il livello dell'acqua sulla carena della barca e separa la parte immersa da quella emersa". In altre parole, più si accresce la velocità più aumentano le resistenze al suo avanzamento, più è lunga la barca maggiore è la sua velocità di navigazione.
Le forme che una carena può assumere sono essenzialmente due: tonda o a spigolo.
La carena tonda, detta carena dislocante, con l'aumentare della velocità, ha una resistenza d'onda superiore a quella d'attrito, perciò si adatta meglio alle basse e medie velocità. Viceversa, la carena a spigolo, raggiunte determinate velocità, ha una resistenza d'attrito che supera in modo sensibile quella d'onda. Quest'ultimo tipo di carena, detta carena planante, risulta quindi adatto alle alte velocità.
X = Velocità Y = Resistenza ____ Resistenza d'onda ____ Resistenza di attrito
XXI Per i mezzi che per peso, lunghezza e velocità hanno caratteristiche intermedie tra una carena tonda ed una planante, si adotta la carena semi-planante o semi-dislocante, usata soprattutto per i mezzi di piccolo tonnellaggio.
III.2 La carena planante e dislocante
La carena planante è piú indicata quando si vogliono raggiungere velocità relative elevate, e uno scafo planante è uno scafo che, in moto, trova il suo principale sostegno nella reazione dinamica dell’acqua. Essenzialmente, esso scivola o sfiora la superficie idrica e ciò lo distingue da un normale scafo, che, viceversa, galleggia solamente e si apre la rotta nell’acqua. L'azione di una superficie planante è simile a quella di un cuneo forzato al di sotto di un peso allo scopo di alzarlo. Una superficie planante inclina il suo piano forzando l'acqua in basso, e questa forza crea un campo di pressione che fa aumentare il livello dell'acqua ai lati della superficie stessa. Quindi è la spinta dinamica che ad alta velocità sostiene quasi l'intero peso delle barche plananti.
Alcune differenze sostanziali tra gli elementi di progetto di uno scafo planante e di uno dislocante sono:
La spinta statica della carena, in una barca a dislocamento, sostiene tutto il peso, mentre in uno scafo planante detta spinta sostiene solo da 1/3 a 1/13 del peso della barca. Il resto è sostenuto dalla spinta dinamica. La forma di carena di una nave a dislocamento interessa la resistenza al moto della nave stessa e le sue qualità manovriere e di tenuta al mare. Nel caso degli scafi plananti si ha una esaltazione di questa influenza. La forma del fondo influisce sulla possibilità di planata, sul comportamento dell'unità durante la planata, sul beccheggio, sul "delfinamento", sulla tenuta al mare, sulla stabilità di rotta e sull'angolo di sbandamento in virata. La resistenza dell'aria per una unità a dislocamento rappresenta una piccola parte della resistenza totale. Per una unità planante, molto veloce, detta resistenza costituisce invece un fattore molto importante e talvolta è accompagnata da notevoli forze verticali. Il progetto della carena di uno scafo planante costituisce dunque non solo un problema notevolmente diverso da quello di una nave a dislocamento, ma anche un problema più intricato e complesso data la interdipendenza e la interazione dei più importanti fattori che lo governano. In nessun'altra branca del progetto navale esiste una così profonda, mutua dipendenza dei vari fattori. Un aumento del 10% nel peso di un'unità a dislocamento significa che quest'ultima s'immerge un poco di più, naviga ad una velocità più ridotta ma
XXII conserva circa le stesse forme di carena ed il suo comportamento rimane essenzialmente lo stesso. Il medesimo aumento di peso in uno scafo planante può impedire a quest'ultimo di planare e/o modificare le sue caratteristiche di stabilità dinamica e quindi di tenuta all’acqua.
Purtroppo le forme di scafo più idonee per la navigazione in planata mal si conciliano con quelle classiche per la navigazione in dislocamento. Quindi a bassa velocità, o per meglio dire, prima che si raggiunga la velocità di planata, lo scafo progettato per una navigazione prevalentemente in regime di planata è soggetto generalmente a forti resistenze d'onda, di vortici ed a noiosi spruzzi d'acqua. Prima di raggiungere la velocità di planata la prua rimane sospesa nell'aria, la resistenza aumenta sensibilmente e, se non si dispone di una riserva di potenza, la barca si trova di fronte ad una barriera per la sua velocità che non potrà superare. Se, viceversa, c'è ancora potenza disponibile, lo scafo supererà la resistenza, la sua velocità aumenterà sensibilmente e la cresta dell'onda di prua si sposterà verso poppa. Con una espressione indovinata molti dicono che, a questo punto, l'imbarcazione sale sulla sua onda di prua, l'angolo di assetto longitudinale diminuisce e la velocità aumenta. Lo scafo aumenta sensibilmente la velocità senza richiedere un ulteriore aumento di potenza; si dice che è stata raggiunta la velocità di planata che, certamente, non è la massima velocità raggiungibile.
Un'imbarcazione è in planata, quando l'acqua è a contatto con il fondo solamente all'interno degli spigoli.
Quanto maggiore è la distanza fra la massima velocità e la velocità di planata, tanto migliore sarà il comportamento dell'imbarcazione, perché l'aumento di resistenza dovuto all'aumento della sporcizia di carena, al mare mosso, ad un eventuale supplemento di carico, al vento contrario, non sarà tale da impedire la planata. Quindi di un'imbarcazione è importante conoscere la velocità massima e la velocità di planata alla condizione di pieno carico massimo.
XXIII
Bibliografia appendice
APHA (1992) “Standard Methods for the Examination of Water and Wastewater”, 18th Ed., Am. Public Health Assoc., Washington
Cozzi R., Protti P. e Ruaro T. (1987) “Analisi chimica: Moderni metodi strumentali”, ESU spa
Custodio E., Llamas M.R. (1976) “Hidrologia subterranea”, Omega, Barcellona
Davis N.S & DeWiest R.J.M (1966) “Hidrogeology”, John Wiley & Sons, Inc., New York
Hach (1988) “DR/2000 Spectrophotometer Handbook Instrument and Procedures manual”, Hach Company
Stumm W.and Morgan J.J.(1996) “Aquatic chemistryi”, 3th Edition, John Wiley and Sons, Inc., pp.1022
Crea A., Falchet L. (1994) “Chimica analitica, analisi quantitativa e qualitativa”, Edizioni Massonscuola, Milano
Appendice III: Natanti da diporto: www.nautica.it www.leganavale.it www.velarossa.it