1. Introduzione
1.1 Fisiopatologia della malattia asmatica
1.1.1 Aspetti generali
Secondo le linee guida GINA (Global INitiative for Asthma) aggiornate al 2006, l’asma è un disordine infiammatorio cronico delle vie aeree che causa iperresponsività della muscolatura respiratoria agli agenti contratturanti, senso di costrizione, respiro sibilante e tosse, soprattutto notturni e mattutini. Negli ultimi anni, infatti, l’asma è considerato sempre più come un complesso di disordini che coinvolgono molte cellule e mediatori cellulari.
L’evento finale che provoca i sintomi dell’asma è l’ostruzione episodica del flusso aereo espiratorio, provocata da numerosi eventi:
Iperreattività bronchiale: anomalia funzionale parzialmente reversibile, caratteristica dell’asma. Determina il restringimento delle vie aeree ed è collegata sia all’infiammazione sia al rimodellamento delle vie aeree che provocano contrazione, proliferazione e secrezione di mediatori infiammatori da parte delle cellule muscolari (Figura 1) (Pelaia et al., 2008).
Edema: dovuto a un’aumentata permeabilità vascolare in risposta a mediatori infiammatori e alla vasodilatazione indotta dalla liberazione di NO (Ricciardolo et al., 2004).
Ipersecrezione di muco: dovuta all’iperplasia delle cellule caliciformi e all’ipertrofia delle ghiandole della sottomucosa. È stato evidenziato che i neutrofili e alcune interleuchine (Dabbagh et al., 1999; Louahed et al., 2000; Takeyama et al., 2000) provocano l’induzione trascrizionale dei geni MUC5AC e MUC2, mentre LTD4 induce il gene MUC2 (Suzuki et al., 2008).
Fibrosi subepiteliale: deriva dalla deposizione di collagene e proteoglicani sotto la membrana basale, durante il fenomeno di rimodellamento, per effetto dei fattori di crescita (FGF) e dei miofibroblasti (Brewster et al., 1990; Hoshino et al., 1998). E’ accompagnata da ipertrofia e iperplasia delle cellule muscolari lisce (Gizycki et al., 1997).
Sfaldamento delle cellule epiteliali: dovuto ai mediatori infiammatori. Studi recenti hanno evidenziato lo sviluppo di giunzioni cellulari incomplete a livello delle cellule apicali dell’epitelio durante il processo di riparazione (De Boer et al., 2008).
Angiogenesi: indotta dall’elevato rilascio di fattori di crescita vascolari (VEGF) (Simcock et al., 2008). E’ stata correlata all’incremento di TGF-β1 dipendente dai cisteinil-leucotrieni (Bossé et al., 2008).
Figura 1. Meccanismi cellulari coinvolti nell’infiammazione acuta e cronica
delle vie aeree.
Esistono numerosi fattori, individuali e ambientali, che influenzano il rischio di sviluppare l’asma. Tra questi è stata individuata una componente genetica che partecipa sia alla patogenesi dell’asma sia alla risposta ai trattamenti antiasmatici.
Tra le più frequenti cause scatenanti gli attacchi vi sono fattori pressoché ubiquitari quali gli allergeni (acari, pollini, muffe, ecc.), gli inquinanti ambientali (fumi, endotossine, ecc.), le infezioni respiratorie (prevalentemente virali), e l’esercizio fisico, che si pensa agisca come stimolo osmotico determinando raffreddamento, disidratazione delle vie aeree con incremento dell’osmolalità del liquido periciliare e conseguente perdita di acqua dovuta all’iperventilazione, specie in presenza di aria fredda e basso tasso di umidità (Tan et al., 1985). Altri agenti meno frequenti sono rappresentati da conservanti (solfiti e benzoato di sodio) e coloranti alimentari, diete povere di antiossidanti (carenza di frutta e verdura) e di acidi grassi n-3 poli-insaturi (prodotti ittici) e ricche di acidi grassi n-6 poli-insaturi (ritrovati nelle margarine e negli oli vegetali) (Devereux et al., 2005), cibi e bevande quali alcool e Coca Cola.
L’asma può essere anche causata da un’intolleranza all’acido acetilsalicilico (aspirina), associata a polimorfismi dei geni codificanti i recettori 1 e 2 dei cisteinil-leucotrieni (Kim et al., 2007; Park et al., 2005) e al polimorfismo del gene LTC4-sintetasi, riscontrato nel 70% dei pazienti con asma da aspirina (Szczeklik et al., 2000). In questi
soggetti, la crisi asmatica è un effetto collaterale “on target” dovuto cioè all’inibizione delle ciclossigenasi, che causa uno spostamento della cascata dell’acido arachidonico verso la via delle lipossigenasi, con maggiore produzione di leucotrieni, che agiscono come potenti broncocostrittori.
L’asma allergico risulta avere una maggiore incidenza nell’infanzia e la sensibilizzazione potrebbe avvenire anche nel periodo prenatale e perinatale (Bousquet et al., 2000; Jones et al., 2000). In questi soggetti la remissione della malattia asmatica prima dell’età adulta si verifica in circa il 50% dei casi (Strachan et al., 1996), mentre negli adulti il processo patologico sembra essere irreversibile e viene associato ad una riduzione progressiva della funzionalità respiratoria (Lange et al., 1998).
1.1.2 Patogenesi dell’asma
Lo sviluppo della sintomatologia asmatica è strettamente correlato all’infiammazione cronica delle vie aeree, secondo un modello che sembra essere simile in tutte le forme cliniche di asma (allergico, non
allergico, o indotto da aspirina) ed è indipendente dall’età dei pazienti. Tuttavia si possono classificare due tipologie di malattia asmatica: estrinseca (indotta da allergeni che scatenano una reazione di ipersensibilità di tipo I, mediata dalle IgE) ed intrinseca (non riconducibile ad una reazione immunitaria).
In aggiunta alla componente infiammatoria sono presenti mutamenti strutturali caratteristici, descritti come “remodelling”, che
possono rappresentare processi riparativi in risposta
all’infiammazione.
Infiammazione
L’asma allergico (atopia) è una patologia caratterizzata da un’iperproduzione di IgE e da un infiltrato infiammatorio particolarmente ricco di eosinofili, basofili, mastociti e linfociti T helper 2 (Th2) che risulta dall’espressione di specifiche chemochine, quali le eotassine, sull’epitelio delle vie aeree, sull’endotelio microvascolare e sui macrofagi tissutali (Komiya et al., 2003). Molti studi hanno dimostrato una correlazione tra la gravità dell’attacco
asmatico e i livelli di eosinofili nei reperti istologici (Wardlaw et al., 2000).
Il passaggio cruciale è rappresentato dalla differenziazione dei linfociti T in linfociti Th2 ad opera delle cellule dendritiche (Kuipers et al., 2004) e dei macrofagi, quali cellule presentanti l’antigene. Le cellule dendritiche della mucosa del tratto respiratorio attivano preferenzialmente i linfociti Th2, liberando citochine e cisteinil-leucotrieni (Machida et al., 2004). Inoltre, è stato dimostrato che anche i linfociti T helper 1 (Th1) partecipano al processo infiammatorio provocando citotossicità, soprattutto nell’asma severo (Abdulamir et al., 2008). Si può anche verificare un aumento delle cellule natural killer (NK), le quali rilasciano citochine stimolanti i linfociti Th1 e Th2 (Akbari et al., 2006).
Nella risposta tardiva all’allergene aumenta anche l’infiltrazione di neutrofili (Fahy et al., 1995). Nelle risposte atopiche e non, acute e croniche, è stata inoltre dimostrata una sensibilizzazione dei neutrofili da parte dei cisteinil-leucotrieni C4 e D4 (Theron et al., 2009).
Nell’infiammazione acuta, il legame tra l’allergene e le IgE legate a mastociti e macrofagi causa degranulazione con liberazione di
istamina, adenosina, specie reattive dell’ossigeno, eparina e proteasi; l’attivazione provoca la sintesi di citochine, chemochine, fattori di crescita, ossido nitrico ed eicosanoidi. I principali eicosanoidi sintetizzati dopo stimolazione sono la PGD2 e il LTC4, potenti broncocostrittori. I basofili risultano importanti nella fase tardiva dell’infiammazione.
Tutti questi mediatori amplificano la risposta infiammatoria sostenendo la produzione di IgE e l’eosinofilia (Larche et al., 2003) e provocano iperresponsività e broncocostrizione, vasodilatazione, ipersecrezione di muco, edema e stravaso proteico.
Remodelling
Nell’infiammazione cronica delle vie aeree, la suscettibilità dell’epitelio agli agenti esterni e ai mediatori infiammatori aumenta e mette in atto un processo continuo di riparazione (Holgate et al., 2007). Non solo le cellule epiteliali, ma anche i miofibroblasti e le cellule infiammatorie producono fattori di crescita ed enzimi
responsabili della fibrosi, dell’angiogenesi, della formazione di nuovi nervi e dell’ipersecrezione di muco.
L’incapacità di un efficiente sistema di riparazione provoca cambiamenti strutturali che si manifestano con iperresponsività e progressiva ostruzione delle vie aeree; ciò è dovuto sia alla produzione di neurotrofine sensibilizzanti e neuropeptidi infiammatori da parte dei nervi sensoriali (Groneberg et al., 2004), che all’aumento di spessore della parete bronchiale (Hirst et al., 2004). Studi sui ratti hanno dimostrato un aumento di responsività agli agenti contratturanti e agli allergeni dovuto all’alterazione del metabolismo del Ca2+
da parte del TNF-α (Amrani et al., 2000), tramite attivazione della fosfolipasi C accoppiata alla proteina G.
Questo fenomeno risulta essere la conseguenza più invalidante della malattia asmatica ed i mediatori e i fattori di crescita coinvolti rappresentano un nuovo target per strategie terapeutiche “anti-remodeling”.
1.1.3 Classificazione dell’asma e terapie utilizzate
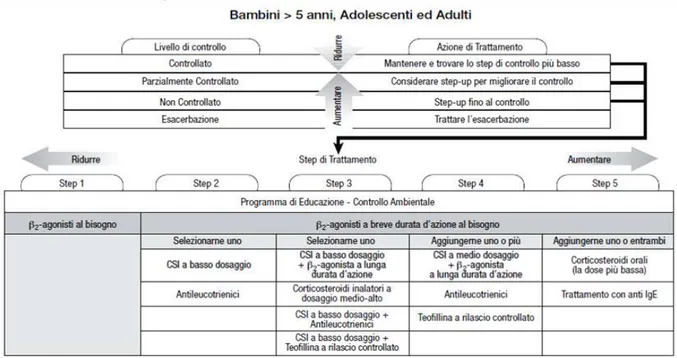
Le linee guida GINA suddividono l’asma in livelli di gravità basati sull’intensità dei sintomi, sull’ostruzione bronchiale e sulla variabilità della funzione respiratoria in quattro categorie: intermittente, lieve persistente, moderato persistente o grave persistente. Tuttavia, la valutazione deve coinvolgere sia la gravità della malattia che la risposta al trattamento.
Asma lieve intermittente. Caratterizzato da sintomi che si presentano al massimo due volte la settimana o meno; gli attacchi sono di breve durata e variano di intensità. In questo stadio viene attuata solo una terapia con farmaci sintomatici broncodilatatori (β2 agonisti short acting per via inalatoria) da assumere al momento degli attacchi.
Asma lieve persistente. Caratterizzato da sintomi che si presentano più di due volte la settimana, ma meno di una volta al giorno. Gli attacchi possono essere abbastanza gravi da compromettere l’attività fisica. Vengono utilizzati farmaci sintomatici associati a corticosteroidi inalatori a basse dosi o antileucotrieni. Non
sono raccomandati come farmaci di fondo di prima scelta la teofillina ed i cromoni, che risultano essere poco efficaci.
Asma moderato persistente. Caratterizzato da sintomi giornalieri e di entità anche tale da influenzare ogni tipo di attività. Sono utilizzate nella terapia, basse dosi di corticosteroidi inalatori combinati con β2 agonisti long acting (Shrewsbury et al., 2000). In alternativa è possibile incrementare i corticosteroidi inalatori ad una dose media, oppure utilizzare una bassa dose di corticosteroidi inalatori in associazione agli antileucotrieni (Price et al., 2003), teofillina a lento rilascio a basso dosaggio o inibitori delle lipossigenasi (Zileuton).
Asma grave persistente. Caratterizzato da sintomi continui e frequenti attacchi. Si utilizzano corticosteroidi inalatori ad alte dosi associati a β2 agonisti long acting e se necessario si aggiungono corticosteroidi orali (nella dose più bassa) o anti-IgE (Omalizumab) (Figura 2).
Figura 2. Linee guida GINA sul trattamento dell'asma
Gli anticorpi monoclonali anti-IgE rappresentano una nuova strategia terapeutica nell’asma indotto da aspirina e risultano efficaci nell’asma allergico grave quando alte dosi di corticosteroidi inalatori o orali risultano essere inefficaci (Humbert et al., 2005).
Le linee guida GINA 2006 raccomandano il vaccino antinfluenzale a soggetti con asma moderato e grave. Recenti studi hanno, infatti, dimostrato l’efficacia del vaccino nel ridurre le esacerbazioni della malattia asmatica (Poole et al., 2006); tuttavia, nei bambini con asma,
il vaccino attenuato offre una maggiore protezione rispetto a quello inattivo (Glezen et al., 2006).
I limiti delle terapie attualmente disponibili stanno orientando la ricerca verso nuovi trattamenti; tra questi, i β2 agonisti ultra-long acting (ultra-LABAs), gli antagonisti muscarinici long acting (LAMA), gli inibitori della PDE4 (roflumilast) e i PPAR-γ agonisti (rosiglitazone e pioglitazone).
Asma in età pediatrica
La diagnosi di asma in età prescolare (<3 anni) è resa complicata dalla presenza di fattori confondenti che possono essere associati ad altre patologie tipicamente infantili. Un sintomo clinico quale il respiro sibilante, associato ad un fattore di rischio maggiore (storia familiare di asma) o a due fattori di rischio minori (eosinofilia, respiro sibilante e rinite allergica) sembra predire lo sviluppo di asma nei bambini più grandi (Castro-Rodriguez et al, 2000). In questi casi l’uso di corticosteroidi per inalazione non si è rivelato utile nel prevenire lo sviluppo della malattia (Guilbert et al., 2006).
I farmaci di scelta nel trattamento dell’asma pediatrica sono i glucocorticoidi per via inalatoria e sistemica, i β2 agonisti a breve e lunga durata d’azione per via inalatoria e sistemica, gli antileucotrieni e i cromoni.
Secondo quanto indicato dalle linee guida GINA, i glucocorticoidi per via inalatoria sono i farmaci di fondo più efficaci e sono raccomandati nel trattamento dell’asma persistente a prescindere dall’età del paziente. Consentono un rapido miglioramento dei sintomi e della funzionalità respiratoria già a basse dosi e riducono la frequenza delle riacutizzazioni e l’iperreattività. Nei bambini di età inferiore ai 5 anni il loro uso determina la remissione dell’asma, ma non sono stati effettuati adeguati studi sulle relazioni dose-risposta.
Un possibile problema nell’uso di alte dosi di glucocorticoidi per via inalatoria è la riduzione della crescita ossea, spesso associata ad un ritardo nell’inizio della pubertà. Tuttavia, studi di questo tipo sono stati condotti solo su bambini di età maggiore di 5 anni, che risultano più sensibili agli effetti avversi dei glucocorticoidi per inalazione (Pedersen, 2001). Pertanto, nei bambini di età inferiore ai 5 anni, questo problema, come anche il rischio di osteoporosi, fratture ossee,
alterazioni comportamentali e inibizione del meccanismo a feed-back dell’asse ipotalamo-ipofisi-surrene, ha una rilevanza ancora incerta.
In questa fascia d’età, i β2 agonisti per via inalatoria a breve durata d’azione, al bisogno, sono utilizzati in associazione con i corticosteroidi nel trattamento dell’asma acuto, mentre i β2 agonisti a lunga durata sono utilizzati soprattutto in bambini di età superiore ai 5 anni. I β2 agonisti long acting, tuttavia, risultano inefficaci nel ridurre la frequenza delle riacutizzazioni (Bisgaard, 2003).
Gli antileucotrieni risultano efficaci nel controllare i sintomi clinici a tutti i livelli di gravità e anche nei bambini di età inferiore ai 5 anni (Garcia et al., 2005; Knorr et al., 2001).
I principali biomarker di asma in età pediatrica sono l’esalazione di ossido nitrico (NO), il ritrovamento di eosinofili nella saliva e di LTE4 nelle urine (Gogate et al., 2008). Il montelukast, oltre ai ben noti effetti sui cisteinil-leucotrieni e sugli eosinofili, riduce significativamente l’esalazione di NO, se somministrato per via sistemica in combinazione ai corticosteroidi (Fritscher et al., 2009).
Attualmente, l’utilizzo del montelukast è approvato nei bambini da 1 a 14 anni in combinazione ai glucocorticoidi per inalazione nel
trattamento dell’asma moderata e severa non controllata con glucocorticoidi per via inalatoria, né con β2 agonisti “al bisogno” (Walia et al., 2006). Da studi recenti è anche emerso che il trattamento con montelukast riduce la richiesta di corticosteroidi orali e β2 agonisti (Knorr et al., 2001). Tuttavia, in genere, gli antileucotrieni determinano effetti minori rispetto ai glucocorticoidi per inalazione a basse dosi.
Montelukast si è rivelato efficace nel ridurre le riacutizzazioni indotte da infezioni virali nei bambini da 2 a 5 anni (Bisgaard et al., 2005) e nella broncocostrizione causata da esercizio fisico (Melo et al., 2003). Inoltre, nel trattamento sintomatico di asma non controllato, il montelukast produce una significativa riduzione delle riacutizzazioni e studi recenti suggeriscono che questo farmaco possa essere utilizzato favorevolmente nell’asma moderato come monoterapia (Wahn et al., 2008).
1.2 Meccanismo d’azione dei farmaci
antiasmatici e loro limitazioni
1.2.1 Glucocorticoidi e polimorfismo recettoriale
L’azione dei glucocorticoidi è mediata da un recettore intracellulare (hGRα) che appartiene alla famiglia dei recettori nucleari per fattori di trascrizione ligando-dipendenti.
Il gene umano del recettore per i glucocorticoidi (hGRα) è localizzato sul cromosoma 5 e consiste di 9 esoni. In base al differente splicing si possono ottenere 3 diversi tipi di mRNA: quelli che codificano i recettori hGRα e hGRβ, che differiscono per l’estremità carbossi-terminale, ovvero per l’esone 9 (9α e 9β), e l’mRNA contenente l’esone 9α, 9β ed una “J region”. Si ritiene che questo mRNA possa essere tradotto sia in hGRα che in hGRβ (Oakley et al., 1996).
Delle due isoforme, solo hGRα ha la capacità di legare i corticosteroidi, mentre hGRβ interagisce con il DNA e risulta avere prevalentemente azione inibitoria nei confronti del legame tra hGRα e DNA (Lewis-Tuffin et al., 2006).
Il meccanismo d’azione dei glucocorticoidi si esplica mediante la diffusione attraverso la membrana citoplasmatica ed il legame del farmaco con il recettore GRα nel citoplasma; questo legame promuove in seguito una cascata di eventi biochimici tra cui:
La dissociazione di GRα dalle proteine dello shock termico (in particolare hsp90)
La fosforilazione e la traslocazione nel nucleo
L’omodimerizzazione ed il legame con il DNA a livello delle sequenze promoter dei Glucocorticoid Responsive Elements (GRE).
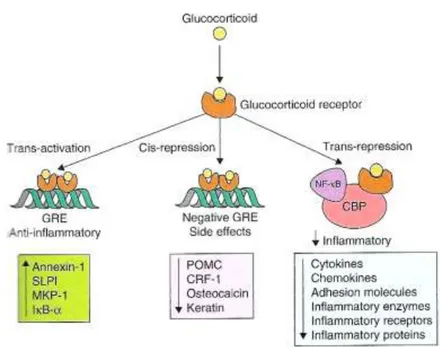
La trans-attivazione, dovuta al legame con positive GREs, che promuove la sintesi del recettore β2-adrenergico, dell’inibitore di NF-kB (INF-kB-α) e di proteine anti-infiammatorie, tra cui la lipocortina-1, che blocca la cPLA2 arrestando la cascata dell’acido arachidonico, precursore di leucotrieni e prostaglandine. L’induzione della trascrizione genica è responsabile degli effetti antinfiammatori.
La cis-repressione, dovuta al legame con negative GREs, che sopprime la trascrizione di alcuni geni ed è responsabile degli effetti avversi dei glucocorticoidi.
La trans-repressione, dovuta all’interazione di GRα attivato con la CREB-binding protein (CBP), coattivatore di NF-kB, di cui inibisce l’attività dell’enzima istone acetilasi. La contemporanea attivazione dell’istone deacetilasi di tipo 2 (HDAC2) provoca la soppressione trascrizionale di tutti i geni pro-infiammatori (Figura 3) (Ito et al., 2000).
Figura 3. Principali meccanismi d’azione dei glucocorticoidi
Inoltre, molti studi rivelano che i glucocorticoidi agiscono anche attraverso un meccanismo non genomico, mediato da recettori di membrana e da interazioni dirette con le membrane cellulari (Stahn e Buttgereit, 2008). Questo potrebbe spiegare le differenze nella rapidità
e durata dell’effetto terapeutico, e nell’attività antinfiammatoria e immunosoppressiva di alcuni glucocorticoidi, in relazione anche al dosaggio ed alla via di somministrazione, aprendo la strada allo sviluppo di agonisti selettivi dei GR.
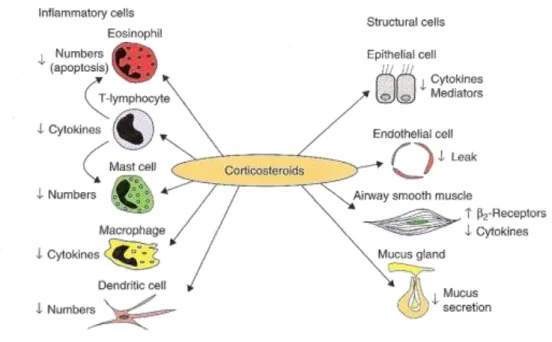
Attraverso il meccanismo della trans-repressione, i glucocorticoidi sopprimono la produzione di citochine, chemochine, molecole di adesione, enzimi infiammatori e recettori (Figura 4) (Barnes et al., 2003). E’ stato anche dimostrato che la riduzione di mediatori infiammatori provocata dai glucocorticoidi contrasta la stabilizzazione del mRNA di geni pro-infiammatori (ad esempio quello del GM-CSF) (Bergmann et al., 2004). Tuttavia, i corticosteroidi non riescono ad inibire la sintesi ed il rilascio dei cisteinil-leucotrieni, potenti broncocostrittori (Dworski et al., 1994).
Figura 4. Effetti cellulari dei glucocorticoidi nell'asma e nell'infiammazione
Nei soggetti resistenti alla terapia con glucocorticoidi, recenti studi hanno rilevato un aumento dell’espressione dell’isoforma hGRβ, che impedisce il legame di hGRα con il DNA (Pujols et al., 2007). Il ruolo di hGRβ risulta tuttavia controverso, dal momento che studi ancora più recenti hanno evidenziato che questa isoforma sopprime la trascrizione genica di IL5 e IL13 tramite reclutamento del complesso istone deacetilasi, in maniera del tutto simile al recettore hGRα (Kelly et al., 2008). Inoltre, non si osserva traslocazione nel nucleo e hGRβ
risulta avere un’attività trascrizionale specifica su geni distinti da quelli su cui agisce hGRα (Kino et al., 2009).
La ridotta attività ed espressione di HDAC2 (Hew et al., 2006), il difetto di traslocazione di hGRα provocato dalla fosforilazione indotta dalle MAP-chinasi (Irusen et al., 2002) e da un’abnorme attività dell’istone acetilasi (Matthews et al., 2004), la trans-repressione mediata da NF-kB e la down-regulation omologa risultano essere meccanismi responsabili della resistenza alla terapia (Schaaf e Cidlowski, 2002).
1.2.2 β
2agonisti e desensibilizzazione recettoriale
I farmaci β2 agonisti utilizzati in terapia sono suddivisi in: Short acting (salbutamolo e terbutalina). Sono i farmaci di scelta per risolvere le riacutizzazioni e per prevenire la broncocostrizione indotta dall’esercizio fisico, mentre l’uso cronico risulta inefficace ed indica un deterioramento della patologia, quindi la necessità di una terapia di fondo con corticosteroidi.
Long acting (salmeterolo e formoterolo). Sono utilizzati in associazione con i glucocorticoidi nella terapia di fondo, poiché migliorano la funzionalità respiratoria e riducono il numero di esacerbazioni in soggetti resistenti ai farmaci sintomatici e ai corticosteroidi in monoterapia. In combinazione con i glucocorticoidi consentono di utilizzare un dosaggio di corticosteroidi inferiore rispetto a quello usato nella monoterapia. In particolare il formoterolo, in associazione con la budesonide, può essere utilizzato sia nel mantenimento che nella prevenzione e risulta essere più efficace della tradizionale terapia che prevede l’uso di β2-agonisti short acting al bisogno in associazione con un corticosteroide (Korn et al., 2008).
I recettori β2 sono localizzati non solo sulla muscolatura liscia, ma anche su cellule epiteliali ed endoteliali e sui mastociti. I β2-agonisti agiscono legandosi al recettore β2 adrenergico (β2AR), caratterizzato da sette domini transmembranari e accoppiato alla proteina Gs. Una volta attivata la proteina Gs, la subunità α promuove una serie di eventi che comprendono l’attivazione dell’adenilato ciclasi, la formazione di AMP ciclico e l’attivazione delle PKA. Le PKA sono direttamente responsabili dell’iperpolarizzazione dei miociti attraverso
la fosforilazione ed attivazione dei canali del K+-ATP dipendenti (Shi et al., 2007), delle Ca2+-ATPasi citoplasmatiche e sarcoplasmatiche e degli scambiatori Na+-K+ e Na+-Ca2+, che contribuiscono all’estrusione del Ca2+
dalla cellula e al rilasciamento (Mueller et al., 1979). L’AMP ciclico, inoltre, attiva le proteine chinasi G (Torphy, 1994) e studi più recenti dimostrano la diretta partecipazione del cGMP alla riduzione del Ca2+ intracellulare ed al rilasciamento muscolare (Carvajal et al., 2000).
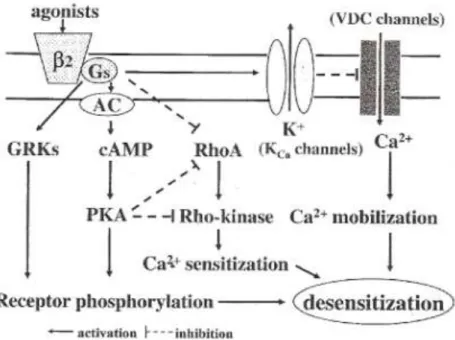
All’azione rilasciante contribuisce anche un meccanismo cAMP-indipendente che consiste nella diretta stimolazione da parte della subunità Gsα dei canali BK dipendenti dal Ca
2+
(Kume et al., 1994). Il recettore β2 non solo antagonizza la mobilizzazione del Ca2+, ma anche l’iperresponsività cellulare, e con meccanismi che prescindono da quelli appena elencati. Infatti, la PKA e la subunità Gs possono entrambe bloccare la proteina RhoA e quindi la RhoA chinasi, responsabile dell’inibizione della miosina fosfatasi (Kume, 2008). Tuttavia, l’inibizione della proteina RhoA e l’attivazione della PKA risultano essere secondari all’attivazione diretta della miosina fosfatasi
da parte della subunità Gs (Figura 5) (Janssen et al., 2004; Oguma et al., 2006).
Figura 5. Meccanismi molecolari coinvolti nella desensitizzazione del
recettore β2 adrenergico.
Inoltre, i β2 agonisti modulano la trascrizione genica tramite l’attivazione della PKA e la fosforilazione del CREB (cAMP response element binding protein), il quale lega il CPB e la proteina p300, che attiva o inibisce la trascrizione di alcuni geni coinvolti nell’infiammazione. Con questo meccanismo, oltre a mediare la broncodilatazione, i β2-agonisti risultano avere altri benefici effetti
nell’asma, tra i quali l’aumento della funzione mucociliare, la diminuzione della permeabilità dell’endotelio vascolare, la stabilizzazione dei mastociti e la modificazione del rilascio di neurotrasmettitori (Broadley et al., 2006). In aggiunta, proteggono le vie aeree dal rimodellamento (Tattersfield, 1987).
La stimolazione del recettore per brevi periodi provoca desensibilizzazione dovuta a fosforilazione, reversibile entro pochi minuti per allontanamento dell’agonista (Johnson, 2006). Tra i fattori responsabili della fosforilazione del recettore β2 e del legame con le β-arrestine, che mediano l’internalizzazione del recettore, sono non solo le PKA, ma anche PKC, e βAR chinasi (Pitcher et al., 1992).
La stimolazione protratta provoca invece down-regulation indipendente dalla fosforilazione, che consiste nel blocco trascrizionale della sintesi del recettore. Si osserva anche una significativa riduzione dei livelli di cAMP, specie in presenza di agonisti puri come l’isoproterenolo (Shore et al., 2003).
Nei mastociti la desensibilizzazione risulta molto più accentuata rispetto alle vie aeree, tanto da ipotizzare che la resistenza alla
broncodilatazione sia mediata largamente dai recettori β2 dei mastociti (Johnson, 2006).
La desensibilizzazione operata da altri recettori o mediatori che utilizzano cAMP come secondo messaggero è anche definita eterologa. E’ questo il caso di agenti contratturanti che agiscono sul recettore M3 attivando la PKC, o delle citochine proinfiammatorie che inducono la COX-2 ed il rilascio di prostanoidi, i quali attivano le PKA. Tra i mediatori anche la fosfolipasi e il fattore di aggregazione piastrinica (PAF).
La desensibilizzazione omologa è, invece, quella provocata dai β2 -agonisti ed è mediata dalla fosforilazione e dall’internalizzazione del recettore.
La resistenza alla terapia con β2-agonisti è dovuta frequentemente alla desensibilizzazione recettoriale, che è stata correlata anche alla presenza di variazioni geniche. Alcuni studi hanno invece riscontrato una riduzione del numero di recettori β ed un aumento di recettori α nelle vie aeree (Agrawal, 1990), suggerendo la presenza di uno squilibrio recettoriale. Ma è anche noto che alcune citochine, tra cui IL-1β, aumentano l’espressione dei recettori M1 accoppiati alle
proteine Gi, le quali inibiscono l’adenilato ciclasi, sia in vivo che in vitro (Hakonarson et al., 1996).
Studi più recenti hanno attribuito un ruolo cruciale al segnale mediato da ERK1/2 nell’attivazione di NF-kB, che determina una diminuzione dell’effetto rilasciante indotto da β2 agonisti e un aumento della responsività a stimoli contratturanti, delineando un fenotipo proasmatico (Shan et al., 2006).
1.2.3 Polimorfismi associati al recettore β
2Tra i polimorfismi a singolo nucleotide (SNP) associati al recettore β2 ne sono stati individuati tre capaci di modulare l’effetto terapeutico degli agonisti: le sostituzioni dell’arginina con la glicina nel codone 16 (Gly16), della glutammina con l’acido glutammico nel codone 27 (Glu27) e della treonina con l’isoleucina nel codone 164 (Ile164).
Il polimorfismo Gly16 risulta avere una frequenza del 65% rispetto ad Arg16 e studi clinici hanno dimostrato un’associazione con l’iperresponsività e fenomeni di riesacerbazione notturna più frequenti (Hall, 1996). Studi in vitro hanno caratterizzato un maggior grado di
desensibilizzazione per internalizzazione del recettore in seguito a trattamenti prolungati con isoproterenolo, pari anche al 96% (Liggett, 2000). Al contrario, lo SNP Glu27 è associato ad una maggiore resistenza alla down-regulation (Weglarz et al., 2003) e di conseguenza ad una migliore risposta terapeutica.
Un recente studio (Stephens et al., 2001) ha associato i singoli polimorfismi ed individuato aplotipi di particolare interesse predittivo della risposta al trattamento e tra questi, l’aplotipo Gly16-Gln27 risulta associato ad una riduzione del numero di recettori β2 espressi (Green et al., 1995).
Il polimorfismo Ile164 è molto più raro dei precedenti (circa 1%), ma interessa il legame con gli agonisti adrenergici e riduce la capacità del recettore di attivare l’adenilato ciclasi (Green et al., 1993). Nonostante la bassa incidenza, tale polimorfismo ha destato interesse in quanto associato ad una riduzione della broncodilatazione in soggetti trattati con farmaci β2 agonisti (Tantisira et al., 2005).
1.2.4 Possibili miglioramenti nella terapia di fondo
I β2 agonisti ed i corticosteroidi sono ritenuti farmaci cardine della terapia dell’asma in tutti gli stadi di malattia.
L’uso cronico di β2 agonisti in regime di monoterapia è associato, oltre che alla desensibilizzazione recettoriale, anche ad un peggioramento dell’iperresponsività bronchiale agli stimoli contratturanti (Lommatzsch et al., 2009). È stato inoltre riscontrato un piccolo ma significativo aumento della mortalità per cause respiratorie con l’impiego di salmeterolo, specie nella popolazione Afro-Americana (Nelson et al., 2006). Inoltre, uno studio ha rilevato che la gravità dell’asma può peggiorare con l’uso regolare di salbutamolo, in soggetti con polimorfismo Gly16 (Wechsler et al., 2006).
La combinazione dei β2 agonisti con i corticosteroidi risulta molto efficace nel ridurre gli svantaggi e gli effetti collaterali dei β2 agonisti, garantendo allo stesso tempo un rapido controllo dei sintomi ed una riduzione delle dosi di glucocorticoidi che verrebbero utilizzate in monoterapia nelle fasi iniziali di trattamento (Bateman et al., 2004). Questo emerge anche dallo studio FACET, che mette a confronto la combinazione formoterolo-budesonide con la somministrazione di una
dose doppia del cortisonico (Pauwels et al., 1997). Inoltre, i glucocorticoidi sembrano ridurre la sopravvivenza degli eosinofili, al contrario dei β2 agonisti (KanKaanranta et al., 2000).
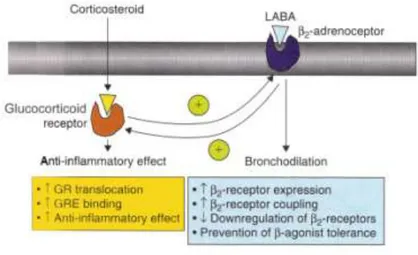
L’efficacia di questa combinazione è dovuta particolarmente ai corticosteroidi, che attraverso la transattivazione provocano un aumento della trascrizione del gene che codifica per i recettori β2, sia in vitro (Mak et al., 1995) che in vivo (Baraniuk et al., 1997). Inoltre promuovono il legame tra il recettore β2 e le proteine Gs, antagonizzando l’effetto delle GPR chinasi (βARK) attivate dall’IL-1β (Mak et al., 2002). Gli agonisti β2, invece, aumentano la traslocazione del complesso glucocorticoide-GR nel nucleo e attivano sinergicamente la C/EBPα, la quale stimola la proteina p21, ad attività antiproliferativa (Figura 6) (Roth et al., 2002).
Un recente studio sui ratti ha anche dimostrato che i glucocorticoidi migliorano l’iperresponsività indotta dall’acetilcolina inibendo l’upregulation della proteina RhoA, così da incrementare l’effetto dei β2 agonisti (Chiba et al., 2008).
Figura 6. Interazione tra glucocorticoidi e LABA
L’utilizzo cronico di farmaci β2 antagonisti ha dimostrato di ridurre l’iperresponsività alla metacolina in un modello animale di asma (Callaerts-Veg et al., 2004). In linea con questo studio, il trattamento cronico con nadololo, un antagonista β aspecifico, comporta la up-regulation dei recettori β2 , l’aumento della risposta broncodilatatoria agli agonisti dei recettori IP ed EP2 e la down-regulation dell’espressione di alcune proteine che causano la contrazione nelle vie aeree, quali Gi, PDE4D e PLCβ1 (Lin et al., 2008).
In questo senso, le nuove strategie terapeutiche si basano sullo sviluppo di β2 agonisti ultra long acting (ultra-LABA) e di corticosteroidi con ridotti effetti sistemici secondari.
Tra gli ultra-LABAs, sono attualmente in sviluppo clinico l’indacaterolo ed il carmoterolo, la cui durata d’azione è di 24 ore. L’indacaterolo sembra non sviluppare tolleranza (Laforce et al., 2008) e risulta un effettivo broncodilatatore delle piccole vie aeree, il che prospetta un suo utilizzo favorevole nell’asma grave (Sturton et al., 2008). E’ plausibile che la loro somministrazione, in associazione con i glucocorticoidi, comporterà un miglioramento della compliance dei pazienti asmatici (Matera et al., 2007).
I nuovi corticosteroidi in sviluppo mirano a ridurre l’assorbimento sistemico a livello polmonare e ad aumentare la biodisponibilità per somministrazione orale. La ciclesonide, un profarmaco di nuova introduzione che viene bioattivato dalle esterasi polmonari, risulta privo di effetti collaterali sistemici. Questa elevata tollerabilità e specificità d’azione sembra essere correlata alla prolungata ritenzione del farmaco a livello polmonare, alla sua ridotta biodisponibilità e
all’elevato grado di legame con le proteine plasmatiche (Nave et al., 2004).
Un altro approccio è rappresentato dalla separazione dell’effetto antinfiammatorio legato alla trans-attivazione e alla trans-repressione, dagli effetti collaterali dovuti alla cis-repressione. Questa strategia è teoricamente possibile, poiché gli effetti collaterali sono mediati principalmente da un meccanismo genomico, mentre l’effetto antinfiammatorio è dovuto all’inibizione dei fattori di trascrizione attraverso un meccanismo non genomico (Schacke et al., 2004). Sono in studio anche attivatori dell’enzima nucleare HDAC2, che andrebbero a coadiuvare l’effetto antinfiammatorio dei corticosteroidi (Barnes e Adcock, 2003).
1.2.5 Ruolo dei PPARs nelle malattie respiratorie
Gli agonisti PPARγ hanno dimostrato di possedere effetti antinfiammatori ed immunomodulatori tali da renderli utilizzabili nel trattamento dell’asma, specie nei casi in cui la terapia convenzionale con corticosteroidi e β2 agonisti sia poco efficace o possa causare
effetti collaterali particolarmente rilevanti, ad esempio in pazienti pediatrici (Spears et al., 2006).
I PPARγ sono recettori citosolici che formano un eterodimero con il recettore dei retinoidi (RXR) e traslocano nel nucleo, dove riconoscono specifiche sequenze nelle regioni promotrici dei geni bersaglio (PPRE-RXRE). Su di essi agiscono i tiazolidindioni, utilizzati nel trattamento del diabete di tipo 2, e tra i ligandi endogeni abbiamo la PGD2 e il suo prodotto 15d-PGJ2 (Scher e Pillinger, 2009), il LTB4 (Obata et al., 1987), gli acidi polinsaturi come l’acido arachidonico e gli acidi eicosapentaenoici (Willson et al., 2000). Esperimenti in vitro hanno dimostrato che anche alcuni antagonisti del recettore CysLT1, tra cui il pranlukast, risultano avere attività agonista nei confronti dei PPARγ (Kojo et al., 2003).
I recettori sono localizzati nella mucosa respiratoria, su eosinofili, macrofagi, epitelio e muscolatura liscia delle vie aeree, ed è stato osservato un incremento dell’espressione di questi recettori nei soggetti asmatici (Benayoun et al., 2001). L’ipotesi è che la sovraespressione dei PPARγ possa rappresentare un meccanismo
compensatorio che facilita l’effetto antinfiammatorio dei ligandi endogeni.
L’effetto antinfiammatorio è mediato principalmente dall’inibizione del rilascio di citochine da parte dei monociti e dall’induzione dell’apoptosi nei linfociti T (Belvisi et al., 2006). Tuttavia, gli agonisti PPARγ agiscono anche con un meccanismo non genomico, antagonizzando fattori trascrizionali quali NF-kB, C/EBP e le proteine STAT e AP-1 (Ricote et al., 1998).
In molti casi resta da chiarire se l’effetto degli agonisti PPAR sia recettore-mediato e quali vantaggi possano avere rispetto ai farmaci in uso. Infatti, è stato recentemente dimostrato che nelle reazioni allergiche di tipo I, i tiazolidindioni, agonisti PPARγ utilizzati nel trattamento del diabete mellito, bloccano la produzione dei cisteinil-leucotrieni nei mastociti in maniera simile allo zileuton, inibitore della 5-lipossigenasi (5-LO). Tuttavia, l’osservazione che la produzione del LTC4 viene inibita anche da parte di un antagonista dei PPARγ rende incerto il contributo di questi recettori nella produzione di CysLTs nei mastociti (Yamashita, 2008).