2
INDICE
Prefazione ... 3
1. La struttura dei mitocondri ... 4
2. Origine e biogenesi ... 8
3. Le funzioni dei mitocondri ... 10
3.1 Il ruolo dei mitocondri nelle ossidazioni cellulari ... 11
3.2 La respirazione cellulare e la fosforilazione ossidativa ... 12
4. Disfunzione mitocondriale primaria e secondaria ... 16
5. Meccanismi d’azione su cui si basa la farmacologia mitocondriale ... 22
5.1 Molecole bioattive che agiscono sui mitocondri ... 23
5.2 Farmaci che modulano bersagli e processi mitocondriali ... 29
5.3 Agenti farmacologici che agiscono indirettamente sui mitocondri ... 32
6. I peptidi Szeto-Shiller (SS) ... 36
6.1 Attività oppioide di SS-02 ... 39
6.2 Sintesi di analoghi SS-02 non oppioidi ... 40
6.3 Assorbimento mitocondriale di SS-31 e SS-20 ... 44
6.4 Attività mitocondriali dei peptidi SS ... 44
7. I composti TP e le cellule tumorali pancreatiche ... 47
Conclusioni ... 59
Glossario ... 61
Bibliografia ... 63
Ringraziamenti ... 70
3
Prefazione
Il termine mitocondrio è stato utilizzato per la prima volta nel 1898 da Carl Benda e deriva da due parole greche, mítos 'filo', e cóndrion 'piccolo grano'. Il mitocondrio è stato definito nel 1907 come organulo presente nelle cellule, a struttura microscopica altamente differenziata, preposto alle funzioni di respirazione e di produzione di energia. Solo dopo le scoperte negli anni 1948-1950 da parte di Eugene Kennedy e Albert Lehninger che il ciclo di Krebs, l'ossidazione degli acidi grassi e la fosforilazione ossidativa hanno luogo nei mitocondri, è stata definita la conoscenza della funzione fondamentale del mitocondrio: la respirazione cellulare nella quale molecole derivate dal metabolismo di zuccheri, proteine e lipidi sono ossidate con formazione di acqua e anidride carbonica e l'energia derivante dalle ossidazioni
biologiche si rende disponibile sotto forma di calore e di ATP. A tutt'oggi la ricerca sulla struttura e funzione dei mitocondri non è conclusa; va
assumendo sempre crescente importanza lo studio sul loro ruolo in molti ambiti della biologia e della medicina.
Negli ultimi dieci anni, è cambiata drasticamente la visione dei biologi sui mitocondri definiti inizialmente come “centrali energetiche della cellula” ora come organuli
intracellulari responsabili non solo della vita ma anche della morte cellulare. I mitocondri hanno un ruolo fondamentale nella morte programmata (apoptosi), ed
essendo l'apoptosi determinante nella cancerogenesi e nell'instaurarsi delle malattie degenerative, ne deriva che la corretta funzionalità dei mitocondri è fondamentale per garantire la salute dell'organismo.
4
1. La struttura dei mitocondri
I mitocondri sono organuli sferici o allungati più o meno delle dimensioni di un batterio,
del diametro di circa 0,5 µm e lunghi circa 1-2 µm. Nella maggior parte delle cellule, ad
eccezione dei batteri, delle alghe blu-verdi e di alcune cellule che nel differenziamento
hanno subito particolari modificazioni (i globuli rossi dei mammiferi) ne sono presenti in media da 500 a 1000; il numero dipende direttamente dall’energia che ogni
cellula/organo deve produrre per svolgere le sue funzioni. I mitocondri si muovono
liberamente nel citoplasma e tendono ad addensarsi nei punti dove è maggiore la
richiesta di energia ( per esempio, nelle fibre muscolari, circondano le miofibrille).
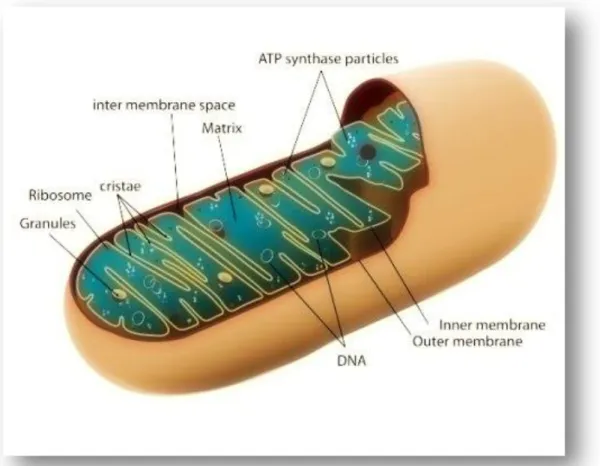
Ogni organulo è delimitato da una doppia membrana, la membrana mitocondriale
esterna (OMM) a contatto con il citoplasma e la membrana mitocondriale interna
(IMM) che si introflette in pieghe dette creste. La forma delle creste varia nei diversi tessuti, a volte sono corte e trasversali rispetto all’asse maggiore del mitocondrio, altre
volte sono allungate e parallele all’asse maggiore del mitocondrio (creste tubulari).
Lo spazio tra la membrana esterna e quella interna è chiamato compartimento o spazio
intermembrana, mentre quello racchiuso dalla IMM è detto matrice.
Nella matrice, di consistenza gelatinosa, sono presenti granuli costituiti
prevalentemente da sali di calcio e ribosomi di struttura simile a quella dei batteri.
Oltre a RNA ribosomiale si trovano RNA messaggero e una piccola frazione di RNA 4S
funzionante come RNA transfer. Gli RNA mitocondriali vengono trascritti da un DNA
circolare, il cromosoma mitocondriale, lungo circa 5 µm e presente in copie multiple.
Il DNA mitocondriale (mtDNA) non ha niente in comune con il DNA nucleare della
cellula; viene trasmesso di generazione in generazione per via materna, dato che i
5
La presenza della catena di trasporto degli elettroni con la sua capacità di produrre
radicali liberi, la mancanza di istoni ed i limitati sistemi di riparo rendono il DNA
mitocondriale dei mammiferi facilmente danneggiabile, subendo mutazioni con una
frequenza 10 volte superiore a quella dei geni nucleari. Il progressivo accumulo di
queste mutazioni contribuisce forse al decadimento con l’età della capacità respiratoria dei muscoli e di altri tessuti. Alcune mutazioni possono causare “malattie
mitocondriali” che colpiscono isolatamente o in combinazione il sistema nervoso, il
cuore, i muscoli scheletrici, il rene e altri organi.
La matrice contiene circa il 50% delle proteine mitocondriali: oltre agli enzimi preposti
ai processi di duplicazione, trascrizione e traduzione del DNA, sono presenti gli enzimi
coinvolti nella liberazione degli atomi di idrogeno da utilizzare per la respirazione
cellulare. Le proteine del mitocondrio sono codificate solo in minima parte dal DNA
mitocondriale; queste sono localizzate prevalentemente sulla parte interna delle creste.
La maggior parte delle proteine mitocondriali, come il citocromo c, sono codificate dal
DNA nucleare, sintetizzate sui ribosomi citoplasmatici e dotate di una sequenza segnale
di 20-28 amminoacidi che le indirizza verso il mitocondrio.
La membrana interna (IMM), molto più estesa di quella esterna per la presenza delle creste, differisce da tutte le altre membrane della cellula per l’assenza di colesterolo nel
suo scheletro lipidico. Il colesterolo è sostituito da un fosfolipide acido, la cardiolipina.
Questa speciale composizione rende la IMM altamente impermeabile a tutte le molecole
inclusi i piccoli cationi. I quattro complessi proteici della catena respiratoria (complessi
I-IV) risiedono sulle creste e inoltre, nello spessore della membrana interna, sono
presenti numerose proteine che funzionano da trasportatori di idrogeno e di elettroni
della catena respiratoria, come i citocromi a, a3, b, c, c1, le ferro-solfoproteine (FeS) e la
6
La faccia della membrana interna rivolta verso la matrice è ricoperta da sferule,
chiamate particelle elementari o particelle F1, ancorate alla membrana con uno stelo
lungo 3-4 nm (particelle Fo; la lettera o significa sensibile alla oligomicina). Queste
strutture costituiscono nel loro insieme la FoF1ATPasi, enzima chiave del processo
della fosforilazione ossidativa. Inoltre è presente una serie di proteine con funzioni di
trasporto per diverse sostanze, come le permeasi per il fosfato inorganico, le proteine
del sistema scambiatore di ATP per ADP che trasporta all’esterno del mitocondrio una molecola di ATP e contemporaneamente una molecola di ADP all’interno e le proteine
trasportatrici degli acidi dicarbossilici e tricarbossilici.
La membrana esterna (OMM) ha una struttura molto simile alla membrana plasmatica
e a quella degli altri organuli cellulari. A differenza di queste, è dotata di grande permeabilità in quanto contiene dei “ pori”, costituiti da proteine del PM di circa 30.000
dalton dette porine. I canali formati dalle porine, conosciuti anche come canali anionici
voltaggio-dipendenti (VDACs), possono essere attraversati da piccole molecole con PM
non superiore a 10.000 dalton.
Lo spazio intermembrana è attraversato da strutture simili alle gap junctions che
riuniscono in qualche tratto le due membrane mitocondriali in modo da garantirne l’integrità strutturale (Chieffi G. et al., 2000; Fig. 1).
7
8
2. Origine e biogenesi dei mitocondri
I mitocondri si riproducono per divisione di mitocondri preesistenti: crescono e si
dividono in un processo coordinato che richiede il contributo di due sistemi genetici separati, quello dell’organulo e quello del nucleo cellulare. I mitocondri sono organuli
“semiautonomi” che contengono un proprio DNA e tutto l’apparato per la sintesi
proteica, inclusi i ribosomi.
Il genoma mitocondriale (mtDNA) contiene 37 geni che codificano per 2 RNA
ribosomiali (rRNA), 22 RNA transfer (tRNA) e 13 polipeptidi che vanno a costituire i complessi enzimatici della catena respiratoria e l’ATPasi. In ogni mitocondrio ci sono
da due a dieci copie di mtDNA. Le altre proteine mitocondriali sono codificate da geni
nucleari, tradotte nel citoplasma e poi importate nei mitocondri attraverso il traslocatore
della membrana interna (TIM) e quello della membrana esterna (TOM) dove
svolgono funzioni di enzimi di matrice, proteine di membrana e fattori e enzimi
coinvolti nella replicazione, trascrizione e sintesi.
La sorprendente somiglianza dei mitocondri ai batteri ha fatto avanzare l’ipotesi dell’origine simbiontica dei mitocondri.
Le principali somiglianze riguardano la sede degli enzimi della catena respiratoria,
localizzati nella membrana plasmatica dei batteri e nella IMM. Inoltre la membrana
plasmatica dei batteri presenta delle introflessioni (mesosomi) simili alle creste dei
mitocondri e il DNA mitocondriale è circolare come quello batterico. Da queste osservazioni è nata l’ipotesi che la IMM e la matrice rappresenterebbero il simbionte di
origine che sarebbe poi stato avvolto dalla membrana plasmatica e inglobato nella
9
Figura 2. Biogenesi mitocondriale (treccani.it)
10
3. Le funzioni dei mitocondri
I mitocondri sono in grado di svolgere molteplici funzioni, la più importante delle quali
è la produzione di energia utilizzando i principali prodotti della glicolisi, il piruvato e il
NADH, rispettivamente attraverso il ciclo di Krebs e la fosforilazione ossidativa
associata al trasporto di elettroni lungo la catena respiratoria durante la respirazione
cellulare. Per questo motivo i mitocondri sono definiti le “centrali energetiche” della cellula. Gli organuli intervengono anche nell’apoptosi o morte cellulare programmata,
un processo fisiologico che porta all’autodistruzione della cellula, nella modulazione dei
flussi di calcio in tutta la cellula e nella regolazione dello stato redox della cellula.
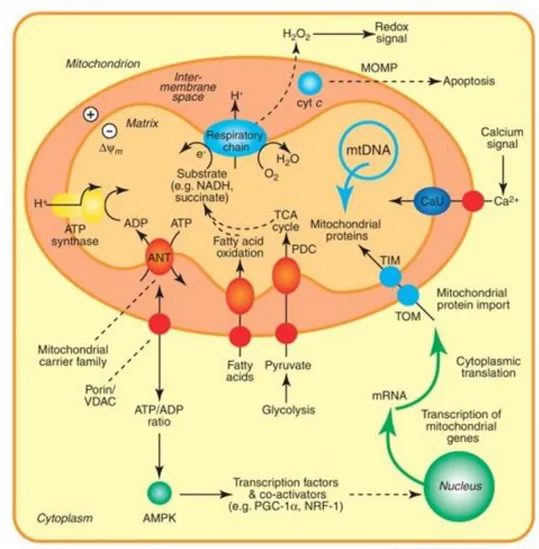
Figura 3. Funzioni dei mitocondri (Smith et al., Mitochondrial pharmacology, Trends in Pharmacological Sciences, 2012)
11
I mitocondri svolgono un ruolo cruciale nell’apoptosi; infatti quando si verificano
segnali apoptotici la membrana esterna viene alterata, attraverso l’apertura di un poro,
determinando la permeabilizzazione della membrana mitocondriale esterna (MOMP)
che porta al rilascio di citocromo c e di molte altre proteine pro-apoptotiche dallo spazio
intermembrana al citosol, dove attivano la morte cellulare per apoptosi.
Gli organuli si muovono intorno alla cellula coordinati dal citoscheletro, e inoltre sono
sottoposti continuamente ai processi di fusione e fissione. L’equilibrio tra questi due
processi è indispensabile perché il loro numero e la loro morfologia all’interno della
cellula siano quelli corretti e le funzioni mitocondriali possano svolgersi normalmente.
La frammentazione o fissione è anche la tappa iniziale del processo di apoptosi ed è
legata alla rimozione dei mitocondri disfunzionali.
In risposta all’aumento della concentrazione di calcio citosolico, i mitocondri
promuovono l’ingresso di calcio nella matrice mitocondriale attraverso il canale a
uniporto del calcio (CaU).
I mitocondri sono anche responsabili della generazione di specie reattive dell’ossigeno
(ROS), principale causa del danneggiamento del DNA che porta all’invecchiamento cellulare. In condizioni normali, durante la respirazione cellulare, una piccola
percentuale di ossigeno può essere ridotta da uno, due o tre elettroni con formazione di
anione superossido, perossido di idrogeno e radicale ossidrile (Smith R.A.J. et al., 2012).
3.1 Il ruolo dei mitocondri nelle ossidazioni cellulari
Nelle cellule eucariotiche, i mitocondri non sono solo la sede della respirazione
cellulare e della sintesi di ATP ad essa collegata; nella matrice mitocondriale sono
12
composti, vie che generano la gran parte dei coenzimi ridotti utilizzati poi nella respirazione cellulare. Gli acidi grassi, derivati dall’idrolisi dei trigliceridi, vengono
trasportati nei mitocondri, dove vanno incontro al processo della β-ossidazione che
ossidando parzialmente le catene carboniose di questi acidi, le frammenta in unità a due
atomi di carbonio (acetili) legate ad un particolare coenzima, il coenzima A,
producendo acetil-coenzima A e coenzimi ridotti. D’altra parte il piruvato, formatosi nel
citoplasma in seguito alla parziale demolizione del glucosio ad opera della glicolisi,
anziché venir ridotto ad acido lattico, come nella fermentazione lattica, può essere
trasportato nei mitocondri, dove viene ulteriormente ossidato (generando NADH) e
decarbossilato ad opera di un complesso enzimatico presente nella matrice
mitocondriale (il complesso della piruvato deidrogenasi) dando origine anch’esso ad
acetil- coenzima A.
L’acetil-coenzima A, prodotto da queste due vie, può infine essere completamente
ossidato attraverso una via ciclica, il ciclo degli acidi tricarbossilici o ciclo di Krebs, i
cui enzimi sono presenti nella matrice mitocondriale. Questa via genera, oltre a 2
molecole di anidride carbonica, anche 3 molecole di NADH e una di FADH2 per ogni
molecola di acetil-coenzima A ossidata.
Attraverso tutte queste vie metaboliche all’interno del mitocondrio si genera una grande
quantità di coenzimi ridotti che, grazie al fatto di essere prodotti nella matrice
mitocondriale, sono immediatamente disponibili per la riossidazione da parte della
catena respiratoria assicurando la massima efficienza a tutto il sistema di produzione dell’energia all’interno della cellula (Chieffi G. et al., 2000).
3.2 La respirazione cellulare e la fosforilazione ossidativa
Il processo di trasferimento degli elettroni dai coenzimi ridotti all’ossigeno prende il
13
enzimi (raggruppati in quattro complessi principali formati da proteine integrali di
membrana indicati come I, II,III,IV) e di molecole trasportatrici di elettroni più piccole
che fanno da spola tra di essi (ubichinone o coenzima Q e citocromo c). Questi elementi
nel loro insieme costituiscono la catena respiratoria o catena di trasporto degli
elettroni (ETC). L’energia totale liberata nel passaggio di una coppia di elettroni dal
NADH all’ossigeno, che funge da accettore finale, è di circa 53 Kcal e questo grosso
salto energetico è suddiviso in una serie di salti più piccoli attraverso una serie di
reazioni di ossidoriduzione che si verificano a carico dei vari componenti della catena respiratoria che si passano gli elettroni e li portano gradualmente all’ossigeno, al quale
vengono infine ceduti (Chieffi G. et al., 2000). Gli elettroni provenienti dall’ossidazione
dei carboidrati attraverso il ciclo di Krebs e dalla scissione degli acidi grassi per
B-ossidazione si accumulano sul vettore NADH. Il NADH quindi viene ossidato a NAD+ a
livello del complesso I ( NADH-ubichinone ossidoreduttasi) e gli elettroni vengono
ceduti al pool del coezima Q, un vettore lipofilo di elettroni che esiste nella forma ossidata ubichinone e nella forma ridotta ubichinolo. L’energia rilasciata viene utilizzata
per pompare protoni (H+) attraverso la IMM. Gli elettroni dal succinato (ciclo di Krebs)
vengono invece ceduti al pool del coenzima Q a livello del complesso II
(succinato-ubichinone ossidoreduttasi). Gli elettroni dal pool del CoQ vengono poi passati attraverso il complesso III (ubichinolo-citocromo c ossidoreduttasi) al citocromo c e
infine dal citocromo c, attraverso il complesso IV (citocromo c ossidasi), gli elettroni
vengono utilizzati per ridurre l’ossigeno (Smith R.A.J. et al., 2012). Durante il passaggio degli elettroni lungo la catena respiratoria, l’energia man mano
liberata nel corso delle ossidoriduzioni, viene utilizzata dai complessi I, III e IV per
pompare protoni fuori dal mitocondrio. Siccome la membrana interna è impermeabile ai
protoni e quindi questi non possono rientrare nel mitocondrio, il passaggio degli
14
protoni tra la matrice mitocondriale , dove la concentrazione diminuisce e il pH quindi diventa basico, e l’esterno del mitocondrio dove la concentrazione dei protoni aumenta
e il pH diventa acido. Si viene quindi a creare un gradiente di concentrazione di protoni
attraverso la IMM che rappresenta una forma di energia potenziale (energia
elettrochimica) utilizzata principalmente per la sintesi di ATP attraverso il processo
della fosforilazione ossidativa. Infatti nella IMM è inserita una proteina, la FoF1
ATPasi. Questa proteina è una pompa che trasporterebbe protoni dall’interno all’esterno del mitocondrio utilizzando l’energia derivata dall’idrolisi dell’ATP (sarebbe
quindi un enzima che idrolizza ATP, cioè una ATPasi). Tuttavia, a causa dell’elevato
gradiente di concentrazione di protoni generato dal trasporto degli elettroni, la FoF1
ATPasi è costretta a funzionare alla “rovescia”: i protoni, sotto la spinta del gradiente rivolto verso l’interno, rientrano nel mitocondrio attraverso la pompa e, così facendo,
forniscono l’energia per la sintesi di ATP a partire da ADP e fosfato inorganico. Il Pi
viene importato dal citosol attraverso un sistema simporto Pi-H+. L’ATP viene poi trasportato dal mitocondrio al citoplasma in cambio di ADP citoplasmatico dall’adenina
nucleotide traslocasi (ANT).
Questo meccanismo viene indicato con il nome di meccanismo chemioosmotico di
sintesi dell’ATP. La sintesi di ATP può continuare finché continua il trasporto degli
elettroni lungo la catena respiratoria che ripristina il gradiente di pH che altrimenti si
esaurirebbe. I due processi della respirazione cellulare e della fosforilazione ossidativa
sono strettamente accoppiati; infatti, da un lato, se termina il trasporto degli elettroni,
cessa anche la sintesi di ATP (in quanto viene a mancare l’energia necessaria, rappresentata dal gradiente di protoni generato dal trasporto di elettroni), ma dall’altro
lato, quando la fosforilazione ossidativa non può procedere per mancanza di ADP ( in
15
Quest’ultimo fenomeno prende il nome di controllo respiratorio ed ha grande
importanza biologica. La carenza di ADP indica infatti che la cellula ha già fatto il
pieno di energia e sarebbe quindi uno spreco riossidare il NADH e il FADH2 dissipando l’energia potenziale in essi contenuta. In queste condizioni i coenzimi delle
ossidoriduzioni si accumulano in forma ridotta e quindi anche le ossidazioni dei vari
composti organici vengono arrestate per mancanza di coenzimi da ridurre. Il tutto rappresenta per l’organismo la garanzia della massima economia di energia (Chieffi G.
et al., 2000).
16
4. Disfunzione mitocondriale primaria e secondaria
La disfunzione mitocondriale può essere suddivisa in due tipi: primaria e secondaria.
La disfunzione primaria è caratterizzata dalla mutazione in un gene del mtDNA o in un
gene nucleare che codificano per una proteina mitocondriale o per una tossina mitocondriale. Un esempio è l’Encefalopatia mitocondriale con acidosi lattica ed
episodi ictus-simili (MELAS) dovuta a una mutazione puntiforme (transizione
nucleotidica A3243G) nel gene mitocondriale del tRNA della Leucina (UUR) che porta
al difettoso assemblaggio dei complessi della fosforilazione ossidativa e a conseguenti
difetti nel metabolismo energetico nei sistemi neuromuscolari (Goto Y. et al., 1990).
Anche le mutazioni di geni nucleari codificanti proteine mitocondriali portano ad una
vasta gamma di difetti mitocondriali primari, come un difetto neonatale nel
metabolismo energetico dovuto alla mutazione di un gene nucleare che codifica per
NDUFAF3, un fattore di assemblaggio per il complesso I della catena respiratoria
(Angelini C., 2009; Saada A., 2009; McKenzie M., 2010).
La disfunzione secondaria invece è causata da eventi patologici provenienti dall'esterno
dei mitocondri. Per esempio, il danno da ischemia / riperfusione (I/R), che si verifica
quando la circolazione sanguigna torna al tessuto dopo un periodo di ischemia, causa
una forte perturbazione mitocondriale secondaria e conseguente danno tissutale
(Murphy E., 2011). Il danno mitocondriale secondario svolge un ruolo significativo
anche in altri disturbi tra cui sepsi, neurodegenerazione, sindrome metabolica, trapianto
di organi, cancro, malattie autoimmuni e diabete (Wallace D.C., 2010). Di conseguenza,
i mitocondri sono un nodo importante di intervento terapeutico, anche se il danno all’effettivo organulo non è l'evento patologico iniziale. Le malattie mitocondriali
primarie sono abbastanza rare. Al contrario, molti dei disturbi che coinvolgono la
disfunzione mitocondriale secondaria, come i danni cardiaci da lesioni I/R, sindrome
17
diffusi nelle società sviluppate. Quindi, c’è la necessità di trattare la disfunzione
mitocondriale in patologie sia primarie che secondarie.
La correzione delle malattie mitocondriali primarie è particolarmente impegnativa
perché, nella maggior parte dei casi, il trattamento per essere efficace richiederebbe la
sostituzione o addirittura la soppressione del gene difettoso. Ciò potrebbe essere valido
per i geni nucleari, ma la prospettiva di terapie geniche efficaci per le malattie dovute a
mutazioni nel mtDNA rimane lontana (Kyriakouli D.S., 2008). Un’eccezione è il deficit
di coenzima Q (CoQ), causato da un difetto nella biosintesi del CoQ, dove appunto il
semplice apporto di CoQ tramite la dieta migliora la malattia. Così, sempre più
interventi farmacologici mirano a migliorare le conseguenze del difetto primario
anziché affrontarne la causa. La situazione terapeutica invece è diversa per le numerose
malattie che determinano disfunzione mitocondriale secondaria, dove i trattamenti non
sono progettati per colpire direttamente i mitocondri. Questo può sembrare un quadro
sconfortante, suggerendo che i mitocondri siano un bersaglio terapeutico poco
promettente. Per fortuna però ci sono meccanismi comuni di danno cellulare nei disturbi
mitocondriali sia primari che secondari, nonostante le loro cause disparate.
La farmacologia mitocondriale allora è valida, perché le terapie che agiscono su alcune
vie dannose comuni sono in grado di trattare pazienti che presentano una vasta gamma
di malattie mitocondriali sia primarie che secondarie.
Ci sono tre aspetti del danno mitocondriale che contribuiscono generalmente alle
patologie mitocondriali primarie e secondarie: il danno ossidativo, la disomeostasi del
18
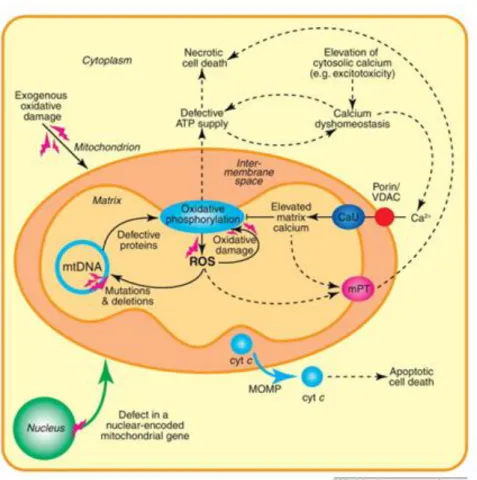
Figura 5. Disfunzioni mitocondriali (Smith et al., Mitochondrial pharmacology, Trends in Pharmacological Sciences, 2012)
La catena respiratoria mitocondriale, soprattutto il complesso I e III, è una delle
principali fonti di anione superossido (O2-) che, a sua volta, forma perossido di
idrogeno (H2O2) e altre specie reattive dell’ossigeno (ROS) dannose (Murphy M.P., 2009). Il complesso I rilascia superossido nella matrice, mentre il complesso III lo
rilascia sia nella matrice che nello spazio intermembrana. In molti scenari patologici,
aumenta la produzione di superossido e il mitocondrio è particolarmente vulnerabile al
danno ossidativo, perché l'organello contiene diversi centri ferro-zolfo (Fe-S), una
membrana interna molto estesa contenente acidi grassi insaturi (cardiolipina), proteine e
molecole di mtDNA che sono essenziali per la funzione mitocondriale. La
19
del citocromo c dalla IMM che, essendo una proteina cationica, si lega con alta affinità
alla cardiolipina anionica. La perdita di Cyt c dalla IMM riduce la sintesi di ATP mentre
aumenta ulteriormente la perdita di elettroni e la produzione di ROS. Le ROS
mitocondriali vengono normalmente rimosse da alcuni enzimi antiossidanti presenti nei
mitocondri come la Mn superossido dismutasi (SOD), che converte il superossido a
perossido di idrogeno, la glutatione perossidasi e la catalasi, che converte il perossido
di idrogeno ad acqua.
Quindi lo stress ossidativo si verifica quando c’è un’eccessiva produzione di ROS
oppure una diminuita capacità antiossidante endogena (i sistemi enzimatici antiossidanti
non funzionano più sotto stress ossidativo; Kono Y. et al.). Il danno ossidativo altera la funzione dell’organulo rendendo più probabile la morte della cellula e contribuendo in
tal modo a diverse patologie come la sepsi, il deterioramento di organi nel trapianto,
danno da I/R, complicanze diabetiche e anche malattie neurodegenerative (Duchen
20
La sintesi di ATP mitocondriale è spesso interrotta da danni alla catena respiratoria, alla membrana interna o all’enzima FoF1ATPasi, contribuendo così alla morte e alla
disfunzione della cellula. La difettosa produzione mitocondriale di ATP porta anche alla disomeostasi del calcio attraverso l’alterazione dell’attività della calcio ATPasi
presente nel reticolo endoplasmatico / sarcoplasmatico e nella membrana plasmatica, determinando l’aumento dei livelli di calcio citosolico al di sopra del normale intervallo
(1-2 µm; Mammucari C., et al, 2011). L'assorbimento di calcio nei mitocondri attraverso l’uniporter del calcio risponde agli aumenti di calcio citosolico, forse per
proteggere la cellula, ma il sostenuto aumento di calcio conduce al suo accumulo
dannoso e cronico all'interno dei mitocondri.
Il danno ossidativo, la difettosa sintesi di ATP e la disomeostasi del calcio si verificano
frequentemente insieme, e siccome ogni tipo di danno conduce agli altre due, poi si
stabilisce un circolo vizioso. Infine tutti e tre inducono la transizione della permeabilità
mitocondriale (mPT) mediante la formazione nella IMM di un poro che causa
rigonfiamento e alterazione della funzione mitocondriale (Rasola A. et al., 2011). Il
poro mPT è un canale a elevata conduttanza che si pensa sia formato dal VDAC sulla
OMM, dall’adenina nucleotide traslocasi sulla IMM e dalla ciclofilina D (CypD) nella matrice. L’ apertura del poro della mPT provoca un repentino aumento della
permeabilità della IMM, con conseguente rigonfiamento della matrice mitocondriale,
rottura della membrana esterna, disaccoppiamento mitocondriale e rilascio di citocromo
c nel citosol. La perdita di citocromo c mitocondriale ostacolerebbe il trasporto degli
elettroni, mentre il citocromo c citosolico è noto per innescare la cascata delle caspasi e l’apoptosi (Crompton M., 1999).
Questi eventi tossici si verificano in molte malattie mitocondriali primarie e secondarie.
21
a molte indicazioni diverse. Ciò è in contrasto con il solito modello di sviluppo dei
farmaci in cui una proteina bersaglio ben definita, che è associata ad una singola
indicazione, può essere modulata da una piccola molecola che si lega selettivamente.
Per cui intervenire terapeuticamente con successo nei processi dannosi generali è
essenziale per la farmacologia mitocondriale al fine di raggiungere il suo pieno
potenziale.
22
5. Meccanismi d’azione su cui si basa la farmacologia
mitocondriale
Ci sono tre meccanismi d’azione su cui si basa la farmacologia mitocondriale (Smith
R.A.J. et al., 2011). Il primo è quello di trovare molecole che si accumulano
selettivamente all'interno dei mitocondri. Il secondo è quello di utilizzare molecole che
legano i bersagli all'interno dei mitocondri. L'approccio finale è quello di modulare i
processi al di fuori dei mitocondri che alterano la funzione mitocondriale.
Figura 6. Tre meccanismi generali di intervento farmacologico
23
5.1 Molecole bioattive che agiscono sui mitocondri
Per riuscire a indirizzare farmaci e molecole bioattive verso i mitocondri in vivo, sono
stati sviluppati alcuni cationi lipofili e alcuni peptidi. Questi derivati infatti consentono una maggiore concentrazione del composto all’interno del mitocondrio, ne aumentano
la potenza e di conseguenza permettono l’utilizzo del composto ad una dose minore;
questa strategia minimizza il metabolismo extramitocondriale che può portare a
inattivazione del farmaco, alla sua escrezione o alla comparsa di effetti collaterali o
tossici. Questo meccanismo di trasporto consente anche a molecole che sono
scarsamente assorbite dai mitocondri per vari motivi (ad es. idrofobicità) di agire a
livello di mitocondri in vivo. Una limitazione del processo, però, è costituita dal fatto
che queste procedure coinvolgono sostanze chimiche che tendono a localizzarsi nella
matrice mitocondriale e sulla superficie della membrana interna rivolta verso la matrice
stessa. Ci sono molti processi che avvengono sulla superficie esterna della membrana
interna, nello spazio intermembrana e nella membrana esterna dei mitocondri, ma,
finora, non sono state sviluppate strategie adatte a interagire con questi compartimenti.
I cationi lipofili, come i derivati del trifenilfosfonio (TPP+), sono rapidamente e
ampiamente assorbiti dai mitocondri in vivo, guidati dal grande potenziale di membrana
mitocondriale [Δψm (interno negativo)];( Smith R.A.J., et al., 2011). Il meccanismo di
assorbimento è determinato dal movimento dei cationi lipofili attraverso la membrana
plasmatica e quella mitocondriale interna che si verifica grazie all'ampia superficie
idrofoba e al grande raggio ionico del catione che abbassa efficacemente l'energia di
attivazione per il passaggio della membrana. L'equazione di Nernst descrive che l’assorbimento di membrana potenziale-dipendente dei cationi lipofili aumenta di 10
volte per ogni ~ 60 mV di Δψm, portando alla centuplicazione del loro assorbimento
24
L'uso di cationi lipofili per facilitare il rilascio della porzione bioattiva, ad essi attaccata,
all'interno delle cellule è stato per la prima volta dimostrato con il catione lipofilo
rodamina 123 che forma un complesso con il farmaco antitumorale cisplatino (Teicher B.A. et al., 1987).
Anche i peptidi possono essere utilizzati per fornire molecole ai mitocondri. I peptidi
Szeto-Schiller (SS) ed i peptidi MPPs (mitocondrial-penetrating peptides), costituiti da una miscela di residui amminoacidici aromatici e alchilici cationici- idrofobici,
vengono assorbiti a livello dei mitocondri e possono essere usati per trasportare
molecole bioattive (Szeto H.H. and Schiller P.W., 2011). I risultati ottenuti con i peptidi
MPPs indicano che carica e idrofobicità ne determinano l’accumulo nella matrice
mitocondriale (Yousif L.F. et al., 2009), anche se non è stato ancora stabilito se il
passaggio dei MPPs attraverso il doppio strato fosfolipidico sia mediato da qualche
meccanismo non ancora chiarito o se l'assorbimento mitocondriale sia semplicemente determinato dal Δψm. Al contrario si pensa che l'assorbimento dei peptidi SS sia
indipendente dal Δψm (vedi capitolo successivo peptidi SS).
Ad oggi il principale obiettivo dei cationi lipofili e dei peptidi mitocondri -mirati è stato
quello di fornire un agente bioattivo, tramite legame covalente, ai mitocondri.
Una variante di questo approccio è stata quella di progettare una molecola in modo che
la porzione di trasporto venga scissa dalla porzione bioattiva all'interno dei mitocondri,
liberando la molecola attiva nella matrice mitocondriale. Esempi recenti di questo approccio sono l’apporto di acido lipoico, temporaneamente attaccato al catione TPP
tramite un legame estereo enzima-scindibile, e il rilascio di ossido nitrico (NO)
25
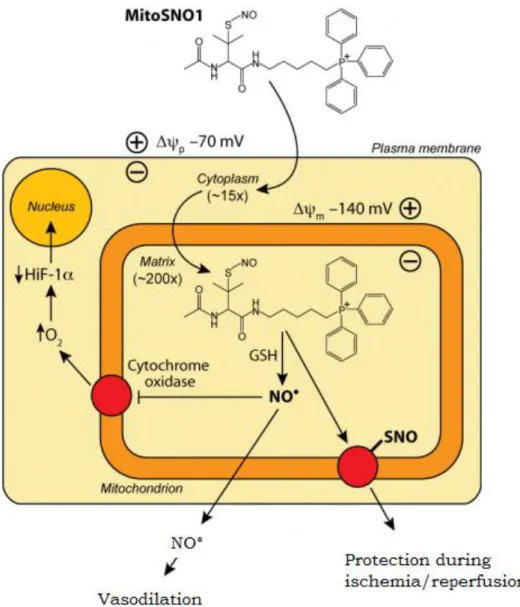
Figura 7. Meccanismo d’azione di MitoSNO (Google immagini)
Un ulteriore esempio è quello di utilizzare TPP collegato ad una porzione bioattiva per
mezzo di un legame fotoscindibile che può essere fotolizzato in situ, fornendo in tal
modo l'agente attivo all'interno dei mitocondri in un particolare tessuto o cellula. Questo
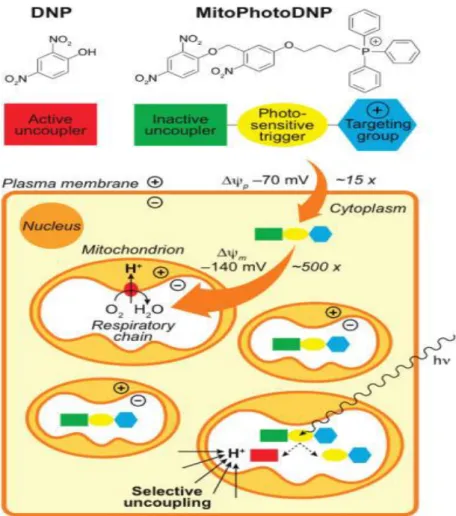
è stato dimostrato con il composto MitoPhotoDNP dove TPP unito a un disaccoppiante,
il 2,4 dinitrofenolo (DNP), tramite un legame fotolabile rilascia DNP all'interno dei
26
Figura 8. Meccanismo d’azione di MitoPhotoDNP (Google immagini)
Il mitocondrio può essere utilizzato anche come camera di reazione intracellulare tra
due composti mitocondri -mirati che reagiscono insieme per formare un nuovo prodotto
o per generare una molecola bioattiva all'interno dei mitocondri che può poi diffondere
fuori dall’organello per agire su obiettivi extramitocondriali.
Queste strategie mitocondrio-mirate hanno dimostrato di funzionare in vivo. Un ampio
lavoro sui composti TPP-coniugati ha dimostrato che possono essere indirizzati ai
mitocondri in vivo dopo somministrazione orale in acqua o tramite tablet, per iniezione
intraperitoneale (IP) o endovenosa (IV),tramite colliri e per infusione in organi ex vivo.
I composti TPP-coniugati sono stati somministrati in modo sicuro a lungo termine a vari
modelli di roditori (McManus M.J. et al., 2011). Essi sono stati anche somministrati per
27
sicurezza (Gane E.J. et al., 2010) I composti TPP possono essere assorbiti dai
mitocondri all'interno delle cellule cardiache entro pochi minuti dopo l'iniezione IV,
consentendo il rapido trattamento della disfunzione mitocondriale acuta. L'assorbimento
dei composti TPP-coniugati, attraverso organi diversi, non è uniforme e sebbene questi
composti attraversino la barriera ematoencefalica (BEE) in quantità sufficiente a
proteggere il cervello dalla degenerazione in vari tipi di malattia (Ghosh A. et al., 2010),
l'assorbimento è inferiore a quella per altri organi. Gli studi fino ad oggi hanno
dimostrato che il metabolismo dei composti TPP-coniugati avviene principalmente
attraverso la reazione della porzione attiva, lasciando la funzione TPP immodificata, e
infine i composti vengono escreti nella bile e nelle urine (Li Y. et al.,2010).
Ad oggi la principale applicazione terapeutica dei cationi lipofili e dei peptidi
mitocondrio-mirati è stata quella di antiossidanti per bloccare il danno ossidativo
mitocondriale. Gli antiossidanti convenzionali (non selettivi), come il CoQ,
l’N-acetilcisteina e la vitamina E hanno avuto scarsa efficacia in studi clinici a causa della
loro limitata distribuzione ai mitocondri. Il maggior accumulo degli antiossidanti
mitocondrio- mirati nella matrice mitocondriale, li rende molto più efficaci nella
protezione contro il danno ossidativo.
L'antiossidante mitocondrio-selettivo più studiato fino ad oggi è stato l'ubichinone
TPP-modificato, MitoQ, che ha dimostrato efficacia in vari tipi di malattie animali ed anche in studi sull’uomo (Smith R.A.J. and Murphy M.P., 2010). E’ stato visto che per
somministrazione orale o endovenosa MitoQ passa dalla circolazione alle cellule per
diffusione attraverso la membrana plasmatica guidato dal potenziale di membrana plasmatica. Nel citosol, la grande Δψm porta all'ulteriore diffusione di MitoQ nella
matrice mitocondriale. All'interno dei mitocondri, MitoQ viene soprattutto adsorbito
dalla superficie della membrana interna rivolta verso la matrice, facilitando così la sua
28
ossidativa. La forma ubichinone di MitoQ viene rapidamente ridotta dal complesso II
alla forma ubichinolo, un antiossidante molto efficace, che inibisce la perossidazione
lipidica mitocondriale e reagisce direttamente con ossidanti come il perossinitrito.
Durante queste reazioni, l'ubichinolo viene convertito in radicale ubisemichinone che si
trasforma in un ubichinolo e un ubichinone, che viene poi ridotto di nuovo ad
ubichinolo dalla catena respiratoria. La forma ubichinone di MitoQ può anche reagire
direttamente con il superossido (Fig. 9).
.
Figura 9. Meccanismo d’azione di MitoQ (Google immagini)
Lo ione TPP+ è stato utilizzato con successo in combinazione con nitrossido,
plastochinone e tocoferolo per sviluppare una gamma di antiossidanti mitocondrio
-selettivi come MitoSNO, SKQ1 e MitoVit E.
Anche il peptide SS31, che ha una capacità antiossidante intrinseca, ha dimostrato essere efficace in vari tipi di animali e viene ora applicato a studi sull’uomo. L'ampia
29
mitocondriale costituisce un importante obiettivo terapeutico.
Sono state sviluppate altre terapie selettive verso i mitocondri. Per esempio, l'aumento di Δψm nelle cellule tumorali ha portato allo sviluppo di diverse tossine mitocondriali
(TP187) destinate ad accumularsi nei mitocondri e ad uccidere quindi selettivamente le
cellule tumorali (Fulda S. et al., 2010).
5.2 Farmaci che modulano bersagli e processi mitocondriali
I mitocondri contengono molti bersagli e processi specifici che possono essere
modulati da farmaci e agenti bioattivi. Tra questi ci sono agenti che funzionano come
farmaci convenzionali legandosi e modulando bersagli proteici specifici all'interno dei
mitocondri. Un esempio è la ciclosporina A (CsA, Fig. 10), un immunosoppressore,
che agisce anche come efficace
inibitore della ciclofilina D all'interno
dei mitocondri, impedendo così la
transizione della permeabilità
mitocondriale( mPT). L'induzione della
mPT è causata da danno ossidativo
elevato, deplezione di ATP e
disomeostasi del calcio, che insieme portano all’ induzione ciclofilina D-dipendente di
un poro nella membrana mitocondriale interna. Una volta formato, il poro provoca
rigonfiamento mitocondriale e interrompe la sintesi di ATP portando alla morte
cellulare necrotica, che è un fattore importante nella lesione da I/R. Per cui la CsA,
legandosi ed inibendo la ciclofilina D, può impedire l'induzione della mPT. La CsA è un
immunosoppressore che è stato utilizzato in uno studio pilota sull’uomo per verificare
se il blocco del mPT potrebbe essere terapeutico dopo infarto miocardico; è stata
30
somministrata a pazienti per via endovenosa immediatamente prima di un’ angioplastica
coronarica ed è stata notata una diminuzione della superficie di tessuto infartuato (Piot
C. et al., 2008). La CsA è risultata efficace anche in due sindromi degenerative correlate
chiamate miopatia di Bethlem e distrofia muscolare congenita di Ullrich che sono
dovute a mutazioni del gene del collagene VI. Sorprendentemente, le mutazioni in questa proteina extracellulare portano all’aumento della disfunzione mitocondriale e alla
morte cellulare nei topi, e questi difetti possono essere migliorati con CsA, suggerendo
che l'induzione della mPT è fondamentale per la morte delle cellule muscolari colpite
dalla patologia (Merlini L. et al., 2008). Gli inibitori della ciclofilina D sono un esempio
molto promettente di farmaci che possono modulare bersagli mitocondriali. Al fine di
evitare gli effetti collaterali immunosoppressori e tossici della CsA a lungo termine,
sono stati sviluppati analoghi della CsA mitocondrio-selettivi che non si legano alle
ciclofiline localizzate al di fuori dei mitocondri (Hansson M.J. et al.,2004).
Un altro interessante processo mitocondriale sul quale intervenire farmacologicamente è
la via intrinseca della morte cellulare per apoptosi caratterizzata dalla
permeabilizzazione della membrana esterna mitocondriale (MOMP), durante la quale
la rottura della membrana esterna rilascia proteine come il citocromo c (cyt c) dallo
spazio intermembrana e quindi impegna la cellula all'apoptosi. Questo processo richiede
l’interazione tra proteine antiapoptotiche, come le cellule B linfoma-2 (BCL-2), e le
relative proteine pro-apoptotiche, come l’ antagonista/killer omologo delle BCL-2 (BAK) . Infatti l’attivazione delle proteine pro-apoptotiche, come le BAK, porta alla
formazione di un poro nella membrana esterna mitocondriale e alla MOMP, che viene
compensata dal sequestro di BAK da parte delle BCL-2. Le piccole molecole che
interrompono questa interazione (ad esempio ABT-737), rendono le cellule più
31
diventate resistenti all'apoptosi a causa di una maggiore espressione di BCL-2 (Fulda
S.et al., 2010).
Altri bersagli mitocondriali per l'intervento farmacologico sono i processi legati alla
fissione, fusione e autofagia mitocondriale che consentono alle cellule di rispondere ai danni derivati dai mitocondri danneggiati o dall’ apoptosi. L'accumulo di mitocondri
danneggiati e difettosi riduce il pool di mitocondri funzionanti correttamente e aumenta
la probabilità di morte cellulare attraverso il rilascio di fattori pro-apoptotici. Questo
può essere determinato dall’inversione dell’attività della FoF1 ATAasi e dall’aumento
dello stress ossidativo cellulare. Migliorare l’eliminazione dei mitocondri danneggiati
permetterebbe così alla cellula di ricostituire il pool dei mitocondri funzionanti e di
normalizzare la sintesi di ATP e l'omeostasi del calcio. Pertanto, l'obiettivo è quello di
progettare piccole molecole, di manipolare questi processi e di eliminare i mitocondri danneggiati tramite upregulation dell’autofagia. Ci sono composti invece, che
interagiscono con il meccanismo della fissione e fusione mitocondriale, come l’
inibitore 1 della divisione mitocondriale (Mdivi-1) che inibisce la dinamina, proteina correlata alla proteina-1 (DRP-1) coinvolta nella fissione o frammentazione
mitocondriale. La modulazione della frammentazione mitocondriale ha potenziale
terapeutico perché la sua inibizione attraverso Mdivi-1 riduce l’apoptosi, la MOMP e
anche la morte cellulare nelle lesioni da I / R cardiache (Cassidy-Stone A. et al., 2008).
Alcuni processi che sono specifici per i mitocondri possono essere indirizzati verso
percorsi meno convenzionali ma con potenziale beneficio terapeutico. Un esempio è il
disaccoppiamento della fosforilazione ossidativa dalla catena di trasporto degli elettroni. L’aumento del passaggio degli ioni H+
attraverso la membrana interna presenta due
potenziali vantaggi: 1) la fosforilazione ossidativa (produzione di energia) è meno
efficiente con conseguente conversione del grasso immagazzinato per produrre calore,
32
attraverso la proteina disaccoppiante chiamata termogenina; 2) il disaccoppiamento
aumenta il consumo di O2 e la velocità con cui i vettori di elettroni come il NADH e il
CoQ si ossidano riducendo lo stress ossidativo. La dissipazione del gradiente
elettrochimico protonico può essere aumentata con l'uso di ionofori o disaccoppianti che
hanno la capacità di trasportare direttamente i protoni attraverso la membrana interna. Il
miglior esempio tra questi è il 2,4- dinitrofenolo (DNP, Fig.11); questo composto è stato
scoperto perché causava la perdita di peso in lavoratori di materiali bellici che
maneggiavano composti nitro-aromatici. Nel 1930 veniva usato come agente
dimagrante provocando però numerose morti, per cui da allora il suo uso è stato
bandito. Nonostante ciò il 2,4-DNP era molto efficace a promuovere la perdita di peso e
quindi nel corso degli anni ci sono stati diversi tentativi di modificare i disaccoppianti
così da poter essere utilizzati in modo sicuro nel trattamento dell'obesità cronica, come
ad esempio lo sviluppo di disaccoppianti autolimitanti (Harper J.A. et al., 2001).
5.3 Agenti farmacologici che agiscono indirettamente sui mitocondri
Un’ulteriore strategia innovativa è quella di usare piccole molecole per modulare i
processi all'esterno dei mitocondri che controllano la biogenesi e l'attività dell’organello. Si tratta di manipolare vie endogene che normalmente consentono al
contenuto e all'attività mitocondriale di rispondere alle esigenze ambientali, come
33
l'aumento del carico di lavoro o cambiamenti nell’apporto di sostanze nutritive.
Attraverso la manipolazione diretta o indiretta di queste vie si altera quindi la
trascrizione dei geni mitocondriali codificati dal nucleo. Il principale regolatore della
biogenesi mitocondriale è il co-attivatore trascrizionale PGC-1α (peroxisome
proliferator-activated receptor- γ coactivator-1α). PGC-1α aumenta l'attività dei fattori
di trascrizione che sono coinvolti nella biogenesi mitocondriale, come NRF -1 (nuclear
respiratory factor), che a sua volta modula l'espressione di altri fattori importanti per la
replicazione e la trascrizione del mtDNA, come TFAM (transcription factor A
mitochondrial); (Ventura-Clapier R. et al., 2008). L’upregulation farmacologica dell’attività di PGC-1α può essere un modo di ripristinare la mitocondriogenesi per
superare un difetto mitocondriale o per rispondere ad un aumento della domanda energetica. L’espressione di PGC-1α è regolata dall'attività del recettore attivato dal
proliferatore dei perossisomi (PPAR) γ. Inizialmente PGC-1α risponde alla chinasi
AMP-dipendente (AMPK) che funziona da sensore ATP / ADP citosolico ed entra in
funzione quando i livelli di energia calano, determinando la fosforilazione di PGC-1α.
A questo punto PGC-1α fosforilata migra verso il nucleo, si unisce al recettore PPARγ e tramite i fattori di trascrizione attiva i geni mitocondriali di origine nucleare. L’attività
di PGC-1α può anche rispondere alla proteina chinasi calcio/calmodulina-dipendente
(CAMK) IV, che richiede la sua attività per rispondere ai segnali di calcio come quelli
coinvolti nella contrazione muscolare. L'attività di PGC-1α è stata modificata
farmacologicamente upregolando la sua espressione attraverso l'uso dell’agonista PPAR
pan, bezafibrato, permettendo di sopprimere un difetto nel fattore di assemblaggio del
complesso IV in un topo in vivo (Wenz T. et al., 2008). In uno studio successivo, il
bezafibrato è risultato invece inefficace nei confronti di una simile mutazione del
complesso IV e, in questo caso, il problema è stato risolto tramite l'attivazione indiretta
34
La manipolazione farmacologica dell’attività di PGC-1α è dunque particolarmente
promettente come trattamento delle disfunzioni mitocondriali.
Un’altra via enzimatica che regola l’attività mitocondriale alterando la trascrizione
genica è rappresentata dalle deacetilasi NAD+ dipendenti appartenenti alla famiglia
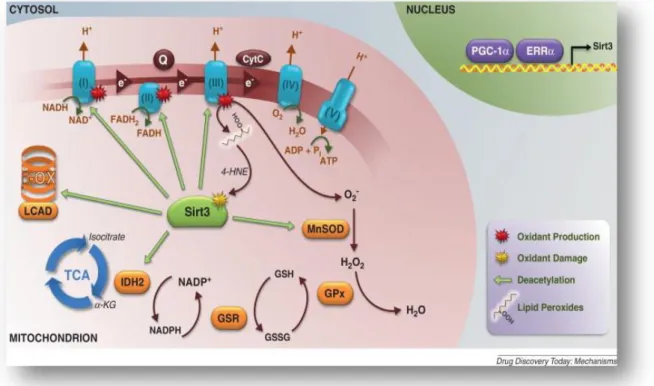
delle Sirtuine (Fernandez-Marcos P.J. and Auwerx, J., 2011) .Ad esempio, il pool nucleare di Sirtuin3 promuove la deacetilazione e quindi l’attivazione del fattore di
trascrizione FOXO3a (appartenente alla famiglia dei fattori di trascrizione denominati forkhead), upregolando l’espressione degli enzimi antiossidanti mitocondriali come
MnSOD (Sundaresan N.R. et al., 2009). Le sirtuine però possono determinare
cambiamenti anche nello stato di acetilazione degli istoni, di altri fattori di trascrizione e
di coattivatori, come PGC-1α, interferendo con la trascrizione dei geni mitocondriali di origine nucleare. Per cui al momento l’interpretazione dell’attività delle sirtuine sui
mitocondri è ancora da chiarire. Nonostante ciò, il fatto che l’attività delle sirtuine
venga inibita dalla nicotinammide e promossa dal resveratrolo (un fenolo non flavonoide ritrovato nella buccia dell’acino d’uva), ha promosso l’interesse sulla
manipolazione farmacologica della funzione mitocondriale attraverso la modulazione dell’attività delle sirtuine (Fig.12).
35
Figura 12. Attività della Sirtuina3 a livello mitocondriale (Google immagini)
36
6. I peptidi Szeto-Schiller (SS)
Come abbiamo precedentemente detto, oltre al loro ruolo principale come fornitori di
energia per i processi cellulari, i mitocondri sono anche i maggiori produttori di specie intracellulari reattive dell’ossigeno (ROS) e sono molto sensibili agli effetti dannosi dei
radicali liberi. Il danno ossidativo mitocondriale determina la riduzione della
produzione di ATP e dunque l'ulteriore aumento della produzione di ROS che possono
portare al declino della funzione cellulare e alla morte della stessa. Lo stress ossidativo
e la disfunzione mitocondriale partecipano anche al processo di invecchiamento e
contribuiscono allo sviluppo di numerose malattie croniche, tra cui diabete, malattia
ischemica cardiaca, insufficienza cardiaca, malattie renali acute e croniche e malattie
neurodegenerative come il morbo di Parkinson e di Alzheimer. Recenti studi infatti
hanno dimostrato che la durata della vita aumentava significativamente in topi
transgenici C57BL/6J che iperesprimevano catalasi umane a livello dei mitocondri del
muscolo cardiaco e scheletrico (topi MCAT), mentre non veniva aumentata in topi
transgenici che iperesprimevano catalasi nei perossisomi o nel nucleo (dove
normalmente questo enzima è localizzato; Schriner S.E. et al., 2005). Oltre alla
longevità, i topi MCAT avevano meno patologie legate all'età (cataratta, sordità,
infiammazione, massa tumorale, lesioni cardiache e resistenza all'insulina; Treuting
P.M., et al., 2008), hanno dimostrato di aver migliorato la loro prestazione all’esercizio
(Li D., et al., 2009) e di essere protetti nei confronti dell’insulino-resistenza indotta da
dieta ricca di grassi, cardiomiopatia causata da zidovudina, e cardiomiopatia ipertensiva.
L'efficacia in vivo dei peptidi SS è stata valutata in diversi tipi di disturbi animali dove
37
In alcuni studi, le azioni protettive di SS-31 erano simili a quelle mostrate nei topi
MCAT, fornendo la prova che SS-31 agisce riducendo lo stress ossidativo
mitocondriale.
Anche molti degli effetti collaterali dei farmaci sono oggi riconosciuti essere la causa di
tossicità mitocondriale (Scatena R. et al., 2007). E’ quindi evidente la necessità di
terapie in grado di proteggere e / o migliorare la funzione mitocondriale. Tuttavia, lo
sviluppo di farmaci mitoprotettivi è stato ostacolato da numerose sfide, e non ci sono al
momento approvate terapie per le malattie mitocondriali.
I due più grandi ostacoli allo sviluppo di farmaci mitoprottettivi sono la mancanza di uno specifico bersaglio molecolare e la difficoltà di indirizzare l’agente terapeutico al
corretto compartimento mitocondriale. La moderna ricerca farmacologica si concentra sullo sviluppo di terapie che colpiscono un’ entità molecolare specifica nel processo
della malattia, come recettori, enzimi, o fattori di trascrizione. In questo caso è difficile
individuare una singola entità molecolare per sviluppare farmaci mitoprottetivi perché
la sintesi di ATP mitocondriale è un processo che richiede molti componenti
mitocondriali per funzionare correttamente (Szeto H.H. and Schiller P.W., 2011).
Negli anni 2000 è stata scoperta una nuova classe di molecole peptidiche, i peptidi
Szeto-Schiller (SS), che agiscono selettivamente sulla membrana mitocondriale interna
(IMM), dove avviene la produzione di ATP e di radicali liberi, e proteggono la funzione
dei mitocondri. I peptidi SS sono stati scoperti mentre Hazel H. Szeto e Peter W.
Schiller stavano lavorando su una famiglia di analoghi della dermorfina che mostravano
elevata affinità e selettività per il recettore oppioide μ (Schiller P.W. et al., 2000)
SS-01 e SS-02 (originariamente abbreviati rispettivamente come DALDA e [ dmt1] DALDA) sono tetrapeptidi idrosolubili altamente polari che portano una carica netta 3+
38
Dmt= 2’6’-dimetiltirosina) in posizione N-terminale, che è necessaria per l’alta affinità
di legame ai recettori oppioidi.
La sostituzione della Tyr con la Dmt in SS-02 ha ulteriormente aumentato l’affinità di legame per il recettore μ.
C’è stata una grande sorpresa quando SS-02, dopo somministrazione sottocutanea (Sc)
in roditori, ha mostrato una potente attività analgesica suggerendo quindi che era in
grado di attraversare la barriera ematoencefalica (BEE) perché generalmente si pensava
che solo i composti altamente lipofili fossero in grado di attraversarla (Zhao G.M. et al.,
2002).
Inoltre successivi studi in colture cellulari hanno dimostrato che SS-02 penetrava
prontamente in vari tipi di cellule senza bisogno di trasportatori o recettori specifici
(Zhao G.M. et al., 2003). Questo era del tutto inaspettato, vista la dimensione
molecolare di SS-02 (640), l’elevata polarità della struttura peptidica e la presenza di
carica netta 3+ data dal gruppo amminico N-terminale e dalle catene laterali di Arg e
Lys. La basicità dell’Arginina è dovuta al gruppo guanidinico che caratterizza la sua
catena laterale, mentre quella della Lisina è dovuta al gruppo amminico.
La scoperta chiave è stata l'osservazione, tramite microscopio a scansione laser
confocale, di un analogo di SS-02 contenente il piccolo anthraniloyl fluorescente
marcato che mostrava una distribuzione intracellulare simile a quella di Mitotracker
39
L’assorbimento mitocondriale di SS-02 è stato confermato usando sia [3
H]-SS 02 che
l'analogo marcato fluorescente in mitocondri isolati. Sorprendentemente, anche se
SS-02 ha una carica netta 3+ a pH fisiologico, il suo assorbimento non dipende dal
potenziale mitocondriale, suggerendo così che non si era distribuito nella matrice mitocondriale. L’assorbimento potenziale-indipendente è un enorme vantaggio quando
si trattano mitocondri danneggiati che probabilmente hanno un potenziale mitocondriale
ridotto. Al contrario, i composti TPP+-coniugati, come MitoQ e SkQ1, non vengono
assorbiti dai mitocondri depolarizzati e vengono rilasciati dalla matrice mitocondriale
quando il potenziale mitocondriale diminuisce (Murphy M.P. et al., 2007). Infine studi di frazionamento mitocondriale hanno rivelato che più dell’ 85% di SS-02 è stato
trovato nella frazione contenente la IMM, rendendolo il primo composto che agisce
selettivamente sulla IMM dove si trova la ETC (Zhao K. et al., 2004). Per cui a
differenza di MitoQ e SkQ1, SS-02 non provoca la depolarizzazione mitocondriale,
neanche ad alte concentrazioni, perché non si distribuisce nella matrice.
6.1 Attività oppioide di SS-02
Prima che SS-02 fosse riconosciuto come antiossidante mitocondrio- mirato, era noto
per la sua attività analgesica di tipo oppioide molto potente ed efficace.
I recettori degli oppioidi sono recettori transmembrana
accoppiati a proteine G. Ci sono tre tipi principali di recettori degli oppioidi: μ, δ e κ. SS-02 è un agonista
di tutti e tre i recettori: si lega con elevata affinità al recettore oppioide μ e con affinità molto inferiore al
recettore δ e κ, rendendolo così altamente selettivo per
il recettore μ (Zhao G.M. et al., 2003). L'affinità di legame di SS-02 per il recettore μ
40
La selettività e l’attività agonista di SS-02 è stata inoltre confermata utilizzando test
biologici standard basati sull'inibizione di contrazioni evocate elettricamente in modelli
sperimentali di ileo di cavia (GPI) e deferente di topo (MVD).
Il saggio GPI è di solito considerato come rappresentativo per le interazioni con i recettori oppioidi μ, anche se l'ileo può anche contenere recettori oppioidi k. Nel saggio
MVD, l'attività è principalmente mediata da recettori oppioidi δ, anche se esistono recettori μ e k anche in questo tessuto. I risultati hanno mostrato che SS-02 è un potente
agonista per il recettore μ (Schiller P.W. et al.,2000).
L'EC50 per SS-02 nel test di GPI è 1,41 ± 0.29 nM, rispetto al 29,3 ± 2,2 nM per la
morfina. In vivo il test analgesico nei topi ha rivelato che SS-02 è 36 volte più potente
della morfina, quando somministrata per via sottocutanea e fino a 833 volte più potente
della morfina quando somministrato nello spazio intratecale (Zhao G.M. et al.,2002).
L'elevata affinità di SS-02 per il recettore oppioide μ lo rende però inadatto come
composto mitoprotettivo perché oltre all’analgesia, il recettore oppioide μ è associato a
diversi effetti collaterali tra cui stipsi, depressione respiratoria, tolleranza e dipendenza. E’ pertanto importante sviluppare altri composti peptidici mitocondrio- mirati che non
abbiano alta affinità per i recettori oppioidi.
6.2 Sintesi di analoghi non-oppioidi di SS-02
I peptidi solitamente non attraversano liberamente la membrana cellulare a causa della tendenza dello scheletro peptidico di formare legami idrogeno con l’ acqua. Il peptide
oppioide endogeno, metencefalina (H-Tyr-Gly-Gly-Phe-Met-OH), è un pentapeptide
che può penetrare nelle cellule tramite assorbimento mediato da recettori o mediante
altri sistemi trasportatori (Hauser M. et al., 2000). SS- 02 è un po’ più piccolo, con
carica netta 3+ a pH fisiologico e inaspettatamente diffonde in modo rapido in diversi
41
permeabilità di SS-02 attraverso un monostrato di cellule epiteliali era 100 volte più
grande rispetto a quello di quattro analoghi di metencefalina, nessuno dei quali
contenente cariche residue (Lang V.B. et al., 1997) . D'altra parte, l’ingresso nelle
cellule è trascurabile anche per oligopeptidi costituiti da meno di sei residui
amminoacidici carichi come Lys o Arg, suggerendo che i residui basici da soli non
possono spiegare la permeabilità cellulare di SS-02. L’unica caratteristica di SS-02 è l’alternanza del motivo strutturale aromatico-cationico, dove residui aromatici (Dmt e
Phe) si alternano a residui basici (Arg e Lys).
Residui aromatici:
Residui basici:
42
Questo motivo consente l’interazione intramolecolare catione- π tra l’anello π ricco di
elettroni (Dmt o Phe) e il catione adiacente (Arg o Lys). L’aggiunta di gruppi metilici su Dmt aumenta ulteriormente la densità di elettroni sull’anello π . L’energia catione-π è
dello stesso ordine di grandezza dell’ energia del legame idrogeno e gli anelli π possono
schermare la carica cationica e migliorare il passaggio attraverso la membrana. Così, è
stato deciso di mantenere questo motivo aromatico-cationico nella sintesi di analoghi
non-oppioidi di SS-02 (Szeto H.H. and Schiller P.W., 2011).
Dagli studi sui peptidi oppioidi si è capito che il gruppo ossidrile sulla Tyr N-terminale
è essenziale per l'attività oppioide e che la sua sostituzione con Dmt aumenta
ulteriormente l’ affinità di legame oppioide.
Un modo relativamente semplice per ridurre l’affinità oppioide è stato allora quello di
sostituire la Dmt di SS-02 con una Phe, sintetizzando così l’analogo SS-20. Tuttavia, la
Dmt è il residuo amminoacidico capace di combattere i radicali liberi e quindi SS-20
non avrebbe tale capacità. Un secondo tentativo è stato allora quello di modificare
l'ordine dei residui amminoacidici in SS-02 in modo che la Dmt non fosse
l'amminoacido N-terminale, ma preservando comunque l'alternanza del motivo
43
L'affinità di legame di SS-20 e SS-31 per i recettori oppioidi μ, δ e κ è stata determinata
mediante spiazzamento competitivo rispettivamente di [3H] DAMGO, [3H] DSLET, e
[3H] U69,593, dalle membrane cerebrali di suino di Guinea. Questi tre radioleganti sono considerati selettivi , rispettivamente, per i recettori oppioidi μ, δ e κ e sono
abitualmente utilizzati per valutare l’affinità di legame (Zhao G.M. et al., 2003). La
sostituzione di Dmt con Phe ha comportato un decremento di 450 volte dell’ affinità per il recettore μ e la mancanza di un legame apprezzabile con i recettori oppioidi δ e κ.
Cambiando invece la Dmt dalla posizione 1 alla posizione 2 ha comportato una affinità
2.000 volte minore di SS-31 per il recettore μ e nessun apprezzabile legame con i recettori δ e κ.
La perdita di attività oppioide è stata confermata usando i biotest GPI e MVD. SS-20 è
più di 100 volte meno potente di SS-02 sia nel saggio GPI che MVD, mentre SS-31 è
almeno 30.000 volte meno potente del SS-02 nel test GPI e 2.000 volte minore nel
saggio MVD. Così, sia SS-20 che SS-31 sono essenzialmente privi di attività oppioide.
Ad ulteriore conferma che SS-31 non aveva attività oppioide, è stato somministrato a
topi per via intratecale ed è stata misurata la sensibilità al dolore (nocicezione) mediante
il classico saggio tail-flick (saggio dello scatto della coda). Questo è un test nocicettivo
44
registrato il tempo di latenza con cui il topo rimuove la coda dalla fonte di calore. Nei
topi, è stato visto che SS-31, neanche a dosi 1.000 volte maggiori l’ED50 di SS-02,
determina nessun effetto antidolorifico (Szeto H.H. and Schiller P.W., 2011).
6.3 Assorbimento mitocondriale di SS-31 e SS-20
La conservazione dell’alternanza del motivo aromatico-cationico sembra essere
sufficiente per la penetrazione cellulare e per l’azione sui mitocondri. L’assorbimento
mitocondriale di [3H] SS-31 è stato dimostrato utilizzando mitocondri isolati da fegato e
cervello di topi con livelli massimali (30-50%) raggiunti entro 2 min (Zhao K. et al.,
2005).
La penetrazione cellulare di SS-20 è stata dimostrata con microscopia confocale a
scansione laser in normali fibroblasti di polmone e in cellule tumorali. SS-20 è stato
colocalizzato con CMXRos Mitotracker, suggerendo la distribuzione mitocondriale.
Inoltre, studi di struttura-attività hanno dimostrato che altri aminoacidi aromatici e
basici possono essere utilizzati per rafforzare la libreria dei Mitochondria-penetrating
peptides (Horton K.L. et al., 2008).
6.4 Attività mitocondriali dei peptidi SS
A differenza di TPP + che ha solo la funzione di vettore di trasporto mitocondriale per
altri composti, i peptidi SS hanno anche attività intrinseca nei mitocondri.
Gli analoghi contenenti Dmt hanno attività antiossidante intrinseca perché la Tyr
elimina gli ossiradicali, formando radicali tirosilici poco reattivi che possono essere
seguiti da accoppiamento radicale-radicale per dare ditirosina oppure reagire con
superossido per formare tirosina idroperossido (Winterbourn C.C. et al., 2004). La Dmt ha una capacità “scavenger” addirittura maggiore a causa della maggiore densità