UNIVERSITÀ DI PISA
CORSO DI LAUREA MAGISTRALE IN CHIMICA
Curriculum Inorganico
Classe: LM-54
Exploitation of strongly fluorescent aggregates of a
perylene bisimide derivative: the case of thermochromic
smart materials and luminescent solar concentrators
Relatori
Dott. Andrea Pucci
Candidato Marco Carlotti Dott. Marco Geppi
Controrelatore
Al secondo principio
e alla neve
Index
Riassunto (Abstract)
1. Introduction_______________________________________________1
1.1 Supramolecular chemistry and non-covalent aggregates …...1
1.2 π-π stacking aggregates …...3
1.3 H and J aggregates and their optical properties ...6
1.4 Luminescent and Smart Materials …...9
1.5 Dye dispersion in polymer blends …...12
References …...14
2. Perylene bisimides: synthesis and characterization of Pery species__17
2.1 Synthesis of RS-Pery and S-Pery …...82.2 Caracterization of RS-Pery and S-Pery …...19
2.3 Crystallographic study …...21
2.3.1 S-Pery …...21
2.3.2 RS-Pery …...25
2.4 Aggregation of Pery species …...29
2.4.1 Aggregation in mixed solvents solutions …...29
2.4.2 Aggregation in high concentrate solutions …...30
References …...33
3. Thermochromic PBI-polymer blends with smart properties_______35
3.1 General considerations …...353.1.1 Thermochromic smart materials …...35
3.1.2 Fluorescence in smart materials …...37
3.1.3 The application of solid state NMR to smart materials …...39
3.2 Preparation of polyolefin-based polymer blends with RS-Pery ...40
3.3 Optical properties of RS-Pery/polymer blends …...41
3.3.1 Survey of the optical properties of a RS-Pery/LLDPE blend …...41
3.3.2 RS-Pery/LDPE blend …...43
3.4 Fluorescence emission temperature dependence of RS-Pery/polymer blends …...47 3.4.1 RS-Pery/LLDPE blend …...48 3.4.2 RS-Pery/LDPE blend …...48 3.4.3 RS-Pery/EVA blends …...51 3.5 PE morphology …...52
3.6 PE crystallinity study by DSC techniques …...53
3.7 Study of PE phase properties by ssNMR spectroscopy …...54
3.7.1 13C-CP-MAS experiments …...54
3.7.2 1H FID analysis …...58
3.7.2.1 LLDPE composites 1H FID analysis 59
3.7.2.2 LDPE composites 1H FID analysis 62
3.8 Final discussion …...64
3.8.1 Model for the thermochromic behavior of RS-Pery/LLDPE blends …...64
3.8.2 Thermochromic behavior of RS-Pery/LDPE blends …...68
References …...70
4. Application of RS-Pery as fluorescent emitter in
Luminescent Solar Concentrators_____________75
Abstract …...754.1 Introduction …...75
4.2 General working principles …...79
4.2.1 Losses and factors influencing ηopt …...81
4.4 Optical properties of the selected dyes …...87
4.4.1 RS-Pery …...87
4.4.2 Rhodamine B (RhB) …...90
4.4.3 Lumogen Red F305 (LR) …...91
4.5 Photostability test of RS-Pery, RhB and LR …...92
4.6 Introduction of an easy method to optimize the efficiency of a single layer thin-film LSC …...96
4.6.1 Effect of the thickness of the polymer layer on the characteristics of the PeryRoom curve …...100
4.7.1 Optical characterization …...107
4.7.2 LSC current output measurement …...109
4.8 Improving the efficiency of RS-Pery LSC devices by enhancement of the light harvesting properties: mixed dye thin films and 'sandwich' devices …...111
4.8.1 Efficiency measurements …...118
References …...124
Appendix A – Summary of experimental ssNMR techinques used in this thesis …...129
References …...134
Appendix B - LLDPE 2D WISE spectroscopy …...135
References …...138
Appendix C - Estimation of ηopt by means of dye concentration in thin film LSC …...139
References …...146
5. Experimental Section_______________________________________147
5.1 Intstrumentation …...1475.2 Materials …...151
5.2.1 Commercial polymer matrices …...152
5.2.2 Synthetic materials …...152
5.3 Synthesis and samples preparation …...153
5.4 Optimization of dye composition in multi-dye LSC devices …...161
5.4.1 Sandwich devices …...161
5.4.2 Mix devices …...161
5.4.3 RS-Pery double layer devices …...162
5.4.4 Mix layer in SandwichMix design …...162
References …...163
Riassunto
Nel lavoro di tesi è stata effettuata la sintesi e la caratterizzazione di due derivati di perilene bisimmide, propriamente N,N'-bis-(1'-feniletil)- perilene-3,4,9,10-tetracarbossildiimide (RS-Pery), ottenuto per condensazione di perilene-3,4,9,10-tetracarbossi-dianidride e 1-feniletilammina in miscela racema, e S,S-N,N'-bis-(1'-feniletil)- perilene-3,4,9,10-tetracarbossildiimide (S-Pery), ottenuto analogamente utilizzando S-1-feniletilammina. Strutture cristalline per S-Pery e per R,S-N,N'-bis-(1'-feniletil)- perilene-3,4,9,10-tetracarbossildiimide sono state ottenute tramite diffrazione di raggi X (XRD). Le proprietà aggregacromiche di RS-Pery in soluzione ed in matrice polimerica sono studiate mediante spettroscopie di assorbimento UV-Vis e di fluorescenza e mediante tecniche di risonanza magnetica nucleare (NMR). Dispersioni di RS-Pery in polietilene lineare a bassa densità (LLDPE), polietilene a bassa densità (LDPE) e in copolimeri etilene-co-vinil acetato in concentrazioni comprese tra 0.01% e 0.5% in peso ottenute per pressofusione, sono investigate come sensori ottici di temperatura nell'intervallo 20-80 °C mediante tecniche spettroscopiche di fluorescenza, calorimetria differenziale a scansione (DSC) e NMR allo stato solido sia con tecniche ad alta risoluzione che tramite l'analisi del decadimento libero dell'induzione del protone (1H-FID). Infine, l'applicazione di RS-Pery in concentratori solari
luminescenti (LSC) a film sottile a base di polimetilmetacrilato (PMMA) e policarbonato (PC) è valutata mediante tecniche innovative e comparata ad altri sistemi presenti in letteratura. Soluzioni per migliorare l'efficienza dei LSC a film sottile mediante l'utilizzo di Quantum Dots (QD) CdSe@ZnS, miscelazione con altri coloranti sia commerciali che di sintesi e nuovi design sono proposte e valutate.
Abstract
In this thesis, two derivatives of perylene diimide, namely N,N'-bis-(1'-phenylethyl)-3,4,9,10-tetracarboxydiimide (RS-Pery), obtained by condensation of perylene-3,4,9,10-tetracarboxydianhydride and 1-phenylethylamine racemate, and S,S-N,N'-bis-(1'-phenylethyl)- perylene-3,4,9,10-tetracarboxydiimide (S-Pery), obtained similarly using S-1-phenylethylamine, are synthetized and characterized. Unit cells for S-Pery and R,S-N,N'-bis-(1'-phenylethyl)- perylene-3,4,9,10-tetracarboxydiimide crystals are obtained by X-ray diffractometry (XRD). Aggregachromic properties of RS-Pery in solution and polymeric matrices are investigated by nuclear magnetic resonance (NMR), UV-Vis and fluorescence
spectroscopies. RS-Pery dispersions in linear low density polyethylene (LLDPE), low density polyethylene (LDPE) and different poly[ethylene-co-(vinylacetate)] copolymers in the concentration range 0.01% and 0.5% by weight obtained by die casting, are investigated as optical thermochromic sensors in the temperature range 20-80 °C by fluorescence spectroscopy, differential scan calorimetry and solid state NMR, by high resolution techniques and proton free induction decay (1H-FID) analysis. Finally, the possible
application of RS-Pery to thin-film luminescent solar concentrator (LSC) is studied with innovative techniques and it is compared to other systems known from the literature. Solutions to improve the LSC efficiency such as doping with CdSe@ZnS quantum dots (QD), mixing with other dyes (both commercial and from laboratory synthesis) and new designs are proposed and evaluated.
1. Introduction
1.1 Supramolecular chemistry and non-covalent aggregates
In Nature, complex systems often show a level of organization that goes beyond the simple molecular model where the atoms are covalently bonded together and molecules are considered to act more or less independently. As a matter of fact molecules can interact together by non-covalent means to form supramolecular entities which may display peculiar proprieties compared to their fundamental components1,2, like the atoms that come together to
form a molecule and lose their elemental qualities!
For instance, this is the case of chlorophyll pigments that can enhance their light harvesting properties by forming molecular aggregates that modifies their optical features3, or
phoshpolipid molecules that compose cell membranes organizing in a dynamic double-layered structure guided by solvophobic interactions4, or the DNA molecule that, through
hydrogen bonding, folds up in the double helix form and can encode a quantity of information that make it even able to repair themself5. Keeping in mind these well-known kinds of
structures it's easy to understand how molecules can interact by means of non-covalent interaction and generate new systems able to accomplish tasks, which require high selectivity and efficiencies, otherwise impossible for the single molecules. As Nature proceeds on its way to higher levels of complexity, on a path which has led from elementary particles to the appearance of life and thinking organisms, new and improved levels of organization have become necessary to face tasks so tangled and elaborated for which the molecular level is not enough.
In this sense, supramolecular chemistry has been defined as “the chemistry beyond the molecule”, “the science of non-covalent, intermolecular interaction”, where two or more chemical species organize together in higher complexity assemblies held together by intermolecular forces and exhibit qualities that could be well defined as those of the molecules themselves6.
Even if the rationalization of supramolecular chemistry's basic ideas it's not even 50 years old2, it is nowadays one of the most active fields in chemistry studying a vast variety of topics
like molecular self-assembly, molecular recognition, folding, catalysis, host-guest chemistry and dynamic chemistry, and shares many common interfaces with other science disciplines like biology and physics7,8.
As already stated, supramolecular chemistry studies the non-covalent interactions between chemical species and that means focusing the attention on interactions that are usually considered weak by ordinary molecular chemistry: this is the case of pure electrostatic ion-ion, ion-dipole and dipole-dipole interactions§, of the hydrogen bond, of the π-interactions
between two π-systems or a π-system and a charged specie, of the der Waals dispersion forces, and of the solvophobic effect. As shown in Table 1.1 all these kind of interactions are very weak compared to bond energy of a covalent compound: for instance the hydrogen bond enthalpy in the bifluoride ion, one of the strongest hydrogen bond known, is -163 kJ/mol9
which is similar in magnitude to bond enthalpy of fluorine or iodine molecules, two elements widely known for their high – and often violent – reactivity, which can also be addressed to their bond's easy cleavage.
Table 1.1 Summary of supramolecular interactions.2
Interaction Strength (kJ/mol)
Ion-Ion 200-300 Ion-dipole 50-200 Dipole-dipole 5-50 Hydrogen bond 4-120 Cation-π 5-80 π-π 0-50
Van der Waals < 5*
Hydrophobic Various
* It depends on the surface extention
Despite the weakness regarding all the interactions just introduced, the formation of strong and stable supramolecular aggregates can be achieved by increasing the number of possible interactions between the components, just like a strong rope is made of twisted textiles, which are quite feeble if taken separately. Moreover the intrinsic reversibility of the non-covalent interactions and the possibility of easily varying the number of binding sites by tuning the conditions can led to the creation of systems showing tunable properties and able to respond efficiently to different stimuli. The same thing happens in most biological systems that we find in Nature, as they do not behave in a simple on-off manner but modulate their response to
§ To be precise electrostatic interactions are quite strong and in magnitude by all meanings comparable to covalent bonds' energy: it's the fact that ions interact mainly by their electric fields, which are non-directional, and arrange themselves in a lattice to minimize the electrostatic attractive/repulsive energy (therefore losing all the informations that are involved in a covalent bond) that put the electric interactions in the background of molecular chemistry.
various external conditions2: a straightforward example could be thinking how precisely our
hands can work when we have to a fine task like putting a thread in the eye of a needle and, at the same time, how strong they can be when lifting an heavy weight or holding tight to something, and all of this become even more incredible considering that the interaction between actin and myosin is mainly based on the “weak” – but in this case extended – hydrogen bond interaction!
Being able to control the non-covalent interactions extensively, the chemist can therefore prepare new beautiful and intricate functional structures with appropriate design and dimensions on the nanometer scale: the ability to work with a bottom-up approach, creating systems able to self-organize, has given new and extraordinary insights into nanochemistry, with the possibility to manage the manipulation and the preparation of nano architectures and components characterized by desirable properties that are not given by the individual units but emerge in the assemblies10.
In this interesting discipline, one of the most appealing topics is the self-assembly of functional π-conjugated systems used as versatile building blocks: peculiar optical and electronic properties arise in the aggregates, due to electronic coupling of transition moments and interaction of the molecular π-orbitals between the components in the spacial arrangement of the assembly11.
This kind of system have long been known: the earliest report of a dye aggregation phenomena is dated almost 130 years ago by Stengrer12 who observed a variation in the
optical absorption and emission proprieties of a dye solution upon temperature variation: since then many functional π-conjugated systems have been discovered – both in Nature or by laboratory synthesis – and investigated. In the recent years, many papers have been published studying the methods and the tools to control the organization and the anisotropy of the spacial arrangement in π-stacked aggregates10,11,13, and that led to amazing improvements in
the emerging field of nanoelectronics14, liquid crystals15, and new interesting approaches to the
preparation of photonic materials built on the nanoscale16.
1.2 π-π stacking aggregates
Supramolecular assemblies are driven by non-covalent interactions. When we focus the attention on the functional aggregates formed by extended π-conjugated systems, the driving force of the aggregation is played in most cases by 'π-π-stacking'17: this is commonly known
as a type of non-covalent interaction that can be found between aromatics entities (or extensive π-conjugated species) that tend to arrange their aromatic planes parallel one to another when they interact18 resembling a graphite-like structure.
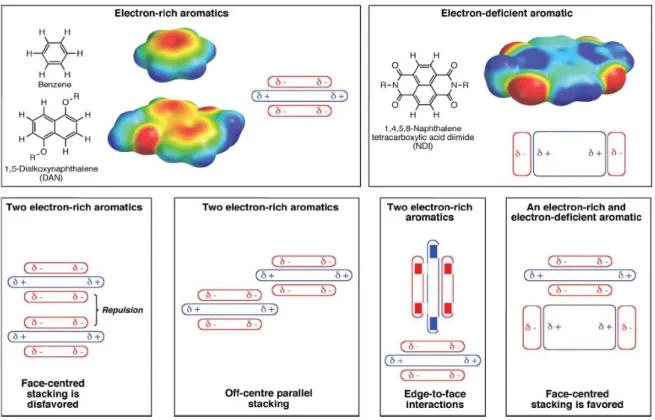
Despite having been extensively studied, the nature and all the forces that contributes to this kind of interaction have not yet been fully understood. For instance, in the case of small aromatic molecules, the sandwich geometry – which is the one that should maximize the interaction between the π-orbitals – is quite rare compared to edge to face or parallel displaced geometries. In the 1990 Hunter and Sanders proposed a model based on electrostatic consideration arising from the fact that the π electrons of an aromatic ring give a large quadrupole moment to the molecule19: this model explains well the experimental data
regarding sandwich-dimer formation between electron-rich and electron-poor small aromatic units like benzene and its substituted derivatives, in which quadrupole moments are large and opposite in sign and their geometry closely resembles the face-to-face stacking of the rings; it also somewhat predicts the distorted or T-shaped benzene dimer being the most stable in gas phase, as two rings with similar electronic proprieties would experience repulsion if put face-to-face (Fig. 1.1).
Anyway, this model has received a lot of critics as the pure electrostatic interaction is not enough to explain all the experimental results. Recent works by Grimme20, Wheeler21, and
many others18 have pointed out the limits of such model and tried to move in a direction
where primary importance is given to polarizability and to the direct interaction between the aromatic host's quadrupole and the electric dipoles of the guest's substituents over the variation of the electron density on the ring.
Also the solvent effect has to be taken into account when rationalizing minimum energy conformations as, being the π-π interaction usually weak, a strongly interactive solvent like water can guide the architecture of the structure22.
A different case is obtained when considering the interaction between extended aromatic systems like many carbons poycyclic aromatic hydrocarbons (PAH) or aromatic molecules dispersed on a graphite-like layer (e.g. graphene, carbon nanotube's walls).
In this case the parallel stacking of the aromatic planes is encountered much more often and the existence of a 'π-π interaction', as commonly intended like a driving force toward a parallel planes stacking geometry, might be a viable model to describe an important interaction between large and highly delocalized systems18.
This change in trend, may be due to the increased polarizability, as the host aromatic carbon sheet is extended and its delocalization increased, and to the wider surface showed to the aromatic guest molecule which maximize the van der Waals attraction. As a result, going from the small benzene units to larger PAHs, the parallel planes geometry become the most common and the resulting aggregates tend to resemble the graphite structure both in inter planar distances and in the off-set of the carbon atoms belonging to consecutive planes. More than that, the interaction itself become even stronger. Measured values for the T-shaped benzene dimer report -11 kJ/mol for the non convalent interaction energy23 (which can go up
to about -20 kJ/mol for substituted benzene rings24), but already for the antracene dimer the
parallel displaced geometry is favored over the T-shaped one with a interaction energy that have been estimated to be about -54 kJ/mol20 (almost five times larger than benzene), and a
further increase in the number of carbons produces even more stabilized entities with calculated values for the interaction energy in the range of -80 kJ/mol for coronene (C24H12)25,
and -270 kJ/mol for the circumcoronene (C54H18)26.
However, it is still difficult to predict which are the effects that dominate the geometry of the interaction depending on the 'size' of the π-conjugated domain: this kind of systems are quite difficult to study, both experimentally, as this type of interaction is weak and controlled by a large number of parameters, and theoretically, being a high number of atoms difficult and time consuming to model25,26.
As already stated when the solvent effect was briefly introduced, the π-π interactions have the same magnitude of other non-covalent interactions, therefore the final shape of the assembly will depend on the competitive average between all the possible interactions: many factors like the structure and the nature of the electronic π-core of the molecule, the number and position of the electron withdrawing/donating groups, the interaction between the side chains, the presence of ions and the possibility of complexation, the solvent effects, all play an important role in the aggregate final geometry and aggregation free energy27. The architecture
of the supramolecular systems will therefore be the result of the nature and the intensity of all the forces, and will create a broad brand of assemblies with different shapes and dimensions (Fig. 1.2), ranging from small dimers or oligomers (in the case of steric hindered species, especially at low concentration), to extended 1D or 2D aggregates28, to liquid crystal phases
which columnar packing – and therefore order proprieties – can be easily modulated29, to
molecular crystals. Moreover, the little spacing between the molecular entities can make their electric transition dipole moments interact and new proprieties can arise in these phases as will be briefly introduced in the next section.
1.3 H and J aggregates and their optical properties
Increasing the concentration of a π-conjugated dye in a solution may induce self-association. The aggregates formed in this way often exhibit significant changes in their absorption and emission spectra compared to the monomer. From the analysis of bands' modification various aggregation geometries have been proposed30: in particular the aggregation can generate in the
absorption spectrum the appearance of a bathochromically shifted band, called J-band (from
Fig. 1.2 Different types of π-stacking: dimers (a), extended aggregates (b) and crystal.like packing (c)
a
b
E.E. Jelley who identified it in cyanine dyes in the '30s31§), or a hypsochromically shifted
band, named H-band (from hypsocrhomic); this phenomenon has been studied in term of molecular exciton coupling theory33 (i.e. coupling of the transition moments of the constituent
molecules) and the resulting effects have been related to two different types of aggregates – named J and H aggregates – characterized by a different orientation of the molecule transition moment compared to the surroundings (Fig. 1.3). Besides the change in absorption properties, J-aggregates are know to be fluorescent with high quantum yields with the appearance of a red-shifted band compared to the monomer emission, while in the H-aggregates, with few exceptions34, fluorescence is strongly quenched35.
It is generally agreed that both J and H aggregates are formed by a two-dimensional structures where the dye molecules are stacked parallel face-to-face (H-aggregates) or head-to-tail (J-aggregates)£; the difference in behavior between these two systems is to be sough in the
different alignment of the molecules' transition moments, as in the H-aggregates the dye molecules are aligned along the π-columnar axis while in J-aggregates are tilted and a non-zero angle between the dipoles can be measured.
According to the simple exciton theory, when molecules are parallel aligned, two new exciton bands are found, one with higher and one with lower energy compared to the single energy level when only monomers are present: in H-aggregates the transition from the fundamental
§ Also G. Sheibe, who observed the aggregation phenomenon and its effects on the dye optical properties in
the same year32, is credited for the discovery of the J-aggregates.
£ It has to be kept in mind that observed stacks in experimental crystals of this kind may show a more complex form where the molecules arrange in a bent, twisted or helix manner, still the H and J differentiation apply to the vast majority of the systems as they show similar optical properties.
Fig. 1.3 Schematic representation of the origin of J and H bands related to different orientation of dipole transition moments according to the exciton theory.
state to the lower lying exciton level is forbidden, anyway the system usually goes through a rapid downward energy relaxation to the lower exciton level that commonly exhibits a small dipole transition moment (from here the low fluorescence yields); on the contrary, when the chromophore transition dipole moments are tilted compared to the columnar stacking as in J-type aggregates, only the transition toward the lower energy exciton level is allowed and so the resulting J-aggregates show a bathochromic shift and high quantum yields36.
For an accurate description of the exciton model and J and H aggregates please refer to the papers herein introduced.
A special case of the aggregation phenomena is the formation of excimer species. An excimer (excited dimer) can be defined as the complex resulting from the interaction between a molecule in its excited state and a nearby molecule in its fundamental state. This specie is short-lived and breaks up in the monomers spontaneously producing a characteristic excimer emission37. Excimer formation has been reported to occur in monoatomic gasses38 or between
aromatic molecule in solution, crystal phase39, and polymeric dispersions37,40.
Compared to the previous J and H aggregates, the fact that excimer formation is consequent to monomer excitation means that no evidence is found in the absorption spectrum of the chromophore: excimer formation is only observable examining the emission properties as they are usually characterized by the appearance of a broad, structureless band shifted to longer wavelength compared to the monomer41.
In the case of small aromatic molecules and PAHs that do not form dimers or other oligomers species in the fundamental state, the excimer formation and emission can be interpreted as an energy minimum in the plot potential energy vs. intermolecular distance of the system composed by a molecule in the fundamental state (M) and a molecule in the excited state (M*): as shown in Figure 1.4, the curve which represents the M+M* system is not only described by a repulsive potential as the entities get closer but also by an attractive potential, arising from charge transfer and dipole-dipole interactions39, which forms an energy minimum
interpreted as the formation of the complex MM*. The radiative decay as MM* split up to
Fig. 1.4 Schamtic representation of red-shifted excimeric emission.
M+M, accruing from a stabilized lower energy level compared to the decay of M+M* to the same fundamental state, is identified by the appearance of a longer wavelength emission band, which is also structureless since no fundamental state of the MM* complex exists.
The excimer formation is usually observed between molecules which are separated by no more than 0.4 nm42: it is interesting to note that this is similar to the distance between the
planes in graphite and as well the distance between the planes of π-π interacting systems that show the parallel planes orientation introduced before.
For large π-conjugated species that tend to form π-π-aggregates is therefore straightforward to seek the optimization of the molecule to guide, by means of control of the environment and the other non-covalent interactions, the π-π-stacking and give the system new desirable properties that may then be even transferred in functional materials16,40,43.
1.4 Luminescent and Smart Materials
Smart materials – also defined 'intelligent', 'functional', 'active' or similar – have been defined as materials that form part of a structural system that has the capability to sense its environment and the effects thereof, and to respond to that external stimulus via an active control mechanism44.
In this sense, smart materials are a class of structural materials able to altering their physical and/or chemical properties when they are solicited by a proper external variation.
The range of stimuli that can possibly trigger a response is large and vary and so is the ensemble of possible feedback responses: for example a mechanochromic material45 can
change is color upon a mechanical stimulus, a photoconducting material46 can transform an
optical stimulation in an elettrical response, and electrostrictive material47 can modify its
shape as an electrical field is applied to it, but there can be many more examples16,48.
Still, in a certain sense, every material is 'functional' under these definitions: to this point for example ice is perfect to know if the temperature of a system is above 0 °C and an iron rod can be used to obtain information about the pH of an unknown solution, just like the canaries were used in mines to detect if there were poisonous gas in the air as the birds would die before the miners. These materials do respond to the external variation but doing that they lose their structural properties and cannot accomplish any task they used to: the ice is no more solid, the iron rod has been dissolved and the canary is dead. They respond to the external
stimulus but in a passive way49.
On the contrary, smart materials have to be thoughtfully designed to respond in an active fashion, in a desirable way, and retain as much as they can their structural function and properties. In this sense the 'smart' qualities are just an improvement of a material which has already got its function: contrary to last examples, a polymer film for smart packaging can show information about the history and the condition of its interior (i.e. food products that shouldn't be warmed, fragile objects that should be handled with care) while still performing its protective function45, a cup colored with a thermochromic dye changes its color when hot
water is poured inside but it's still a cup, it senses the temperature without melting or breaking. These materials are 'smart' because they can do what similar materials – a sheet of the same polymer without the dye, a cup made from the same clay but no smart coloration – can do but more than that they can actively modify their properties and show a measurable and/or observable response to a specific stimulation. In other words, they show qualities that the original material did not have and it has been designed for.
Among the vast field of smart material, the class of smart systems based on commodity materials such as refined plastics from oil or renewable resources has been drawing more and more attention in the last decades thanks to their excellent thermo-mechanical properties, chemical stability, easy processing and low cost50. The growing knowledge of the
photophysics of chromophores and the advances of the synthetic techniques, paved the way for stimuli-responsive materials able to detect thermal, mechanical, optical and chemical solicitations that have been used effectively to develop sensor probes and information displays which still possess the polymer's mechanical characteristic51.
When talking about smart polymers, the new properties are usually obtained by embedding of dopants in the polymer matrix. This can be achieved according to two different routes based either on covalent binding of the responsive unit to the polymer chain or by its dispersion in the polymer bulk48. In the latter case, which is convenient to the point that no preparation of
any new structure is necessary, new materials are generally obtained by properly combining chemical functional species in a system which is a multiphase one and which distinct responses could be effectively modulated in terms of intensity and selectivity by controlling interphase interactions52.
Organic and inorganic chromophores such as dyes with available delocalized electrons or metal derivatives like metal nanoparticles53 or nanocrystals54, constitute the basis of the sensor
properties as a consequence of an environmental variation. Additionally, high-conjugated dyes supplied with electron-donating and electron-withdrawing groups often display high sensitivity to changes in the polarity and/or viscosity of their immediate environment at the molecular scale55 thus affording a further way to sense external induced changes in materials
with high resolution.
Anyway, functional material doesn't necessarily equal sensor behavior. All the molecules capable to enhance a properties of the host may be used to realize a functional material: for example, addition of carbon nanotubes can improve the electrical conductivity56, or clay
dispersion in packaging polymers can modify the permeability to gasses and liquids57. Such
materials have improved structural properties that makes them available to application for which their non-functional equivalent would not be completely effective.
Of particular interest is the dispersion fluorescent molecules characterized by a high quantum yield: in proper matrices this can lead to the production of luminescent materials that find application in laser dyes gain mediums58, Organic Light Emitting Diodes (OLED)59 and
Luminescent Solar Concentrators (LSC)60. These latter are devices able to concentrate solar
radiation by only meaning of light trapping inside a waveguide without needing any lens or mirror, and they will be investigated further in Chapter 4 of this thesis.
Among the possible dopants, also supramolecular systems are often investigated as they are able to show characteristic and tunable properties. Their use in chromic-responsible materials has been one of the main research topic in our research group40,43,45,61, with particular concern
about π-π aggregates and excimers and the emerging of new opto-electronic properties from such entities.
The dispersion of such species in suitable polymer matrices is a straightforward route to the design and preparation of polymers with peculiar optical responses40,43: depending on both
processing conditions and dye-polymer phase interactions (which are related to the chemical nature of both guest and host components), aggregation-disaggregation phenomena con occur in the polymer phases to different extent, thus modulating the overall optical result in a precise manner62.
Beside the exploitation in controlled optical responses to external stimuli, aggregachromic substances can be embedded in functional matrices to obtain new properties that are hard to find when using single molecules as dopant.
conversion applications as the aggregation phenomena can enhance electronic properties, light collection efficiencies and fluorescence Stoke's shift63. Studies have been published where
stacked molecules are used in photovoltaic as bulk hetero-junctions64, as absorber in dye
synthesized cells65 and as active component in LSC66. More than that, self-assembly can guide
the access to nano devices with an efficient bottom-up approach, that grants a finer control on the properties compared to standard devices67.
1.5 Dye dispersion in polymer blends
The properties of commodity plastics are ubiquitously known and have already been reported in the introduction. It is possible to add more properties to these systems by dispersion of organic and inorganic molecules as dopant: the types of dopants used and the way they are dispersed in the polymer matrix are often the main reasons for the commercial success of a polymeric material. Of particular interest are optical dopants, like dyes and pigments, because commodity plastics, besides exhibiting noteworthy thermo-mechanical properties, usually show no absorption in the visible region of light spectrum, limiting their absorption to the near-UV region (< 300 nm).
Obtaining a good homogeneous distribution of the dopant in the polymer is of course of primary importance for the properties and the application of the blend. Usually, the thermodynamic solubility of most dopants (i.e. a phase segregation does not occur) is around 0.1-2%68 by weight and it is inversely proportional to the dopant molecular weight. Higher
concentrations may stimulate phase-separation phenomena (even if kinetically hindered by the high viscosity of the polymer matrix) with segregation of the dopant.
For thermoplastic polymers – as the type of materials presented herein – dopant dispersion can be achieved either by solution methods or by mixing in the molten polymer mass.
The first method is preferred for polymers composed by functional repeating units compatible with the chemical structure of the guest molecule, and homogeneous dopant-polymer solid mixtures can be obtained by film casting techniques (Fig. 1.5). These latter consist in dissolving both the dopant and the polymer pellets or powder in the same solution, pouring it in a mold and then allowing the solvent to evaporate to obtain the solid film69. Polymers of the
described type, which are convenient to handle this way, are for example poly(methyl methacrylate), polycarbonate, poly(ethylene terephtalate) and polystyrene.
More apolar and non-interacting polymers, which is the case of polyolefins like polyethylene and polypropylene, tend to show limiting miscibility and compatibility with dyes and other dopants and this may produce phase separation during casting and drying70: in this case
dopant-polymer blends can be obtained by means of continuous mechanical mixers which are able to disperse thoroughly the polymer bulk. The mixing is performed at a temperature higher than the polymer melting point, also under inert atmosphere (to prevent the formation of radicals with the oxygen in the air which may degrade the polymer), in a mechanical mixer which shearing forces are able to overcome the interfacial tension and break the dopant aggregates (Fig. 1.6). Rapid cooling
of the melt after the mixing process prevents the aggregation of dopant particles because of the high viscosity of the polymer matrix. In industrial processes this method is preferred over film casting techniques.
In this thesis, the dispersion in various polymeric matrices of a high-π-conjugated perylene bisimide derivative (PBI), namely N,N'-bis-(1'-phenylethyl)-perylene-3,4,9,10-tetracarboxidiimide (RS-Pery), is studied and application for the optical properties arising from its aggregates and excimers are proposed by meaning of thermochromic materials and Luminescent Solar Concetrators (LSC).
In Chapter 2, PBIs are introduced as versatile π-conjugated units; RS-Pery synthesis and characterization are presented with special regards to its crystal structure we have obtained. In addition, few insights will be given about the aggregation properties and the dye optical features in solution.
Fig. 1.6 Melt extrusion process Fig. 1.5 Film casting in open mold
In Chapter 3, thermochromic polymer materials are studied by means of RS-Pery dispersions in thermoplastic matrices such as polyethylene (LLDPE, LDPE) and poly[ethylene-co-(vinyl acetate)] copolymer (EVA) with different compositions. The smart properties shown by these systems are then analyzed by means of UV-Vis and fluorescence spectroscopy and solid state NMR investigations.
In Chapter 4, the application of the high Stoke's shift excimer emission of RS-Pery to LSC devices is investigated and accounts for dye dispersions in poly(methyl methacrylate) (PMMA) and polycarbonate (PC) polymer matrices.
References
1 J.M. Lehn, Supramolecular Chemistry; 1995, Wiley-VCH, Weinheim
2 J.W. Steed, D.R. Turner, K. Wallace, Core Concenpts in Supramolecular Chemistry and Nanochemistry, 2007, Wiley, Weinheim
3 A.A. Krasnovsky, M.I. Bystrova, Biosystems, 1980, 12, 181
4 D.L. Nelson, M.M Cox, Lehninger Principles of Biochemistry, 5th ed., 2008, W.H. Freeman and Company,
New York
5 T. Lindahl, R.D. Wood, Science, 1999, 286, 1897 6 J.M. Lehn, Angew. Chem. Int. Ed. Engl., 1988, 27, 89
7 G.. Oshovsky, D.N. Reinhoudt, W. Verboom; Angew. Chem. Int. Ed. 2007, 46, 2366 8 J.M. Lehn; PNAS, 2002, 99, 4763
9 T. Steiner; Angew. Chem. Int. Ed., 2002, 41, 48
10 J.L. Brédas, D. Beljonne, V. Coropceanu, J. Cornil, Chem. Rev., 2004, 104, 4971 11 F. Wurthner, Chem. Commun. 2004, 1564
12 F. Stenger, Ann. Phys. Chem., 1888, 33, 577
13 K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao, L. Zang, J.Am. Chem. Soc., 2006, 128, 7290
14 A.P.H.J. Schenning, E.W. Meijer, Chem Commun, 2005, 3245; J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimoruka, K. Ito, T. Hashizume, N. Ishii, T. Aida, Science, 2004, 116, 1249; W. Jin, T. Fukushima, M. Niki, N. Ishii, T. Aida, J. Am. Chem. Soc., 2005, 127, 8284
15 J.M. Warman, A.M. Van der Craats, Mol. Cryst. Liq. Cryst., 2003, 396, 41; W.E. Banjamin, D.R. Veit, M.J.erkins, E. Bain, K. Scharnhorst, S. McDowall, D.L. Patrick, J.D. Gilbertson, Chem. Mater., 2014, 26, 1291
16 A. Seeboth, D. Lotzsch, R. Ruhmann, O. Muehling, Chem. Rev., 2013, online 17 Z. Chen, A. Lohr, C.R. Saha-Moller, F. Wurthner, Chem. Soc. Rev., 2009, 38, 564
18 C.R. Martinez, B.L. Iverson, Chem. Sci., 2012, online
19 C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 20 S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430
21 J. W. G. Bloom, S. E. Wheeler, Angew. Chem., Int. Ed., 2011, 50, 7847
22 E. A. Meyer, R. K. Castellano, F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210
23 D.J. Wales, Intramoleculat Forces and Clusters I, 2005, Structure and Bonding, 115, Springer-Verlag 24 S.E. Wheeler, A.J. McNeil, P. Muller, T.M Swager, K.N. Houk, J. Am. Chem. Soc., 2010, 132, 3304 25 Y. Zhao, D.G. Truhlar, J. Phys. Chem. C, 2008, 112, 4061
26 S. Grimme, C. Muck-Lichtenfeld, J. Antony, J. Phys. Chem. C, 2007, 111. 11199 27 A.W. Snow, N.L. Jarvis, J. Am. Chem. Soc., 1984, 106, 4706
28 V. Perec, H.J. Sun, P. Leowanawat, M. Peterca, R. Graf, H.W. Spiess, X. Zeng, G. Ungar, P.A. Heiney, J. Am.
Chem. Soc., 2013, 135, 4129
29 M.L. Bushey, T.Q. Nguyen, W. Zhang, D. Horoszewsky, C. Nuckollos, Angew. Chem. Int. Ed., 2004, 43, 5446
30 A. Mishra, R.K. Behera, P.K. Behera, B.K. Mishra, G.B. Behera, Chemical Reviews 2000, 100, 1973 31 E.E. Jelly, Nature, 1936, 138, 1009; E.E. Jelly, Nature, 1936, 139, 631
32 G. Scheibe, Angewandte chemie 1936, 49, 563.
33 M. Kasha, H. R. Rawls, Pure and Applied Chemistry 1965, 11, 371; A.S. Davydov, Theory of molecular
excitons, 1971, Plenum - New York
34 J. Gierschner, L. Luer, B. Milian-Medina, D. Oelkrug, H.-J. Egelhaaf, J. Phys. Chem. Lett., 2013, 4, 2686 35 A. Eisfeld, J.S. Briggs, Chemical Physics, 2006, 324, 376
36 B. Valeur, Molecular Fluorescence Principles and Application, 2002, Wiley-VCH, Weinheim 37 M.A. Slifkin, Nature, 1963, 200, 767
38 T. Forster, Fifth Europ, Cong. Mol. Spec. 121 (London 1962) 39 J.B. Birks, Nature, 1967, 214, 1187
40 A. Pucci, F. Donati, S. Nazzi, G.U. Barretta, G. Pescitelli, L. Di Bari, G. Ruggeri, Reactive & Functional
Polymers, 2010, 70, 951
41 J.B. Birks, Rep. Prog. Phys., 1975, 38, 903
42 J.B. Birks, L.G. Christophorou, Proc. Roy. Soc. A, 1964, 277, 571; J.B. Birks, J.B. Aladekomo, Spectrochim. Acta, 1964, 20, 15; J. Ferguson, J. Chem. Phys., 1965, 43, 306
43 A. Pucci, F. Donati, G. Ruggeri, F. Ciardelli, e-Polymers , 2009, 58, online
44 G. Davies, Smart Materials for the 21st century, http://www.iom3.org/
45 A. Pucci, R. Bizzarri, G. Ruggeri, Soft Matter, 2011, 7, 3689 46 Law K.Y.; Chem. Rev., 1993, 93, 449-486
47 L. Sun, W.M. Huang, Z. Ding, Y. Zhao, C.C. Wang, H. Purnawali, C.Tang, Materials and Desing, 2012, 33, 577
48 F. Ciardelli, G. Ruggeri, A. Pucci, Chem. Soc. Rev., 2013, 42, 857 49 A. Thomas, Angew. Chem. Int. Ed., 2010, 49, 8328
50 J. Harmsen, J. B. Powell, Sustainable Development in the Process Industries: Cases and Impact, 2010, John Wiley & Sons, Inc., Hoboken, New Jersey
51 D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278; F. M. Winnik, D. G. Whitten, M. W. Urban and G. Lopez, Langmuir, 2007, 23, 1; R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480–4487
52 A. Pucci, C. Cappelli, S. Bronco and G. Ruggeri, J. Phys. Chem. B, 2006, 110, 3127; N. Tirelli, S. Amabile, C. Cellai, A. Pucci, L. Regoli, G. Ruggeri and F. Ciardelli, Macromolecules, 2001, 34, 2129; F. Martini, S. Borsacchi, M. Geppi, G. Ruggeri, A. Pucci, Polymer Chemistry, in the press
53 A. Pucci, G. Ruggeri and F. Ciardelli, in Adv. Nanomater, 2010, 1, pp. 379–401, Wiley-VCH Verlag GmbH & Co., Weinheim
54 A. Pucci, M. Boccia, F. Galembeck, C. A. d. P. Leite, N. Tirelli and G. Ruggeri, React. Funct. Polym., 2008, 68, 1144–1151
55 M. A. Haidekker, E. A. Theodorakis, J. Biol. Eng., 2010, 4; G. Signore, R. Nifosì, L. Albertazzi, B. Storti, R. Bizzarri, J. Am. Chem. Soc., 2010, 132, 1276
56 N. Grossiord, J. Loos, O. Regev, C.E. Koning, Chem. Mater., 2006, 18, 1089
57 G. Gorrasi, M. Tortora, V. Vittoria, E. Pollet, B. Lepoittevin, M. Alexandre, P. Dubois, Polymer, 2003, 44, 2271
58 P.J. Sebastian, K. Sathianandan, Optics Communications, 1980, 35, 133
59 C.-G. Zhen, Y.-F. Dai, W.-J. Zeng, Z. Ma, Z.-K.Chen, J. Kieffer, Adv. Funct. Mater., 2011, 21, 699; N. Giebink, S. Forrest, Phys Rev B, 2008, 77, 235215
60 M.G. Debije, P.C. Verbunt, Adv. Energy Mater., 2012, 2, 12
61 F. Donati, A. Pucci, C. Cappelli, B. Mennucci, G. Ruggeri, J. Phys. Chem. B, 2008, 112, 3668
62 D. Phillips, Polymer Photophysics: Luminescence, Energy Migration, and Molecular Motion in Synthetic
Polymers, 1985, Chapman and Hall Ltd, London
63 C. Li, H, Wonneberger, Adv. Mater., 2012, 24, 613
64 L. J. Bu, X. Y. Guo, B. Yu, Y. Qu, Z. Y. Xie, D. H. Yan, Y. H. Geng, F. S. Wang, J. Am. Chem. Soc. 2009, 131, 13242
65 C. Li, Z. H. Liu, J. Schoneboom, F. Eickemeyer, N. G. Pschirer, P. Erk, A. Herrmann, K. Müllen, J. Mater.
Chem. 2009, 19, 5405
66 B.J. Walker, V. Bulovic, M.G. Bawendi, Nano Lett., 2010, 10, 3995; C. Haines, M. Chen, K.P. Ghiggino, Solar Energy Materilas & Solar Cells, 2012, 105, 287
67 Troshin, Pavel A.; Koeppe, Robert; Peregudov, Alexander S.; Peregudova, Svetlana M.; Egginger, Martin; Lyubovskaya, Rimma N.; Sariciftci, N. Serdar, Chemistry of Materials, 2007, 19, 5363
68 F. Donati, Master Thesis, 2006
69 T.L. Richardson, E. Lokensgard, Industrial Plastics: Thoery and Applications, Delmar Publishers Inc., 1997, Albany NY
2. Perylene bisimides: synthesis and characterization
of Pery species
The chemistry of perylene bisimides (PBI) started with the work of Kardos in 19131 who first
described the condensation of naphthalene-1,8-dicarboximide to perylene-3,4,9,10-tetracarboxi-diimide in molten alkali. The obtained products find use as pigments and dyes still nowadays with colors ranging from red to black, the color being dependent on particle size and crystal packing of the dye molecules2: some of the crhomphores are commercial
products (e.g. Lumogen Red, Indanthrene Red LGG, Indanthrene Scarlet R, Perylene Bordeaux, Pigment Red 178, 179 and 148) and depending on their properties find application as vat dyes, highly fast pigments for plastics and coatings and fluorescent dyes with high photostability or as charge-creating layers in electrophotography3; their production was
estimated to be about 100 t/y for the dyes and 1000 t/y for the pigments at 20004. Beside the
optical properties (i.e. strong absorption in the visible region, high quantum yields, excellent photostability), PBIs also feature a low reduction potential5 which enables their use as
electron-acceptor and n-type semiconductor in photoinduced charge-transfer reactions. For these appealing properties perylene derivatives have been extensively study for applications as field-effect transistors6, luminescent solar concentrators7, laser dyes8, photovoltaic cells9,
and light-emitting diodes (OLEDs)10.
Since the work of Kardos, many other elegant synthetic routes have been proposed for the preparation of more and more complex molecules, introducing new starting materials and new synthetic procedures, making possible and easy the syinthesis of symmetrically and unsymmetrically substituted imides as well the opportunity to introduce sobstituent in the perylene bay-area and ortho-position (meaning positions 1, 5, 6, 12 and 2, 5 , 8, 11 respectively)11.
The most common laboratory procedure to achieve the symmetrically substituted PBI is by condensation reaction between perylene-3,4,9,10-tetracarboxi dihanydride 1 and a primary amine carried at high temperature in high boiling solvents like quinoline or imidazole12.
Synthesis in water13 and benzene14 has been reported but require high pressures. Zinc salts
catalyze the reaction: it is believed that they might help the solubilization of 1 in the reaction medium on the basis of anhydride complexation as 1 is generally insoluble in the organic solvents used for the synthesis12,15.
In this thesis perylene bisimide derivatives of 1 and the chiral 1-phenylethylamine 2 will be prepared and characterized by means of IR spectroscopy, X-ray crystallography, liquid and solid state NMR spectroscopy, DOSY measurement. These molecules have long known in our laboratory16 for their ability to form highly fluorescent aggregates17. A brief summary of the
aggregation behavior in solution and simple polymeric matrices (PE, PMMA, PC) will then be proposed with no intention to investigate the thermodynamics and kinetics of aggregation.
2.1 Synthesis of RS-Pery and S-Pery
The synthesis of the N,N'-bis-(1'phenylethyl)-perylene-3,4,9,10-tetracarboxidiimide 3 (from now on named RS-Pery) using the raceme mixture of the chiral amine 2 has been carried out, on the basis of a literature procedure12, in molten imidazole at 180°C with no catalyst (Fig.
2.1): considering that the chiral center in the benzil position of the amine is retained, this reaction should produce a mixture of two enantiomers (the S,S and R,R) and a meso form (R,S) with a statistical composition of 25%, 25% and 50% respectively. The purification was carried out with a series of basic aqueous extractions of a dichloromethane solution after washing with diluted HCl, like described in the Experimental Section, to remove the solvent, the unreacted anhydride and amine, and the product of the monocondensation. RS-Pery was obtained as a dark red powder. We had no intention to separate the mixture's components as it is our intention to evaluate the proprieties of the RS-Pery mixture.
To have a comparison, a synthesis of the same Pery derivate was carried out with the enantio-pure S form of 2, therefore producing only the S,S enantiomer (from now on named S-Pery) using similar reaction condition and purification methods. S-Pery was obtained as a red
N O O N O O N O O N O O N O O N O O O O O O O O NH2 NH2 + Imidazole 4 h, 175 °C - H2O
powder which color is much brighter than RS-Pery§.
The reactions yields after purification are 57% for RS-Pery and 80% for S-Pery. The lower yield of the first is due to ongoing optimization of the purification procedure and the lower solubility in the solvent chosen for the extraction.
2.2 Caracterization of RS-Pery and S-Pery
Characterization of the products was carried by FT-IR, liquid state 1H-NMR and X-ray
diffraction analysis (XRD).
The FT-IR spectrum for RPery is showed in Fig. 2.2 and it resemble very well the one of S-Pery.
The main features of the spectrum are summarized in Table 2.1. The absence of the ahydride CO signals at 1770 and 1741 cm-1 (see Experimental Section) indicates the formation of the
bisimide and the absence of the reagent anyhdride or mono condensed by-product.
§ This difference is manteined in the crystal form and if the products are grinded in fine powders.
1800 1600 1400 1200 1000 800 600 55 60 65 70 75 80 85 90 95 100 Wavenumber (cm-1) T (% )
Table 2.1 FT-IR signals and associated motions
Vibration Signal (cm-1)
υCH aromatic 3064, 3030
υCH alifatic 2982, 2942
υC=O 1697, 1655, 1593, 1575 1H-NMR measurement have been carried out in CDCl
3 (Fig. 2.3) and confirm the molecular
structure proposed. The found chemical shifts as well as the molteplicity of each signal is shown in Table 2.2 for a 1 mM solution of RS-Pery. It's interesting to notice that in 1H-NMR
as well no trace of differentiation between S-Pery, R-Pery and R,S-Pery is found as the chiral centers are distant one from the other and they don't interact.
Table 2.1 1H-NMR (600 MHz, CDCl
3) signals for a RS-Pery 1 mM
solution. Proton label are shown in Fig. 2.4
H Signal (ppm) H Signal (ppm)
1 2.00 (d) 2 6.5 (q)
3, 7 7.51 (d) 4, 6 7.32 (t)
5 7.23 8, 11* 8.58 (d)
9, 10* 8.63 (d)
*Assigned using predictions obtained with MestreNova software (v6.0.2)
N O O 1 2 3 4 5 6 7 8 9 10 11
Fig. 2.4 1H labels for Pery molecule
(only half of the symmtric moelcule is shown)
Fig. 2.3 1H-NMR (CDCL
2.3 Crystallographic study
Crystals of the various Pery species have been obtained by slow cooling of hot saturated nitrobenzene solution of both RPery and S-Pery. In those conditions the dyes tend to form red-brown elongated needle-shaped crystals with a parallelogram-like section (Fig. 2.5). Finding a proper crystal for diffraction was not
a trivial task as they usually grow quite small, tend to twin and are often too brittle to cut. Sublimation at 300°C and 0.01mmHg, despite being reported in the literature for similar compounds2b,c, was not effective to obtain any crystal in both cases.
The structures of the crystals herein presented were determined by Prof. F. Marchetti.
2.3.1 S-Pery
The unit cell found for the compound S-Pery is showed in Fig. 2.6. It is similar to other PBI dye crystal structures found in the literature2a,c.
The unit cell contains two molecules, is triclinc and belongs to the P1 group its parameters being a= 7.8401(5), b= 11.4530(6), c= 17.9555(9), α= 86.487(3), β= 82.168(3), γ= 87.107(3)§.
A solvent molecule is also present in it even if we weren't able to fit its position properly. As a matter of fact, the obtained diffractogram shows many electron-density maxima in that part of the cell, all comparable with a nitrobenzene molecule in different positions: this may arise from the fact that the interactions between the crystallization solvent and the dye molecule are not particularly strong and the nitrobenzene is found in different geometries (although around similar position) in different unit cells. Still the structural role of nitrobenzene is undeniable as a batch of crystals from which the solvent was removed by heating in vacuum showed no diffracting properties. Furthermore XRD powder analysis (see Experimental Section) showed no crystallinity for S-Pery powder obtained via precipitation from chloroform.
In its crystal form, S-Pery tends to stack in a quasi-parallel planes fashion to form 1D columnar packing along the a-axis of the unit cell (Fig. 2.7) with an average distance between the planes of 0.34nm. The planes in the cell are not perfectly parallel as the mean planes of
§ Other crystallographic parameters are given in the Experimental Section.
Fig. 2.6 Unit cell of S-Pery (left) showing the three cell axes a (red), b (green), c (blue) and molecular arrangement (right). Ellipsoids in the bottom right image are defined to 90% probability.
the perylene cores are inclined by almost 5°. As we can notice from the unit cell, the perylene planes tend to twist and translate in the growing of the column so it is possible to measure an angle of 34° between the axises of two consecutive perylene planes and a direction of developing of the column displaced from the normal to perylene planes by 30° (Fig. 2.8). Focusing the attention on the dye molecules in the unit cell, it is interesting to notice how they are particularly distorted as showed in Figure 2.9 where is highlighted for each molecule the mean plane calculated from the carbon and hydrogen atoms of the perylene core: this may arise from a serie of C=O···H
interactions between the carbonyls of the imino groups and the aromatic hydrogens of nearby molecules side chain phenyl groups and lateral molecules perylene planes. In fact the O···H distances of the possible interactions presented in Figure 2.10 are all in the range between 2.4 and 2.7 nm, while the COH angles can be measured to range from 130° to 150°: all of this data are congruous with the presence of a moderate hydrogen bond19.§
§ To confirm the presence of an hydrogen bond a lengthening of the C-H bond should be observed, anyway in XRD analysis, where the electron density is observed, the little hydrogen atom with no closed shells it's hard to be detected (sometimes it's even invisible!). The position of hydrogen atoms in the obtained unit cell is found during the refining depending on the model used: usually hydrogen positions are computed (i.e. where and to which atom are they bonded) and then refined by difference Fourier methods.
Fig. 2.8 Coloumnar packing in S-Pery crystal
Fig. 2.9 Distorsions of S-Pery molecules from the mean perylene plane (light blue and orange)
Fig. 2.11 S-Pery packing with highlighted CH-π distances regarding side chain phenyl groups. The orange asterisk indicate the only benzyl CH found on the mean molecular plane (see Text).
More than that, another clue of this interaction may be seen in the fact thatthe most distorted imino groups are the ones for which the hydrogen interaction is more evident. Also the structural nitrobenzene may interact the same way with the dye.
Moving the attention the the side chains, is possible to notice that also for the phenyl rings the pi-pi-interaction could play a role in the stacking conformation (see Chapter 1): as Figure 2.11 shows, many of the phenyl rings are found to resemble a T-shaped geometry which is know to minimize the quadrupolar interaction energy between small aromatic units20. The distances
between the aromatic CH bonds and the center of the various phenyl groups are similar to other crystal structures reported in the literature where the CH-π interaction is claimed21.
Interestingly also one out of the four benzyl protons in the unit cell seems to take part in an interaction similar to the latter: the CH-pi distance is 0.32 nm, not different from the aromatic CH-pi interaction described before, and surprisingly it is the only proton to be aligned with the mean plane of the perylene core with an eclipsed geometry, while the other three protons are tilted by 15°-20°.
2.3.2 R,S-Pery
Remarkably the measured crystal obtained by the nitrobenzene solution of the RS-Pery mixture revealed to be composed only by the meso R,S form of the Pery species (this form is going to be named R,S-Pery in this section and must not be confused with RS-Pery, which represent the mixture of R,R, S,S and R,S Pery molecules). This is not completely unexpected as the R,S-Pery isomer should be the dominant specie in a statistical mixture: anyway the low quality of the refining obtained (R= 24%) may as well arise from the other components – S-and R-Pery – rather high concentration, S-and the fact that they are chemically very similar to R,S-Pery may be responsible for them to enter the lattice, creating defects and twin ramifications. Recrystallization didn't show any significant improvement in the diffracting properties.
Anyway the found R factor of 24% is too high to be certain of the cell parameters and the accurate position of the atoms in the cell: the proposed results are therefore an estimation of the possible crystal structure.
Compared to S- and R-Pery, the R,S-Pery molecule belongs to Ci symmetry group and has
therefore got an inversion center placed in the central ring of the perylene system: the symmetry operator seems to be retained in the proposed unit cell. The latter, showed in Fig. 2.12, contains one molecule, is triclinc and belongs to the P space group, with its parameters
being a= 9.223(6) b= 12.439(6) c= 12.947(9) α=91.19(5) β= 100.05(5) γ= 92.38(4).
As in S-Pery, the perylene cores stack in a parallel planes and form a 1D alternate columnar packing where the planes are once again separated by 0.35 nm. Also the arrange of the columns along the a-axis in the lattice resemble the one in S-Pery (Fig. 2.13). The differences arise when we focus on the characteristic of these columns: the orientation of the long axis of one perylene plane compared to the consecutive is twisted by 75°, 40° more than in S-Pery, also the columns in the lattice evolve inclined by 40° from the normal to the perylene planes (Fig. 2.14).
Despite the overall conformation being similar, the differences in the packing, even if small, are responsible for a garish change in the color of the crystals being bright red and red-violet the ones from S-Pery and R,S-Pery respectively.
The nitrobenzene, although not present in the unit cell is determinant for the crystal structure, in the case of R,S-Pery as well, as crystals heated under vacuum – again – do not diffract. Looking for the side chain interactions as was done for S-Pery, they seem to be less and, considering the longer distances (Fig. 2.15), weaker. Anyway, differently form the S-Pery structure, the side chains interaction seems to be directed to the perylene core: the phenyl palettes, now further apart, looks like interacting with the perylene CH, while the carbonyls show longer C=O···H distances (0.27-0.26nm) and the angles are more twisted. Still, with a R factor of 24%, it is a little pretentious to hold a discussion over the atomic distances in the lattice and the previous assumptions have to be considered qualitative and based on the similarity with S-Pery crystal structure.
Fig. 2.12 Unit cell of R,S-Pery (left) showing the three cell axes a (red), b (green), c (blue) and molecular arrangement (right). Ellipsoids in the bottom right image are defined to 90% probability (absence of the ellipsoid means that the thermal factor did not refine).
Fig. 2.15 Side chanis interaction and relative ditances in RS-Pery crystal. Fig. 2.14 Columnar packing (left) and schematic representation (left) of RS-Pery
2.4 Aggregation of Pery species
Aggregation of PBI derivatives both in solution and in solid matrices is well documented5,22.
If the perylene core is not particularly hindered, this is usually achieved by a pi-pi stacking of the aromatic planes to form columnar arrays of the same kind of those observed for the crystals' structures.
Aggregation phenomena for the R,R enantiomer of the Pery molecule (R-Pery) in mixed solvents, concentrate solutions and linear low density polyethylene have been reported17.
RS-Pery show a similar behavior although, compared to S- and R-Pery, its solubility properties are somewhat different.
2.4.1 Aggregation in mixed solvents solutions
RS-Pery aggregation was obtained in dilute acetone solution by adding an increased amount of water which act as non solvent. The aggregation was followed by absorption and fluorescence spectroscopy.
Absorption of RS-Pery acetone 5x10-6 M solutions containing different amount of water is
presented in Fig. 2.16. The presence of 50% of water in the solution triggered the aggregation of RS-Pery as it is possible to observe by the strong variation in absorption lineshape and intensity: the appearance of a new band at 560 nm, in accord to fluorescence diminished intensity (Fig. 2.17), is an evidence of the formation of J-aggregates according to the exciton theory. Contrarily to what reported for R-Pery, no clue for the presence of H-aggregates was obtained.
Fig. 2.16 Absorption spectra of a RS-Pery 5x10-6 M
solution in a acetone/water mixture with different amounts of water.
Fig. 2.17 Absorption spectra of a RS-Pery 5x10-6 M
solution in a acetone/water mixture with different
Similar behavior was obtained for a more dilute acetone solution (2x10-6 M) where,
anyway, the aggregation occurred at higher water concentrations (Fig. 2.18).
Other solvents mixture were tried, i.e. chloroform/acetonitrile,
acetone/acetonitrile, but no aggregation occurred (see Experimental Section, Chapter 5).
2.4.2 Aggregation in high concentrate solutions
UV-Vis and fluorescence spectroscopy are powerful tools to analyze aggregation processes, anyway they are limited to low concentrate solution. Liquid state NMR gives to possibility to study more concentrated system (i.e. up to 50 mM) and hence the aggregation process without the presence of a non-solvent.
Chloroform solution of S-Pery undergo a serie of color changes when increased concentration, being green for very dilute solution (<10-5 M), yellow-ornage in the millimolar
range and bright red when >15 mM. The study of this behavior is of particular interest as, compared to the aggregates obtained in the acetone/water solution, the latter red aggregates were found to be highly fluorescent.
RS-Pery in the same solvent showed a similar coloration pattern but, because of its lower solubility limited at about 0.7 mg/mL (1.2 mM), it was impossible to obtain solutions more concentrated than those showing the yellow-orange coloration. Red solutions of RS-Pery have been obtained in nitrobenzene and m-cresol.
The chemical shifts in 1H-NMR spectra of S-Pery in CDCl
3 solution with different
concentrations, showed to be concentration dependent (Fig. 2.19): in particular, on increasing the concentration, the signals of the perylene core and the benzyl proton are shifted to lower fields while the side chain phenyl rings somewhat upshifted. The behavior of the methyl group is unclear. This behavior could be attributed to anisotropic effects of aromatic nuclei which come closer during the aggregation phenomenon18. In Table 2.3 are presented the
chemical shift obtained at different concentration for the various S-Pery solutions.
Fig. 2.18 Absorption spectra of a RS-Pery 2x10-6 M
solution in a acetone/water mixture with different amounts of water.
Table 2.3 Chemical shifts in S-Pery CDCl3 solution at different concentrations. The
protons are labelled as described in Fig. 2.5
[S-Pery] (mM) 8, 11 9, 10 3, 7 4, 6 5 2 1
20 8.41 8.18 7.56 7.53 7.25 6.53 2.03
2 8.65 8.60 7.53 7.34 7.25 6.56 2.02
1 8.62 8.55 7.51 7.32 7.23 6.54 2.02
Confrontation of 1H-NMR spectra of RS-Pery and S-Pery both 1 mM in CDCl
3 revealed them
to be similar (i.e. similar aggregation pattern) except for slight differences for the methyl
Fig. 2.19 Comparison of 1H-NMR (600 MHz) spectra of S-Pery CDCl
3 solution at diffrente concentrations. From
top: 1 mM, 2 mM, 20 mM. N O O 1 2 3 4 5 6 7 8 9 10 11
Fig. 2.20 Comparison of 1H-NMR (600 MHz) spectra of 1 mM RS-Pery (top) and S-Pery