INTRODUZIONE
Il Morbo di Behcet è una patologia multisistemica ad eziologia sconosciuta, causata da un processo flogistico cronico che può coinvolgere vasi arteriosi e venosi di qualsiasi calibro. Il quadro clinico è classicamente rappresentato dalla presenza di aftosi orale ricorrente, aftosi genitale, lesioni cutanee e impegno oculare, a cui si associano con frequenza variabile altre manifestazioni cliniche estremamente eterogenee, che coinvolgono le articolazioni, l’apparato gastroenterico, l’apparato vascolare, il SNC e il SNP. Il coinvolgimento neurologico rappresenta una delle complicanze più temibili della malattia, tanto da costituire, se non precocemente riconosciuto, una delle principali cause di morbilità e mortalità(1, 2).
Epidemiologia. La distribuzione geografica della malattia ha un aspetto estremamente peculiare, assumendo proporzioni “endemiche” in Giappone (15/100.000) e in Turchia (80/100.000); seppur in misura minore, la prevalenza risulta comunque abbastanza elevata in tutti i paesi del bacino del Mediterraneo, mentre è rara negli Stati Uniti (0,1/100.000) e nell’Europa settentrionale (0,6/100.000). Nelle aree ad elevata incidenza di malattia è stata riscontrata una frequente associazione con l’antigene HLA B51, associazione peraltro confermata anche in aree non endemiche, dove la prevalenza è superiore al 50%. L’età di esordio è tipicamente compresa tra la seconda e la quarta decade di vita; più raramente le lesioni
muco-cutanee possono presentarsi in età infantile, seguite da altre manifestazioni cliniche patognomoniche solo in fase post-adolescenziale. Il sesso maschile sembra maggiormente colpito, seppur con grado variabile di predominanza anche nelle aree geografiche ad elevata incidenza (9:1 in Medio Oriente, 2:1 in Giappone)(3,4).
Eziopatogenesi. L’eziologia del M. di Behcet, che è a tutt’oggi sconosciuta, verosimilmente multifattoriale, chiama in causa agenti infettivi, predisposizione genetica e alterazioni individuali dell’assetto immunologico. L’ipotesi patogenetica attualmente più accreditata prevede che un determinato antigene, di verosimile natura batterica, in soggetti geneticamente predisposti, una volta captato, processato e presentato dai macrofagi o dalle cellule di Langherans della cute ai linfociti T CD4+, sia in grado di attivare le cellule di tipo Th0 e di indirizzare la differenziazione verso il profilo funzionale di tipo Th1. Tale selezione sembrerebbe dipendente dalla presenza di IL-12 e di IFNα, prodotti dai macrofagi al momento della presentazione antigenica. Il TNFα prodotto dai linfociti Th1 e l’IL-1 prodotta dai macrofagi sarebbero responsabili dell’up-regulation delle molecole di adesione a livello delle cellule endoteliali, con conseguente afflusso di linfociti e granulociti a livello delle lesioni. Nelle fasi più tardive sono presenti abbondanti quote di granulociti e numerose citochine, tra cui l’IL-8, che possono richiamare ulteriormente tali elementi cellulari agendo da fattori chemiotattici, aumentando l’affinità di legame di alcune molecole di adesione e favorendo la migrazione trans-endoteliale dei granulociti stessi. Numerosi sono gli agenti infettivi
finora chiamati in causa nella patogenesi del morbo di Behcet, ma per nessuno fino ad ora è stato documentato un ruolo eziologico certo. I microrganismi maggiormente studiati sono l’ Herpes Simplex Virus, lo Streptococcus Sanguis e la Bordetella Pertussis(5). Recentemente è stata inoltre ipotizzata la possibilità che l’HLA B51 possa non solo essere il marker della predisposizione genetica dell’individuo, ma possa avere anche un ruolo attivo nella patogenesi del danno immunomediato della malattia, attraverso meccanismi di molecular mimicry tra antigeni molecolari dell’ HLA B51 e antigeni organo-specifici. E’ stato inoltre recentemente ipotizzato un ruolo patogenetico dell gene MICA, appartenente ad una famiglia di geni nell’ambito dell’ MHC di classe I e localizzato in prossimità del locus-B(6-7).
Istopatologia. Dal punto di vista anatomo-patologico, le lesioni principali della malattia, seppur aspecifiche, sono riscontrabili prevalentemente a carico del sistema vascolare: le pareti dei vasi di piccolo calibro (arteriole e venule) si presentano infiltrate da cellule linfo-monocitarie, a cui si può associare, in misura variabile, necrosi di tipo fibrinoide; i vasi di medio e grosso calibro sono più frequentemente interessati dalla presenza di stenosi, dilatazioni aneurismatiche e, in misura ancora maggiore, trombosi endoluminale(8,9).
Manifestazioni cliniche(10). Aftosi oro-genitale. In accordo con i criteri classificativi, l’aftosi orale rappresenta una “conditio sine qua non” per la diagnosi di malattia di Behcet. Le ulcere orali sono generalmente il primo segno della malattia e possono precedere di molti anni l’esordio delle altre manifestazioni. Le lesioni, singole o multiple, sono più
frequentemente localizzate a livello della lingua, delle labbra, della mucosa gengivale e vestibolare; più raramente possono presentarsi anche a carico del palato, dell’orofaringe e delle tonsille. Le afte orali, tipicamente dolenti, vengono classificate in maggiori e minori in base alle dimensioni (superiori o inferiori a 10 mm); l’aspetto morfologico può talora mimare le classiche lesioni erpetiche, localizzandosi anche a livello della mucosa nasale o del solco naso-labiale. Frequentemente possono comparire nella sede di un’estrazione dentaria o di un micro-trauma. Numerosi studi hanno dimostrato il ruolo protettivo del fumo di tabacco sulle lesioni orali, tanto da suggerire l’ipotesi di un potenziale utilizzo della nicotina come trattamento topico dell’aftosi(11).
Le ulcere genitali sono presenti nel 70-95% dei casi e sono morfologicamente simili a quelle orali, differenziandosi da esse per la maggiore tendenza alla cicatrizzazione in fase di guarigione. Nei maschi le lesioni più frequenti sono a carico dello scroto e, più raramente, del pene. Nelle donne le ulcere possono comparire a carico della vulva, vagina e cervice, causando spesso dispareunia. Impegno oculare. Il coinvolgimento oculare è presente nel 30-70% dei pazienti con M. di Behcet, risultando più comune e severo nei maschi. L’impegno è più frequentemente bilaterale e compare entro 2-3 anni dall’esordio della malattia. L’uveite anteriore, nella quale l’essudato infiammatorio è ben visibile nella camera anteriore come materiale cellulato, è un segno caratteristico della malattia, ma è presente solo in un terzo dei casi. L’uveite posteriore e la vasculite retinica possono entrambe causare diminuizione dell’acuità visiva di grado variabile in
più del 25% dei pazienti, evolvendo nei casi a prognosi peggiore in cecità. Altre manifestazioni oculari sono: iridociclite, sclerite, cheratite, neurite ottica, trombosi retinica; la congiuntivite è rara(12).
Impegno cutaneo. Presenti in circa l’80% dei pazienti con M. di Behcet, le lesioni cutanee possono assumere aspetto estremamente eterogeneo. L’eritema nodoso è frequente, soprattutto nelle donne, localizzandosi tipicamente agli arti inferiori e residuando in aree ipercromiche alla remissione. Lesioni papulo-pustolari e noduli acneiformi sono altre possibili manifestazioni; le ultime sono simili alle lesioni acneiformi della pubertà, ma differiscono da esse per estensione e sede, localizzandosi anche a livello degli arti e dei glutei. In un terzo dei casi, differenti lesioni cutanee possono coesistere nello stesso paziente(13).
Patergia. Caratteristica abbastanza peculiare della malattia è la patergia, presente nel 40% circa dei pazienti. Si tratta di una ipereattività cutanea secondaria a stimoli meccanici di natura minima, come le punture di insetti o le iniezioni. Il test che comunemente viene utilizzato per valutarne la presenza, si basa su un’iniezione intradermica con ago sterile di soluzione fisiologica; la reazione che ne consegue viene valutata entro le 48 ore successive; macroscopicamente il test è considerato positivo se compare nel sito di iniezione una lesione eritemato-papulare; istologicamente è presente infiltrazione di neutrofili e linfociti, associata ad infiltrato infiammatorio perivascolare di entità variabile(14).
Impegno articolare. L’impegno delle articolazioni periferiche è presente in più di un terzo dei pazienti. Occasionalmente artralgie e oligo- o
poli-artrite possono precedere le manifestazioni muco-cutanee e spesso non pongono il sospetto diagnostico; più frequentemente tali segni si presentano nei 2-3 anni successivi all’esordio della malattia. L’impegno articolare tipico è caratterizzato da oligoartrite, non erosiva e più frequentemente localizzata a livello di polsi, ginocchia e caviglie; la sacroileite e l’impegno assiale sono quasi del tutto assenti(15). Impegno cardiovascolare. Il coinvolgimento dei vasi di medio e grosso calibro è presente in percentuale variabile dal 10% al 50% dei casi. Le trombosi venose risultano essere più frequenti di quelle arteriose e possono coinvolgere con uguale frequenza il distretto venoso superficiale e quello profondo. Possono verificarsi anche trombosi della vena cava inferiore e superiore, delle vene epatiche e sovraepatiche (S. di Budd-Chiari) e del distretto venoso cerebrale; tali manifestazioni sono associate ad una prognosi peggiore. Dilatazioni aneurismatiche e coinvolgimento trombotico del distretto arterioso sono manifestazioni spesso associate alla presenza di trombosi venose. L’aneurisma dell’arteria polmonare, che rappresenta una causa di mortalità, è presente in circa l’1% dei casi, mentre l'embolia polmonare è una evenienza ancora più rara. L’impegno cardiaco, estremamente raro, può essere caratterizzato da pericardite, trombosi intracardiaca, fibrosi endomiocardica e trombosi delle coronarie (secondarie ad un processo flogistico perivascolare o allo stato di ipercoagulabilità)(16).
Impegno gastrointestinale. Il coinvolgimento del tratto gastroenterico si presenta con diversa prevalenza a seconda delle aree geografiche colpite, risultando molto comune nelle aree endemiche (20-40%). Lo
spettro delle manifestazioni cliniche include: anoressia, dispepsia, vomito, diarrea, spesso accompagnata da muco e sangue e dolore addominale. Le manifestazioni gastroenteriche in corso di M. di Behcet possono ricordare quelle presenti nelle malattie infiammatorie croniche intestinali e il quadro istologico può essere a volte sovrapponibile a quello presente nel M. di Crohn; la mucosa intestinale si presenta infiammata, ulcerata, con lesioni molto simili alle afte oro-genitali e la regione più frequentemente colpita è il tratto ileocecale.
Impegno neurologico. L’impegno neurologico è presente in percentuale variabile dal 5% al 20%. Le manifestazioni solitamente si presentano entro i primi 5 anni dall’esordio e risultano essere più frequenti negli uomini. Convenzionalmente l’impegno neurologico viene classificato in base alle sedi coinvolte(17), in:
- danno parenchimale (Neuro-Behçet), presente nell’ 80% dei casi; il liquor è caratterizzato da una pressione normale, mentre risulta incrementata la quota di proteine, neutrofili e linfociti. Le strutture più frequentemente coinvolte sono, in ordine decrescente, il troncoencefalico, gli emisferi cerebrali e il midollo;
- danno non-parenchimale (Vasculo-Behcet), presente nel 20% dei casi e caratterizzato da una pressione aumentata del liquor, con normale quota di proteine, neutrofili e linfociti; può coinvolgere i grossi vasi arteriosi e/o venosi e le meningi, attraverso la formazione di trombi endoluminali.
La distribuzione della prevalenza delle lesioni parenchimali in corso di impegno neurologico, supporta l’ipotesi di un processo vasculitico dei piccoli vasi, per lo più a carico del distretto venulare e la distribuzione anatomica del sistema venoso nel SNC, spiega il predominante coinvolgimento delle strutture troncoencefaliche. Nella pratica clinica, tuttavia, tale classificazione assume un aspetto puramente didattico, in quanto risulta spesso estremamente difficoltoso attribuire ad un unico tipo di danno l’origine delle manifestazioni neurologiche presenti in un paziente con M. di Behçet; frequente è peraltro il riscontro di manifestazioni neurologiche anche in assenza di danno del SNC dimostrabile da tecniche di imaging(18,19).
La cefalea rappresenta uno fra i sintomi più frequenti anche in assenza di alterazioni organiche obiettivabili ed è spesso presente in fasi di apparente “non attività di malattia”; meno frequentemente può rappresentare l’unico sintomo in pazienti asintomatici ma con quadro RM postivo.(20,21)
La meningite asettica è una delle manifestazioni più tipiche (12-26%). Altre sedi frequenti di lesione sono la capsula interna, i nuclei della base ed il cervelletto; a seconda delle strutture interessate, ne derivano quadri clinici diversi, tra cui sindromi piramidali (tetraplegia, emiplegia, raramente paraplegia), sindromi cerebellari, turbe della deglutizione, sindromi cocleo-vestibolari, convulsioni focali o generalizzate, sindromi extrapiramidali, turbe sfinteriche, sindromi del tronco dell’encefalo e paralisi dei nervi cranici (oculomotore, faciale, cocleo-vestibolare, sindromi pseudo-bulbari). (22)
L’interessamento del sistema nervoso periferico è piuttosto raro (1.5%) e compare generalmente nel contesto di un coinvolgimento neurologico più esteso; esso è generalmente rappresentato da mononeurite multipla (con deficit puramente sensitivo o di tipo misto) e da polineuropatia a carico degli arti inferiori o dei 4 arti. Frequenti sono anche le manifestazioni psichiatriche in particolare psicosi, s.ansioso-depressive, disturbi del comportamento, insonnia, deficit cognitivo-mnesici e disturbi della libido. Non è del tutto chiaro se tali manifestazioni siano da considerarsi secondarie alla presenza di una patologia cronica o se, più verosimilmente, siano una espressione primitiva di malattia. Nonostante la terapia adeguata e precoce, l’impegno neurologico continua a rappresentare la più importante causa di morbilità, cui si associa una mortalità del 5-10%.(23,24)
Diagnosi. Non esistono markers sierologici specifici di malattia e pertanto la diagnosi di M.di Behçet si basa prevalentemente su reperti clinici. La positività dell’antigene HLA-B51, pur non appartenendo ai criteri classificativi della malattia, può talvolta considerarsi un utile elemento nell’iter diagnostico. I dati bioumorali più rilevanti, quali l’aumento degli indici aspecifici di flogosi e il dosaggio sierico di alcune citochine proinfiammatorie, possiedono un ruolo rilevante nel monitorare l’attività di malattia, mentre le indagini di diagnostica strumentale risultano fondamentali per valutare l’entità dell’impegno d’organo all’esordio e nel follow-up. Nel 1990 l’”International Study Group for Behcet’s disease” ha proposto criteri clinici classificativi tuttora in uso. Tali criteri prevedono la presenza di almeno un criterio maggiore, caratterizzato dalla presenza di aftosi orale ricorrente, in
associazione ad almeno due criteri minori, che comprendono la presenza di ulcere genitali, impegno oculare, impegno cutaneo e la patergia. L’estrema variabilità di presentazione della malattia e la diagnosi differenziale con altre patologie, quali le malattie infiammatorie croniche intestinali e le spondiloartriti sieronegative (con le quali il M. di Behçet condivide alcune manifestazioni cliniche), ha condotto nell’ultima decade alla crescente necessità di disporre di criteri diagnostici più precisi, al momento in attesa di validazione. Allo stato attuale, quindi, una corretta diagnosi di M. di Behçet prevede l’integrazione dei criteri classificativi con l’evidenza dei segni e sintomi presenti nel singolo paziente e l’esperienza clinica del medico esaminatore(25).
Terapia(26). La gestione di un paziente con malattia di Behçet richiede un approccio multidisciplinare; la scelta del trattamento risulta pertanto condizionata dalla prevalenza delle manifestazioni cliniche, dalla gravità e dall’estensione dell’impegno d’organo nel singolo paziente.
Terapia topica. Il trattamento locale, da utilizzare preferibilmente in associazione a terapia sistemica, si basa sull'applicazione di gel o pomate di lidocaina 1 o 2.5% o di triamcinolone allo 0.1%. Le ulcere orali e genitali rispondono inoltre all’applicazione topica di steroidi, ciclosporina o tetracicline. Anche le uveiti possono essere trattate inizialmente con l’applicazione locale di steroidi e midriatici. Occasionalmente è possibile l’uso intraoculare di cortisone.
Terapia sistemica. Il trattamento sistemico del M.di Behçet prevede principalmente l’utilizzo di steroidi e farmaci immunosoppressori. Lo
steroide viene generalmente utilizzato a dosi medio-basse nelle fasi di mantenimento della malattia, mentre nell’impegno neurologico severo e nell’impegno oculare recidivante viene utilizzato in boli ad alte dosi (1-0,5 g die per 3 giorni consecutivi), seguiti da progressiva riduzione e in associazione a boli di Ciclofosfamide a dosaggi di 500-750 mg die (effettuati per le prime due somministrazioni a distanza di 7 e 15 giorni dal primo bolo di steroide e successivamente con frequenza mensile, fino ad una dose totale di circa 12 g) o Ciclofosfamide per os; L’impegno oculare è generalmente ben controllato dall’uso della ciclosporina (3-5 mg/kg die) e dall’interferone- 2a (3.000.000-Ɣ 6.000.000 UI die). Nell’impegno neurologico e oculare l’utilizzo di anti-TNF (27,28,29) non solo può risultare utile nei casi di fallimento di altri farmaci immunosoppressori, ma è molto efficace anche in fase iniziale di malattia per l’induzione della remissione e la prevenzione dell’insorgenza di recidive a lungo termine. La scelta del farmaco di fondo da inserire subito dopo lo steroide può essere condizionata dal tipo di manifestazioni cliniche prevalenti e dalla gravità. Nell’impegno muco-cutaneo recidivante risultano efficaci la colchicina (utilizzata a dosaggi 1.5 mg/die), la ciclosporina (3mg/kg/die), l’azatioprina (2 mg/Kg/die) e il micofenolato (1,5-2 g die); In alternativa le ulcere orali e/o genitali severe possono essere trattate con talidomide (100-400 mg/die) o dapsone (100 mg/die). L’impegno articolare è generalmente ben controllato dal methotrexate (10-15 mg settimanali) o dalla azatioprina. Nell’impegno gastroenterico viene utilizzata la salazopirina (2 g die) e la ciclofosfamide per os, utilizzata a dosaggi di 1-2 mg/kg die. Nelle trombosi venose profonde sono utili anche gli
anticoagulanti e gli agenti antiaggreganti piastrinici, in associazione con dosi intermedie di steroidi. I nuovi approcci terapeutici, attualmente approvati esclusivamente per il trattamento dell’impegno oculare, prevedono l’utilizzo di un trattamento leucoaferetico (Adacolumn®). La metodica consiste in una leucocitoaferesi selettiva, il cui filtro è costituito da una cartuccia contenente sfere di acetato di cellulosa; esse determinano deplezione selettiva di granulociti e monociti/macrofagi, riducendo pertanto i fattori proinfiammatori, stimolando il midollo a produrre cellule che, essendo meno mature, riducono di per se l’infiammazione e favorendo conseguentemente una modulazione dei mediatori immunogeni. La leucocitoaferesi, da associare preferibilmente a farmaci immunosoppressori steroido-risparmiatori, viene effettuata con frequenza settimanale (per un totale di 5-10 sedute) e risulta efficace sia nel trattamento dell’evento oculare acuto che delle recidive a lungo termine.
CAPITOLO 1
MANIFESTAZIONI OCULARI DELLA MALLATIA
DI BEHÇET
Il coinvolgimento oculare nella malattia di Behçet rappresenta tipicamente la manifestazione di esordio nel 25% dei casi, generalmente si verifica in un periodo di tempo compreso tra i 2 e i 4 anni dall’esordio di malattia(30). Si osserva nel 50% dei pazienti con M. di Behçet fino a raggiunge i 70–90% nel sesso maschile, il coinvolgimento oculare può essere unilaterale (20%) o bilaterale (80%): e può essere classificato come come uveite anteriore, posteriore o pan uveite(31,32). Il decorso della M. di Behçet è cronico e ricorrente. Nelle popolazioni giapponese e turca dove la malattia è endemica il coinvolgimento oculare è molto più comune che nella razza caucasica e spesso ha un decorso più grave(32). In generale, inizialmente il coinvolgimento oculare è uniliterale, ed anteriore: mentre le recidive interessano il segmento posteriore con vitreiti e vasculiti e spesso diventano bilaterali. Il coinvolgimento posteriore è associato con un decorso e una prognosi più severa per il paziente. L’uveite anteriore è più frequente nel sesso femminile, mentre nel sesso maschile sono più tipiche le panuveiti.
La sintomatologia lamentata dai pazienti è in relazione alla sede principale di coinvolgimento.
Coinvolgimento del segmento anteriore
L’uveite anteriore si manifesta tipicamente con episodi ricorrenti e spesso bilaterali di grado da lieve a moderatocalo di annebbiamento visivo associato a dolore periorbitario, fotofobia, miosi reattiva e lacrimazione. L’iperemia congiuntivale insorge rapidamente nell’arco di poche ore con vasodilatazione e iniezione pericheratica di colore rosso-violaceo per anastomosi della circolazione sclero-congiuntivale. L’uveite è classicamente caratterizzata da attacchi ricorrenti per un periodo di 2-3 mesi, che di norma non regrediscono mai completamente nelle fasi di remissione. All’esame biomicroscopico si possono osservare numerose cellule mobili in camera anteriore (leucociti), per una alterata permeabilità dell’endotelio vascolare danneggiato. L’effetto è analogo a quello della polvere illuminata da un raggio di sole ed è un indice di infiammazione oculare. Ugualmente l’osservazione dell’iride è ridotta per il flare (effetto Tyndall), secondario all’essudazione di proteine per diffusione da vasi ematici intraoculari che ricorda l’effetto dei fari di una automobile in una notte nebbiosa. Il flare di norma si osserva in presenza di cellule in camera anteriore ma può persistere anche dopo la scomparsa delle stesse come indice di un danno vascolare persistente più che di una flogosi in atto che meriti un trattamento. La presenza di cellule della serie bianca nell’acqueo può essere considerato un marker di malattia attiva. Inoltre, nella parte inferiore della cornea, possono essere presenti piccoli precipitati cheratici per l’agglutinazione di cellule dell’infiammazione, soprattutto linfociti e polimorfonucleati, depositati sulla superficie corneale endoteliale. Nelle uveiti croniche anteriori
l’iperemia è di grado medio e persistente, il paziente lamenta lieve dolore e la percezione di miodesopsie. Il margine pupillare e/o la faccia posteriore dell’iride si possono sinechiare con la superficie anteriore della lente determinando una irregolarità e distorsione della pupilla che appare fissa(Fig.2). La severità del quadro è legata al livello degli essudati fibrinosi. Una infiammazione severa che dura da lungo tempo può portare anche alla formazione di membrane ciclitiche, seclusio pupillare con blocco pupillare e glaucoma secondario ad angolo chiuso o atrofia iridea e sinechie iridee periferiche anteriori.
Fig.2 Sinechie irido-lenticolari
Talvolta una massiva risposta leucocitaria può dare anche un quadro di ipopion, soprattutto nelle donne(Fig.3). L’ipopion è caratteristicamente mobile con la gravità e la postura per la bassa concentrazione di essudati fibrinosi e spesso è talmente piccolo da essere visibile solo all’esame gonioscopico e scomparire rapidamente senza lasciare sequele(33).
. Fig.3 Ipopion
Coinvolgimento del segmento posteriore
Il paziente si presenta classicamente con calo visivo senza dolore e visione di miodesopsie. Sebbene l’infiltrazione cellulare in camera vitrea possa essere l’unico reperto clinico è opportuno ricercare attentamente anche infiltrati corioretinici, emorragie o infarti retinici. Infatti il reperto più comune è sicuramente la vitreite, che può essere grave e persistente, ma non bisogna trascurare la possibilità di una vasculite retinica, più frequentemente periflebite ma anche periarterite.
L’infiltrazione cellulare si può estendere alla cavità vitrea, dietro la lente, e andrebbe paragonata per entità a quella presente in camera anteriore. Nelle iriti, per esempio, la cellularità in camera anteriore supera di gran lunga quella vitreale, viceversa nelle uveiti intermedie. Nelle vitreiti o nelle uveiti posteriori l’infiltrazione cellulare in camera vitrea è distribuita in tutto il gel vitreale o più posteriormente e anche il movimento delle cellule è molto più ridotto per la viscosità del gel vitreale rispetto all’umore acqueo.
L’aspetto caratteristico di una periflebite attiva è uno sbiancamento a manicotto perivascolare con estensione irregolare. Si può complicare con fenomeni di occlusione venosa trombotica, con o senza ischemia retinica(Fig.4).
Fig.4 Trombosi della vena centrale della retina e periflebite attiva(aspetto fluorangiografico)
Un altro reperto comune è la retinite, caratterizzata da un infiltrato solitario bianco-giallastro superficiale o da infiltrati multifocali della retina interna con margini sfumati. Alcune di queste lesioni sono transitorie e guariscono anche senza esiti cicatriziali. In caso invece di essudazione massiva degli strati retinici esterni si può avere diffusione diffusa dai vasi retinici con edema retinico o del disco ottico, obliterazione vascolare con tortuosità dell’albero venoso ed emorragie retiniche.
La severità e il numero di recidive determinano modificazioni permanenti a livello delle strutture oculari con conseguente calo visivo più o meno definitivo.
Complicanze oculari
La complicanza oculare più comune del M. di Behçet è l'edema maculare cistoide, che si osserva in circa la metà dei pazienti. Si può risolvere con una terapia adeguata o progredire fino ad un danno cronico persistente che coinvolge la regione maculare con formazione di spazi cistici intra-retinici che possono unirsi fino a formare un foro maculare a tutto spessore(34).
La pressione intraoculare nei pazienti con M. di Behçet può essere elevata come risultato di una molteplicità di meccanismi quali l’ostruzione del trabecolato da parte di cellule infiammatorie, la trabeculite, la formazione di sinechie anteriori periferiche o posteriori fino alla seclusio pupillare (iris bombe), il glaucoma neovascolare o il prolungato usato topico e sistemico di corticosteroidi(35).
La sviluppo di cataratta può verificarsi sia durante il corso della malattia infiammatoria come conseguenza dell’uso di corticosteroidi sistemici o topici durante il trattamento. La maggior parte delle cataratte sono subcapsulari posteriori, meno frequenti le opacità sottocapsulare anteriore e corticale.
Nei quadri avanzati di malattia, la periflebite può provocare una occlusione vascolare (ad esempio, occlusione di un ramo della vena retinica), deformità o atrofia iridea, degenerazione maculare con
modifiche a carico dell’epitelio pigmentato, formazione di membrane epiretiniche, ischemia retinica, papillite, e ottica atrofia . In alcuni casi si può osservare neovascolarizazzione del nervo ottico o della retina che è spesso seguita da un vitreo-retinopatia emorragica che può portare a distacco di retina trazionale(36). Infine gli episodi ripetuti di infiammazione del segmento posteriore e le successive complicanze possono portare a cecità completa e, in alcuni casi, se non trattate, si può arrivare fino alla ftisi bulbare che richiede l’enucleazione(37).
CAPITOLO 2
SCOPO DELLO STUDIO
Lo scopo dello studio è stato di effettuare un’analisi retrospettiva delle principali caratteristiche epidemiologiche, della prevalenza delle manifestazioni cliniche e delle misure terapeutiche adottate, in una coorte di pazienti affetti da MB.
I dati sono stati ottenuti attraverso analisi delle cartelle ambulatoriali e di ricovero di 110 pazienti seguiti presso l’U.O. Reumatologia di Pisa dal 1998 al 2010.
CAPITOLO 3
PAZIENTI E METODI
La coorte studiata comprende 110 pazienti con diagnosi di MB, di cui 63 maschi (M) e 47 femmine (F); l’età media dei pazienti è di circa 42 anni (min:18, max:77). L’ età media all’esordio delle prime manifestazioni cliniche è risultata essere di circa 30 anni (min:13, max:58), con una durata media di malattia di circa 11 anni (min:1, max:31). 98/110 pazienti (51 M, 47 F) sono stati seguiti per un periodo uguale o superiore a 12 mesi, con una durata media del follow-up di circa 6 anni (min: 1, max: 16).
Le principali caratteristiche epidemiologiche dei pazienti sono illustrate nella Tabella 1.
Pazienti 110
M/F 63/47
Età media
(anni) 42 (18-77)
Età media all’esordio
(anni) 30 (13-58)
Durata media di
malattia (anni) 11 (1-31)
Durata media del
follow-up (anni) 6 (1-16)
Follow-up ≥1 anno 98
23 pazienti provenivano dal nord Italia, 54 dal centro, 29 dal sud e dalle isole; 4 pazienti non erano di origine italiana.
E’ stata valutata la prevalenza delle manifestazioni cliniche e dell’HLA B51 sulla popolazione in toto, in riferimento al sesso e ai dati forniti dalla letteratura mondiale. Le principali manifestazioni cliniche valutate in termini di prevalenza sono le seguenti: aftosi orale (almeno 3 episodi in 12 mesi), aftosi genitale, impegno articolare (oligo- o poliartrite periferica), manifestazioni cutanee (eritema nodoso, follicolite), sintomi costituzionali (febbre, astenia, perdita di peso, inappetenza), impegno oculare (uveite posteriore, uveite anteriore, vasculite retinica, diplopia, neurite ottica, cecità), impegno neurologico (cefalea, ipoacusia, eventi cerebrovascolari ischemici e/o emorragici, manifestazioni neuropsichiatriche, neuropatia periferica, ipertensione endocranica), eventi vascolari trombotici periferici (arteriosi e/o venosi), coinvolgimento dell’apparato gastro-enterico (impegno flogistico del tenue e/o del colon dimostrato da metodiche endoscopiche o indagini di imaging).
Al fine di valutare eventuali correlazioni tra grado di severità dell’impegno d’organo e caratteristiche epidemiologiche dei pazienti, la coorte è stata convenzionalmente suddivisa in due “gruppi di gravità” (gruppo A: impegno severo di malattia; gruppo B: impegno moderato-lieve di malattia) in base alla presenza/assenza di almeno uno tra i principali interessamenti d’organo che più comunemente condizionano la prognosi: impegno neurologico, oculare e vascolare trombotico periferico.
E’ stata inoltre effettuata una valutazione trasversale della terapia farmacologia assunta dai pazienti all’ultima visita. La scarsa possibilità di utilizzare misure di outcome standardizzate nell’ambito dell’analisi retrospettiva dei dati, non ha tuttavia consentito di effettuare una valutazione dettagliata della durata e dell’efficacia dei trattamenti; la disponibilità di maggiori dati (clinici e strumentali), forniti dalle cartelle di follow-up utilizzate nell’ambito del Day Hospital infusionale, ha consentito di valutare tali aspetti in modo più dettagliato per i pazienti con MB in trattamento con farmaci biologici.
CAPITOLO 4
RISULTATI
3.1 HLA B51
La positività dell’HLA B51 è stata riscontrata nel 66% dei pazienti studiati (73/110, di cui 49/73 maschi e 24/73 femmine).
3.2 Prevalenza manifestazioni cliniche
La prevalenza delle manifestazioni cliniche osservate nei 98 pazienti ha seguito la seguente distribuzione: aftosi orale ricorrente 80% (M: 45%; F: 35%), aftosi genitale 72% (M: 43%; F: 29%), impegno articolare 49% (M: 36%; F: 13%; p<0.0001), manifestazioni cutanee 47% (M: 27%; F: 20%), sintomi costituzionali 45% (M: 26%; F: 19%), impegno oculare 40% (M: 18%; F: 22%), impegno neurologico 38% (M: 22%; F: 16%), eventi vascolari trombotici periferici 22% (M: 7%; F: 15%; p<0.03), coinvolgimento gastroenterico 22% (M: 10%; F: 12%). I risultati sono mostrati nella tabella sottostante (Tab. 2)
Tab.2 Prevalenza manifestazioni cliniche
3.3 Impegno oculare
Nell’ambito dell’impegno oculare, è stata osservata una prevalenza del 40%, con un coinvolgimento del sesso femminile superiore del sesso maschile (22% verso 18%); l’età media di esordio dei sintomi
nei pazienti con coinvolgimento oculare era di 25,8 ± 9.3 anni, con una età media alla diagnosi di 30 anni (min 13 max 58). Il tempo medio tra la diagnosi di malattia e la comparsa delle lesioni oculari in media era pari 3±2 anni. Un coinvolgimento oculare bilaterale si è verificato nel 64% dei casi (Tab. 3).
0 5 1 0 1 5 2 0 2 5 3 0 3 5 4 0 4 5 5 0 Af t e Or a li Aft e Ge n i t Ar t i c ola z C u t e C. N e ur o l C .O c ul a r e P r e v a le n z a M a s c h i P r e v a le n z a F e m m i n e
Prevalenza 40% 22% 18%♀ ♂ Età media esordio 25,8 ± 9
Età media diagnosi 30
min13 max58 Tempo diagnosi/smi oculari 3± 2 anni
Coinvolgimento bilaterale 64% Tab.3 Caratteristiche dei pazienti con coinvolgimento oculare
La classificazione delle uveiti è stata effettuata secondo i criteri dell’ International Uveitis Study Group/Standardization of Uveitis Nomenclature (IUGS/SUN) (38). Le manifestazioni cliniche valutate sono state caratterizzate dalla seguente distribuzione: uveiti anteriori 27%, uveiti posteriori 32%, panuveiti 17%(Fig. 5). Sono state osservateninoltre diplopia in 7 pazienti (16%) e neurite ottica in 4 pazienti(9%)
Sinechie posteriori erano presenti nel 12,5% delle uveiti anteriori, e nell’8% dei pazienti con uveite posteriore al momento della prima visita presso il nostro centro.
Negli uomini è stata osservata una maggior prevalenza di uveiti posteriori (26/29 verso 6/18 nelle donne, p<0.0001) e nell’ambito delle uveiti posteriori si è osservata nel sesso maschile anche una maggior prevalenza delle vasculiti retiniche (15/29 uomini verso 6/18 donne p<0.01) (Fig. 6). Nei pazienti con un coinvolgimento posteriore, circa il 10% è stato sottoposto a chirurgia della cataratta verso un 6% dei pazienti con solo coinvolgimento anteriore (P=.75). Mentre una pressione intraoculare > 21 mmHg è stata osservata nel 19% degli
occhi con uveite anteriore, e solo nel 13% di occhi con uveite posteriore o panuveite (P=.41).
Una ridotta acuità visiva era comune in entrambi i gruppi, con una maggior frequenza negli occhi caratterizzati da coinvolgimento posteriore: con una acuità visiva < 25/50 all’esordio rispettivamente nel 25% delle uveiti anteriori e nel 60% delle uveiti posteriori (P= 0.0001) fino ad arrivare a cecità nel 2% dei casi (Fig. 7).
Fig. 5 Manifestazioni cliniche nella popolazione di studio
0 1 0 2 0 3 0 4 0
U v e i t i A n t
U v e i t i P o s t
Fig. 6 Prevalenza uveiti posteriori nel sesso
Fig 7. Complicanze oculari dall’esordio al follow-up
0 5 1 0 1 5 2 0 2 5 3 0 3 5 U v e i t i p o s t V a s c u li t i ♂ ♀ 0 2 0 4 0 6 0 8 0 1 0 0 sinechie post cataratta ↑TEO ↓Acuità Visiva U V E IT I A N T U V E IT I P O S T
3.4 Impegno neurologico
Nell’ambito dell’impegno neurologico, le principali manifestazioni cliniche osservate hanno riportato la seguente prevalenza: cefalea 31%, ipoacusia 14%, eventi cerebrovascolari 12%, manifestazioni neuropsichiatriche 11%, neuropatia periferica 7%, ipertensione endocranica 4% (Fig. 7)
3.5 Prevalenza delle manifestazioni cliniche e distribuzione geografica
Effettuando un confronto tra la prevalenza delle manifestazioni cliniche nella coorte studiata e quelle di altre casistiche di differente nazionalità, è emersa una minore frequenza di impegno cutaneo e oculare e un più frequente impegno neurologico anche rispetto a zone ad elevata incidenza della malattia.
3.6 Severità di malattia
Nell’ambito dei due “gruppi di gravità”, selezionati in base ai criteri precedentemente menzionati, il 62% dei pazienti (68 pz: 43 M, 25 F) è stato caratterizzato da un impegno severo di malattia, il 38% (42 pz: 20 M, 22 F) da un impegno moderato lieve. I pazienti di sesso maschile appartenenti al gruppo A risultavano significativamente positivi per HLA B51 (88%). Non sono emerse altre correlazioni statisticamente significative nell’ambito degli altri sottogruppi.
3.7 Terapia
Al momento dell’ultima visita il 24% dei pazienti assumeva ciclosporina, il 17% colchicina, il 14% ciclosporina in associazione a colchicina, l’11% methotrexate, il 10% ciclofosfamide, il 10% antiTNF, il 9% azatioprina; il 5% dei pazienti inoltre non assumeva alcuna terapia di fondo (fig. 8).
0 5 10 15 20 25 30 Ciclosporina MTX Colchicina Ciclofosfamide Anti TNF Azatioprina Ciclo+ Colch
Fig.8: Terapia al momento dell’ultima visita
3.8 Terapia con biologici
11 pazienti (7 M, 4 F; età media 30 anni, min: 19, max: 41; durata media di malattia: 7 anni) hanno assunto o tuttora assumono terapia con farmaci biologici, di cui 2 con adalimumab (1 paziente attualmente in trattamento) e 9 con infliximab (6 pazienti attualmente in trattamento).
Lo schema terapeutico per i pazienti che hanno assunto terapia con infliximab, ha previsto un dosaggio di 3-5 mg/kg per infusione,
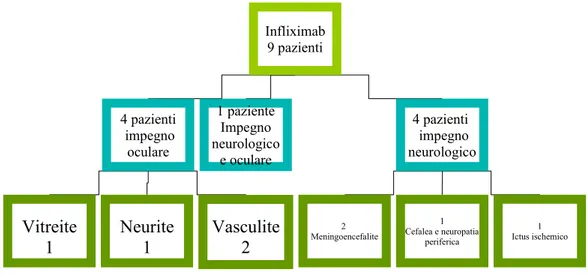
effettuate a tempo 0, dopo 2 e 4 settimane, per le prime tre infusioni e successivamente ogni 6-8 settimane. La durata media del trattamento con infliximab è risultata di circa 7 mesi (min: 2, max 16), con una latenza media di risposta alla 2/3 infusione. 7 pazienti hanno assunto terapia di fondo concomitante con methotrexate o azatioprina. Le manifestazioni cliniche recidivanti non responsive ai precedenti trattamenti, che hanno pertanto orientato la scelta di intraprendere terapia con infliximab, sono state costituite in 4 pazienti da severo impegno oculare (vasculite retinica 2, vitreite 1, neurite ottica 1), in 4 da impegno del SNC e/o SNP (meningoencefalite 2, ictus ischemico 1, cefalea e neuropatia periferica 1 ) e in 1 caso da contemporaneo impegno oculare e neurologico (vasculite retinica e cefalea)(Fig.9).
Fig. 9: Pazienti in terapia con biologici
La terapia è stata efficace in 8/9 casi; l’unico paziente in cui il trattamento è risultato del tutto inefficace presentava un impegno oculare severo di malattia, peraltro di lunga durata, caratterizzato da uveite posteriore bilaterale, refrattaria e recidivante, con acuità visiva
Infliximab 9 pazienti 4 pazienti impegno oculare 4 pazienti impegno neurologico 2 Meningoencefalite 1 paziente Impegno neurologico e oculare 1 Ictus ischemico 1 Cefalea e neuropatia periferica Neurite 1 Vitreite 1 Vasculite 2
residua di 2/10 anche prima di intraprendere la terapia infusionale. Le misure di outcome utilizzate per valutare l’efficacia del farmaco sono state rappresentate da: miglioramento soggettivo del quadro clinico, visita oculistica (valutazione con lampada a fessura), campo visivo, PEV (potenziali evocati visivi), RM encefalo. Nessun paziente ha riportato effetti collaterali durante il trattamento.
CONCLUSIONI
In accordo con i criteri classificativi, l’aftosi orale rappresenta una “conditio sine qua non” per la diagnosi di malattia di Behcet; tuttavia la prevalenza del 100% non è sempre presente anche nell’ambito di casistiche ampie e in zone ad alta incidenza, tanto che negli studi clinici randomizzati la selezione dei pazienti prevede l’esclusione dei casi che non soddisfino i criteri classificativi dell’”ISG for Behçet’s disease”. Tale dato riflette la non assoluta sensibilità dei criteri classificativi nel porre diagnosi di M. di Behçet e la sempre maggiore necessità di disporre di criteri diagnostici che includano altre manifestazioni cliniche, come l’impegno neurologico. Da un’analisi comparativa della prevalenza delle manifestazioni mucocutanee nella nostra coorte, rispetto ai dati della letteratura, emerge una minore frequenza e gravità di impegno rispetto ad aree endemiche.
Come riportato in letteratura, sono state riscontrate differenze nella prevalenza delle manifestazioni in relazione al genere; in particolare, l’impegno articolare è risultato più frequente nei M (36% vs 13%, p<0,0001) e gli eventi vascolari trombotici periferici nelle F (15% vs7%, p<0,03)(39). La correlazione tra prevalenza delle singole manifestazioni cliniche e il sesso è tuttavia caratterizzata da un’ampia variabilità nell’ambito delle casistiche, anche nelle stesse aree geografiche. Dati emersi da studi retrospettivi effettuati in Turchia evidenziano differenze statisticamente significative limitate
all’impegno mucocutaneo (maggiore nelle donne), e all’impegno vascolare trombotico (maggiormente correlato al sesso maschile); allo stesso modo i dati risultano spesso contraddittori anche per quanto riguarda l’impegno neurologico; questo sembra riflettere in parte l’ampia variabilità di presentazione della malattia e in parte i diversi criteri di selezione.dei pazienti studiati. La maggiore gravità di malattia correla con il sesso maschile nelle varie casistiche esaminate e nella coorte dei nostri pazienti(40,41).
Esiste una dimostrata associazione tra HLA-B51 e M. di Behçet, che varia tuttavia in base all’area geografica. La positività per HLA B51 si associa al sesso maschile e correla con una maggiore gravità di malattia, riscontrata nel 62% dei nostri pazienti. La prevalenza di positività per HLA B51 emersa nella nostra casistica (66%), appare sovrapponibile a quelle riportate in altre nazioni europee, mentre risulta inferiore a quanto descritto in aree ad alta incidenza. I dati della letteratura riconoscono fino ad ora all’HLA B51 un ruolo prognostico negativo, mentre l’ipotesi che possa avere un ruolo patogenetico nella malattia non è confermato dai dati più recenti; è stata, infatti, dimostrata l’esistenza di una famiglia di geni nell’ambito dell’ MHC di classe I, in particolare due geni funzionali a livello della regione13: MICA e MICB, localizzati tra il locus-B e il locus dei geni per il TNF. Il gene MICA è espresso prevalentemente a livello delle cellule epiteliali, dei cheratinociti e dei monociti, mentre i geni dell’MHC I risultano ubiquitari. Tali caratteristiche hanno suggerito l’ipotesi che MICA (presente in più dell’80% dei casi) possa avere un reale ruolo patogenetico nel M. di Behcet. La variabilità di prevalenza dell’HLA
B51, rispetto al MICA, giustificata in parte da rapporti di contiguità con tale antigene, da un’immagine riflessa delle alterazioni geniche che potrebbero avere un ruolo patogenetico nella malattia ed essere verosimilmente più correlabile con il MICA(7).
Il coinvolgimento oculare nella M. di Behçet ha implicazioni significative per il paziente, dal momento che si presenta come una grave infiammazione caratterizzata da attacchi acuti con numerose recidive in grado di causare danni significativi e permanenti alle strutture intraoculari. Tuttavia, esistono variazioni individuali in termini di durata severità e decorso della malattia in grado di influenzare la prognosi visiva. Studi su grandi numeri di pazienti possono aiutare a migliorare la nostra conoscenza sulla natura di questa complessa malattia.
Dall’analisi dei nostri dati è emerso, in accordo con la letteratura esistente, come l'età media di insorgenza di uveite fosse di circa 25 anni(42). All’interno della nostra coorte il sesso maschile presentava una maggiore incidenza di coinvolgimento del segmento posteriore con una prognosi visiva peggiore(43). Vasculite retinica e vitreite sono state osservate con maggiore frequenza nei pazienti con uveite posteriore e panuveite. Le complicanze osservate con maggior frequenza sono state rispettivamente l’insorgenza di sinechie posteriori, l’ipertono, la cataratta e l’edema maculare. Globalmente è stata osservata una riduzione importante dell’acuità visiva; pari al 25% nei pazienti con uveiti anteriori e al 60% dei pazienti con uveite posteriore.
La frequenza di impegno bilaterale nella coorte di studio era pari al 60% mentre è compresa in letteratura tra il 78% e il 95%. Colvard et al.(44) e BenEzra e Cohen(45) descrivono un coinvolgimento bilaterale in tutti i loro pazienti. Differenze nel numero di pazienti analizzati e nel follow-up potrebbero aver determinato questa variazione nella percentuale di coinvolgimento bilaterale.
L’Ipopion, una volta considerato un segno distintivo della M. di Behçet viene descritto con una frequenza nettamente inferiore rispetto alle lesioni del fundus, questo può essere dovuto alla natura transitoria dell’ ipopion. Vasculite retinica e vitreite rappresentano entrambe le manifestazioni di un coinvolgimento del segmento posteriore. La vasculite retinica di solito è facilmente rilevabile all’esame biomicroscopico come un rivestimento infiammatorio o gliotico dei vasi retinici. La fluorangiografia è l’esame cardine per dimostrare la presenza di una vasculite anche in occhi senza segni clinici evidenti(46). Dopo la vasculite la seconda forma di coinvolgimento del segmento posteriore nella M. di Behçet è rappresentata dalla retinite che, come l’ipopion a causa della sua natura transitoria, viene spesso misconosciuto; anche perché spesso la presenza di haze vitreale non ne permette una chiara visualizzazione.
Il coinvolgimento oculare nella malattia di Behçet caratteristicamente è associato ad un esordio improvviso con riduzione, anche grave, dell’acuità visiva che può gradualmente migliorare nelle fasi di remissione. Dall’analisi delle curve di sopravvivenza emerge come il rischio di perdita visiva aumenti progressivamente, fino a raggiungere il 25% a 10 anni(47).
La prevalenza delle manifestazioni cliniche nell’ambito dell’impegno neurologico, è risultata non del tutto sovrapponibile alla distribuzione descritta nelle casistiche di diversa nazionalità; tale dato potrebbe essere attribuibile ad un differente profilo geografico di malattia o ad una diversa selezione dei pazienti; in particolare è verosimile che esista un bias di selezione, in quanto i pazienti che più frequentemente afferiscono alla nostra U.O. in regime di ricovero hanno un’attività di malattia più severa. Esistono tuttavia grosse divergenze in letteratura per quello che riguarda la prevalenza dell’impegno neurologico in funzione dei criteri di scelta e di valutazione dei sintomi, delle manifestazioni cliniche e degli esami strumentali(48). La mancanza di parametri standardizzabili e quantificabili, e il non accordo sull’inclusione o esclusione di sintomi, quali la cefalea e l’impegno psichiatrico, in assenza di alterazioni organiche obiettivabili, rendono ulteriormente disomogenei i dati da valutare.
La disponibilità di indici di attività di malattia standardizzati, al momento peraltro assenti, potrebbe avere un ruolo, non solo nella gestione quotidiana dei pazienti, ma anche nello studio dei differenti profili di presentazione clinica della malattia.
Per quanto riguarda la terapia, la maggior parte dei pazienti risponde alle terapie più tradizionalmente utilizzate; i pazienti che effettuano terapia con farmaci biologici(49), che allo stato attuale sono quelli non responsivi ad altri trattamenti e con impegno più grave, hanno mostrato buona risposta al trattamento, sia in termini di efficacia che di tollerabilità.
BIBLIOGRAFIA
1. Yazici H., Behcet's syndrome: an update. Curr Rheumatol Rep. 2003 Jun;5(3):195-9.
2. Chang HK et al, Validation of the classification criteria commonly used in Korea and a modified set of preliminary criteria for Behcet's disease: a multi-center study. Clin Exp Rheumatol. 2004Jul-Aug;22(4 Suppl 34):S21-6.
3. Muhaya M et al, Behcet's disease in Japan and in Great Bri-tain: a comparative study. Ocul Immunol Inflamm. 2000 Sep;8(3):141-8.
4. Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mor-tality and morbidity of Behçet’s syndrome: a 2-decade outcome sur-vey of 387 patients followed at a dedicated center. Medicine 82:60–76, 2003.
5. Takeno M, Ishigatsubo Y., Behcet's disease and familial Medi-terranean fever. Intern Med. 2006;45(13):805-6.
6. Fietta P., Behcet's disease: familial clustering and immuno-genetics. Clin Exp Rheumatol. 2005 Jul-Aug;23(4 Suppl 38):S96-105.
7. Mizuki N et al, Molecular genetics (HLA) of Behcet's disease. Yonsei Med J. 1997 Dec;38(6):333-49.
8. Yazici H, Fresko I., Behcet's disease and other autoinflammat-ory conditions: what's in a name? Clin Exp Rheumatol. 2005 Jul-Aug;23(4 Suppl 38):S1-2.
9. Duzgun N, Characteristics of vascular involvement in Behcet's disease. Scand J Rheumatol. 2006 Jan-Feb;35(1):65-8
10. B'chir Hamzaoui S, Behcet's disease in Tunisia. Clinical study of 519 cases. Rev Med Interne. 2006 Aug 18.
11. Eguia A et al, Adamantiades-Behcet disease: an enigmatic process with oral manifestations. Med Oral Patol Oral Cir Bucal. 2006 Jan 1;11(1):E6-11.
12. Wakefield D, McCluskey P., Behcet's syndrome: ocular fea-tures in an Australian population. Aust N Z J Ophthalmol. 1990 May;18(2):129.
13. Mangelsdorf HC et al., Behcet's disease. Report of twenty-five patients from the United States with prominent mucocutaneous in-volvement. J Am Acad Dermatol. 1996 May;34(5 Pt 1):745-50.
14. Chang HK et al, The prevalence of atopy and atopic diseases in Behcet's disease. Clin Exp Rheumatol. 2003 Jul-Aug;21(4 Suppl 30):S31-4.
15. Zouboulis CC et al, Onset signs, clinical course, prognosis, treatment and outcome of adult patients with Adamantiades-Behcet's disease in Greece. Clin Exp Rheumatol. 2003 Jul-Aug;21(4 Suppl 30):S19-26
16. Yucel A et al, Clinical evaluation of Behcet's disease: a five year follow-up study. J Dermatol. 2005 May;32(5):365-70.
17. Akman-Demir G et al, Clinical patterns of neurological involve-ment in Behcet's disease: evaluation of 200 patients. The Neuro-Be-hcet Study Group. Brain. 1999 Nov;122 ( Pt 11):2171-82.
18. Pearce JM., Neurological symptoms of Adamantiades-Behcet'-s Adamantiades-Behcet'-syndrome. J Neurol NeuroAdamantiades-Behcet'-surg PAdamantiades-Behcet'-sychiatry. 2006 Aug;77(8):956-7.
19. Lo Monaco A. et al, Neurological involvement in North Italian patients with Behcet disease. Rheumatol Int. 2006 Oct;26(12):1113-9.
20. Kidd D. et al, The prevalence of headache in Behcet's syn-drome. Rheumatology (Oxford). 2006 May;45(5):621-3.
21. Saip S et al, Headache in Behcet's syndrome. Headache. 2005 Jul-Aug;45(7):911-9.
22. Akman-Demir G, Serdaroglu P, Tasci B., Clinical patterns of neurological involvement in Behcet's disease: evaluation of 200 pa-tients. The Neuro-Behcet Study Group. Brain. 1999 Nov;122
23. Borhani-Haghighi A et al, Neurological manifestations of Be-hcet s disease. Saudi Med J. 2006 Oct;27(10):1542-6.
24. Serdaroglu P. , Behcet's disease and the nervous system. J Neurol. 1998 Apr;245(4):197-205.
25. Yazici Y et al, Behcet's disease: does lack of knowledge result in under-diagnosis? Clin Exp Rheumatol. 2004 Jul-Aug;22(4 Suppl 34):81-2.
26. Vidaller Palacin A, Robert Olalla J, Sanuy Jimenez B, Rufi Rigau G, Folch Civit J, Charte Gonzalez A, Behcet's disease therapy review. An Med Interna. 2002 Nov;19(11):594-8.
27. Sfikakis PP. et al, Behcet's disease: a new target for anti-tumour necrosis factor treatment. Rheum Dis. 2002 Nov;61 Suppl 2:ii51-3.
28. Triolo G et al, Anti-tumour necrosis factor monoclonal antibody treatment for ocular Behcet's disease. Ann Rheum Dis. 2002 Jun;61(6):560
29. Licata G et al, Anti-tumour necrosis factor alpha monoclonal antibody therapy for recalcitrant cerebral vasculitis in a patient with Behcet's syndrome. Ann Rheum Dis. 2003 Mar;62(3):280-1.
30. Evereklioglu C. Current concepts in the etiology and treatment of Behçet disease. Surv Ophthalmol 2005; 50:297-350
31. Tugal-Tutkun I, Önal S, Altan
Yaycioglu R, et al. Uveitis in Behc et’s disease:̧ an analysis of 880 patients. Am J Ophthalmol 2004; 138:373–80.
32. Peizeng Y, Wang F, Quianli M, et al. Clinical features of Chinese patients with Behçet’s disease. Ophthalmol 2008; 115:312-318.
33. Ramsay A, Lightman S. Hypopyon uveitis. Surv Ophthalmol 46:1–18, 2001.Yates PA, Michelson JB., Behcet disease. Int Ophthal-mol Clin. 2006 Spring;46(2):209-33
34. Sheu SJ, Yang CA. Macular hole in Behçet’s disease. J Med Sci 2004; 20:558–62.
35. Yalvac ̧ IS, Sungur G, Turhan E, et
al. Trabeculectomy with
mitomy-cin-C in uveitic glaucoma associated with Behc et disease. J̧ Glaucoma 2004; 13:450–3.
36. Sakamoto M, Akazawa K, Nishioka Y, et al. Prognostic factor of vision in patients with Behçet’s disease. Ophthalmology 1995; 102:317–21.
37. Evereklioglu C. Managing the symptoms of Behc et’s dis- ease. Expert Opin Pharmacother ̧ 2004; 5:317–28.
38. Jabs DA, Nussenblatt RB, Rosenbaum Jt, et al. Standardiza-tion of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509-516. 39. Pipitone N et al, Clinical manifestations of Behcet's disease in 137 Italian patients: results of a multicenter study. Clin Exp Rheumat-ol. 2004;22(6 Suppl 36):S46-51.
40. Yoshida A et al, Comparison of patients with Behcet's disease in the 1980s and 1990s. Ophthalmology. 2004 Apr;111(4):810-5.
41. Ambresin A, Tran T, Spertini F, Herbort C., Behcet's disease in Western Switzerland: epidemiology and analysis of ocular involve-ment. Ocul Immunol Inflamm. 2002 Mar;10(1):53-63.
42. Demiroglu H, Barista I, Dundar S. Risk factor assessment and prognosis of eye involvement in Behçet’s disease in Turkey. Ophthal-mology 1997;104:701–705.
43. Ozdal PC, Ortac S, Taskintuna I, Firat E. Posterior segment involvement in ocular Behçet’s disease. Eur J Ophthalmol 2002;12:424–431.
44. Colvard DM, Robertson DM, O’Duffy JD. The ocular manifest-ations of Behçet’s disease. Arch Ophthalmol 1977;95:1813–1817.
45. BenEzra D, Cohen E. Treatment and visual prognosis in Be-hçet’s disease. Br J Ophthalmol 1986;70:589–592.
46. Mishima S, Masuda K, Izawa Y, Mochizuki M, Namba K. Be-hçet’s disease in Japan: ophthalmological aspects. Trans Am Oph-thalmol Soc 1979;77:225–279.
47. Ilknur T, Sumru O, Rana A et al. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol 2004;138:373-380.
48. Ozcan C. et al, Proton MRS in Behcet's disease with and without neurological findings. Neuroradiology. 2003 Dec;45(12):860-4.
49. Sfikakis PP. et al, Behcet's disease: a new target for anti-tumour necrosis factor treatment. Rheum Dis. 2002 Nov;61 Suppl 2:ii51-3.