2
3 RIASSUNTO
ABBREVIAZIONI 1. INTRODUZIONE
1.1. Il nitrossido (NO)………...………..14
1.2. Enzimi che sintetizzano il NO: nitrossido sintasi (NOS)……...21
1.2.1. Struttura e meccanismo catalitico della NOS……….23
1.3. La NOS endoteliale (eNOS)………29
1.3.1. Generalità……….29
1.3.2. Gene della eNOS: regolazione trascrizionale e post-trascrizionale………..30
1.3.3. Regolazione dell’attività della eNOS...32
1.3.3.1. Miristoilazione e palmitoilazione……….32
1.3.3.2. Fosforilazione………...35
1.3.3.3. S-Nitrosilazione………41
1.3.3.4. Acetilazione………...44
1.3.3.5. Interazioni proteine-proteine………...45
1.3.4. Localizzazione della eNOS………..57
1.3.4.1. Localizzazione della eNOS nelle caveolae………...59
1.3.4.2. Significato della eNOS golgiana………..61
1.3.4.3. Interazioni con il citoscheletro……….62
1.4. Ruoli fisiologici della eNOS………...65
1.4.1. eNOS e vasodilatazione………...65
1.4.2. Protezione dall’apoptosi………..69
1.4.2.1. Caratteristiche generali dell’apoptosi………..69
1.4.2.2. NO, eNOS ed apoptosi………..70
1.4.3. Angiogenesi e migrazione………74
4
1.4.3.2. Ruolo della eNOS e del NO da esso prodotto nella
migrazione delle cellule endoteliali………...76
1.5. Scopo della tesi………....83
2. MATERIALE E METODI 2.1. Colture cellulari………...86
2.2. Lisi cellulare – quantificazione delle proteine e del DNA…….87
2.3. Immunoblot……….88
2.4. Saggio di proliferazione………..90
2.5. Saggio di adesione………..91
2.6. Saggio di migrazione di Boyden……….92
2.7. Trattamento con i farmaci………...96
2.8. Immunofluorescenza………..97
2.9. Misura del potenziale di membrana mitocondriale…………...98
2.10. Proteomica………...100
2.10.1. Preparazione dei campioni e separazione in 2-D………....100
2.10.2. Acquisizione ed analisi delle immagini………...103
2.11. Analisi statistica………...103
3. RISULTATI 3.1. Linee eNOS-HeLa -TetOff……… .106
3.2. L’espressione cronica della eNOS diminuisce la migrazione di cellule HeLa-TetOff……….108
3.3. Inibizione della capacità migratoria delle cellule HeLa dopo trattamento cronico con concentrazioni nanomolari di donatori di NO……….…113
3.4. Aumento della migrazione delle cellule HUVEC dopo trattamento cronico con L-NAME………..………119
5
3.5. Possibili meccanismi d’azione del NO basale sulla migrazione………..………..124 3.5.1. Mancanza di effetto dell’espressione della eNOS sulla adesione……….…….……..124 3.5.2. Mancanza di effetto dell’espressione della eNOS su Hypoxia Inducibile Factor 1α (HIF-1α)………..………..…....126 3.5.3. Mancanza di effetto dell’espressione della eNOS sull’organizzazione della rete mitocondriale………..…….128 3.5.4. Mancanza di effetto dell’espressione della eNOS sulla funzionalità energetica dei mitocondri………..……..128 3.5.5. L’espressione prolungata della eNOS diminuisce la fosforilazione di Akt in risposta al siero………..……131 3.5.6. Effetto di concentrazioni nanomolari di DETA-NO sulla fosforilazione di Akt in risposta al siero………..…134 3.6. Analisi proteomica delle cellule esposte a NO basale……..…137 4. DISCUSSIONE
4.1. L’esposizione cronica a basse concentrazioni di NO attenua la capacità migratoria di cellule HeLa ed endoteliali……….146 4.2. Possibili meccanismi alla base dell’effetto del NO basale sulla migrazione………149 4.3 L’effetto bifasico del NO………152 4.4. Analisi del proteoma di cellule esposte cronicamente a NO basale………153 4.5. CONCLUSIONI………154 PRODUZIONE SCIENTIFICA DEL LAVORO DI TESI
6
7
Il nitrossido (NO), prodotto nelle cellule endoteliali dall’enzima NO sintasi endoteliale (eNOS) è di fondamentale importanza nella fisiologia del sistema cardiovascolare non soltanto perché causa il rilassamento della muscolatura liscia vasale ma anche per i suoi effetti sulle cellule endoteliali stesse. In particolare, è ben conosciuto che la generazione di NO è richiesto per la risposta migratoria delle cellule endoteliali al Vascular Endothelial Growth Factor (VEGF) e ad altri fattori motogeni; la chemiotassi delle cellule endoteliali verso questi fattori costituisce una componente essenziale del processo dell’angiogenesi. Tuttavia, il NO non è prodotto esclusivamente in risposta a un dato stimolo, ma vi è anche una produzione cronica di bassi livelli del gas, dovuta all’attività basale della eNOS. Mentre moltissimi studi sono stati eseguiti con lo scopo di comprendere gli effetti del NO prodotto acutamente in risposta a svariati stimoli, molto meno studiati sono gli effetti della produzione cronica basale di basse concentrazioni del gas sul fenotipo cellulare.
Lo scopo della mia tesi è stato di valutare gli effetti della esposizione cronica a bassi livelli di NO sulla migrazione chemiotattica e sul signaling che genera la risposta migratoria. A questo scopo, ho utilizzato come modello cellulare cloni HeLa-TetOff con espressione inducibile dell’enzima eNOS. A complemento di questi studi, ho condotto esperimenti su cellule HeLa non transfettate, che non esprimono eNOS endogena, trattate cronicamente con basse concentrazioni di un donatore lento di NO (DETA-NO) e su cellule endoteliali (HUVEC) trattate cronicamente con l’inibitore competitivo
8
La migrazione delle cellule HeLa è stata analizzata in camerette di Boyden, utilizzando il siero bovino come sorgente di fattori motogeni. Inaspettamente, ho osservato che l’espressione della eNOS inibisce potentemente (>80%) la migrazione chemiotattica; l’effetto
viene parzialmente bloccato dal trattamento cronico con l’L-NAME, e
anche da un inibitore specifico (ODQ) dell’enzima guanilato ciclasi, effettore del NO. Coerentemente con l’effetto ottenuto con la espressione della eNOS, il pretrattamento cronico di cellule HeLa non transfettate con basse concentrazioni (10-100 nM) di DETA-NO ha inibito la migrazione eseguita in assenza del donatore. Un effetto simile è stato ottenuto mediante trattamento con un analogo del ciclico-GMP, mentre l’effetto del DETA-NO è stato bloccato dal trattamento con l’ODQ. Infine, cellule HUVEC, trattatate
cronicamente con L-NAME per 48 h e poi soggette a saggio di
migrazione in assenza dell’inibitore, mostravano una capacità migratoria aumentata rispetto ai controlli non trattati, sia verso siero che verso VEGF. Così, in tre sistemi diversi ho dimostrato che il NO basale, a differenza di quello prodotto acutamente, abbassa la capacità migratoria delle cellule esposte al gas.
Per chiarire il meccanismo tramite il quale la esposizione cronica a bassi livelli di NO diminuisce la capacità migratoria delle cellule ho indagato l’effetto dell’espressione della eNOS: sulla
adesione; su Hypoxia inducible Factor 1α (HIF-1α);
sull’organizzazione della rete mitocondriale; sulla funzionalità energetica dei mitocondri; sulla attivazione della via PI3K/Akt in risposta al siero. Tra queste indagini solo l’ultima ha dato un risultato positivo.
9
Per valutare la responsività della via PI3K/Akt, ho mantenuto le cellule senza siero per tutta la notte, e ho poi esposto le celllule a siero per 15 minuti. Il rapporto tra Akt fosforilato (pAkt) e Akt totale è stato determinato prima e dopo l’aggiunta di siero ed è una misura della capacità delle cellule di attivare questa via di signalling. Ho osservato che le cellule indotte ad esprimere eNOS hanno una risposta Akt attenuata in confronto alle cellule che non sono indotte a esprimere l’enzima. Un risultato simile è stato osservato con cellule HeLa non
transfettate esposte cronicamente a 0,1 M DETA-NO. Questi risultati
suggeriscono che il NO basale porti a una desensitizzazione della via PI3K/Akt; poiché questa via è di fondamentale importanza per la risposta chemiotattica, la sua attenuazione potrebbe essere la causa della diminuita migrazione da me osservata in cellule che producono o che sono esposte cronicamente a basse concentrazioni di NO.
Per generare un quadro globale degli effetti della NO basale sul fenotipo cellulare, mi sono avvalsa di tecniche proteomiche. Ho analizzato mediante elettroforesi bi-dimensionale estratti ottenuti da cellule indotte o non-indotte ad esprimere la eNOS, e da cellule non transfettate, trattate o non trattate con DETA-NO. Ho identificato alcuni cambiamenti comuni alle cellule trattate con DETA-NO e a quelle che esprimono la eNOS ma al momento della stesura di questa tesi, gli spot individuati devono essere ancora identificati mediante spettrometria di massa.
In conclusione, i miei risultati hanno evidenziato un inatteso effetto inibitorio del NO basale sul fenotipo migratorio e sul PI3K/Akt signalling di cellule HeLa ed endoteliali, e illustrano come la quantità
10
e le modalità di rilascio del gas possano influenzare in modo drammtico la risposta della cellula.
11 ABBREVIAZIONI
ACh, acetilcolina Akt, protein chinasi B
AMPK, proteina chinasi AMP-dipendente ATP, adenosina-trifosfato
BAECs cellule endoteliali di aorta bovina
BH4, la tetraidrobiopterina
Ca2+, calcio
Ca2+/CaM, complesso calcio/calmodulina
CaMKII, protein chinasi Ca2+/CaM-dipendente
cav-1, caveolina-1 cav-2, caveolina-2 cav-3, caveolina-3
CcO, citocromo c ossidasi
cellule eNOS-HeLa-TetOff, cellule HeLa-TetOff stabilmente
transfette con eNOS bovina
cGMP, guanosin-monofosfato ciclico
CoCl2, cloruro di cobalto
CsH, ciclosporina H
CTGF, connective tissue growth factor Cys, cisteina
DETA-NO, 2,2’-(Hydroxynitrosohydrazino)bis-ethanamine DETA-NO, donatore lento di NO
+dox, cellule eNOS-HeLa-TetOff non indotte ad esprimere la eNOS -dox o +dox 0,05 ng/ml, cellule eNOS-HeLa-TetOff indotte ad esprimere la eNOS
dox, doxiciclina
EDHF, Endothelium Derived Hyperpolarizing Factor eNOS, NO sintasi endoteliale
FAD, flavin adenina dinucleotide
FCCP, idrazone fenilico del cianuro di carbonile di p-trifluoromethossile
FGF, fibroblast growth factor FMN, flavin mononucleotide HDL, lipoproteine ad alta densità
HeLa-TetOff NT, HeLa-TetOff non transfettate HGF, Hepatocyte growth factor
HIF, Hypoxia Induced Factors Hsp90, Heat shock protein 90
12
HUVEC, Human Umbilical Vein Endothelial Cells IGF-1, fattore di crescita insulino-simile 1
IL-8, interleuchina-8 iNOS, NOS inducibile
K+, potassio
LDL, lipoproteine a bassa densità
L-NAME, Nω-Nitro-L-arginine methyl ester
Met, metionina
MMP-13, metalloproteinasi mtNOS, NOS mitocondriale
NADPH, nicotinammide adenina dinucleotide fosfato ridotto NF-kB, Nuclear Factor-Kappa B
NMT, N-miristoil transferasi nNOS, NOS neuronale NO, nitrossido
NO2, diossido di azoto
NOS, ossido nitrico sintasi
NOSIP, eNOS-Interacting Protein NOSTRIN, eNOS TRafficking INducer
N-WASP, neuronal Wiskott-Aldrich syndrome protein
O2-, anione superossido
O2, ossigeno
ODQ, 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one
ONOO-, perossinitrito
PAEC, pulmonary artery endothelial cells PDGF, platelet derived-growth factor
PEDF, pigment epithelium-derived growth factor
PI3K, Phosphatidylinositol 3’-kinase
PKA, proteina chinasi A PKC, protein chinasi C PKG, protein-chinasi G PP1, protein fosfatasi 1 PP2A, fosfatasi 2
PP2A, protein fosfatasi 2 PP2B, calcineurina
RNS, specie reattive dell’ossido nitrico ROS, specie reattive dell’ossigeno S1P, sfingosina 1-fosfato
Ser, serina
13
siRNA, short interfering RNA
TGF- 1, fattore di crescita trasformante-β1 TGF-α, trasforming growth factor alpha
TMRM, TetraMethyl Rhodamine Methylesther TNF-α, tumor necrosis factor alpha
TSP-1, la trombospondina Tyr, tirosina
VEGF, Vascular Endothelial Growth Factor
VEGFR1 o FLT1, Vascular Endothelial Growth Factor Receptor-1 VEGFR2 o KDR Vascular Endothelial Growth Factor Receptor-2
Zn2+, ione zinco
PLCγ, fosfolipasi Cγ
14
15 1.1. IL NITROSSIDO (NO)
Il NO è una molecola ad elevata reattività che, pur essendo potenzialmente tossica, è implicata in una vasta gamma di processi fisiologici. Scoperto inizialmente per la sua proprietà di potente vasodilatatore, è stato successivamente implicato in un grande numero di fenomeni fisiologici e patologici (Moncada et al., 1991).
Il NO ha una funzione importante già all’inizio della vita, quando la sua produzione nei gameti maschili è richiesta per l’attivazione delle uova immediatamente dopo la fecondazione (Kuo et al., 2000). Nell’adulto le funzioni del NO sono molteplici e dipendono dall’organismo considerato e dal tipo di cellula in cui viene prodotto. Esso è il più importante vasodilatatore endogeno. Quando la pressione arteriosa aumenta eccessivamente, l'organismo si difende sintetizzando NO che, dilatando le pareti dei vasi, contribuisce all'abbassamento della pressione. Al contrario, l'inibizione della sintesi del NO determina un aumento delle resistenze periferiche ed un conseguente innalzamento della pressione arteriosa. Esso è anche responsabile della permeabilità vasale; inoltre protegge l’endotelio da fattori aterogeni inibendo l’aggregazione piastrinica e l’adesione delle piastrine e dei monociti circolanti. Il NO gioca importanti ruoli anche all’interno delle cellule endoteliali stesse, regolando l’apoptosi, la crescita cellulare, l’angiogenesi e in particolar modo la migrazione (Murohara et al., 1998; Dimmeler et al., 2000).
Oltre ai suoi molteplici ruoli nel sistema circolatorio, il NO è parte integrante della risposta infiammatoria e nel sistema nervoso centrale ha un ruolo fondamentale come neuromodulatore (Moncada et al., 1991; Duda et al., 2004; Belvisi et al., 1991; Gibbs, 2003).
16
La poliedricità del NO nella modulazione di diversi processi fisiologici e patologici deriva fondamentalmente dalla sua complessa chimica. Il NO è una molecola che presenta un elettrone spaiato nel
suo orbitale 2p-(NO.) (Marletta et al., 1990) e può quindi
trasformarsi, attraverso la rimozione o l’aggiunta di un elettrone in:
NO+ (catione nitroso), NO- (anione nitrossido).
Molte delle azioni biologiche attivate dal NO dipendono dalla sua capacità di reagire con metalli di transizione o di modificare residui amminoacidici delle proteine. Alcune di queste modificazioni (nitrazione della tirosina (Tyr), ossidazione della metionina (Met)) sono irreversibili e potenzialmente dannose; altre invece, come la S-nitrosilazione di residui cisteinici (Cys) o la S-nitrosilazione del gruppo eme, sono reversibili e possono regolare finemente l’attività delle proteine.
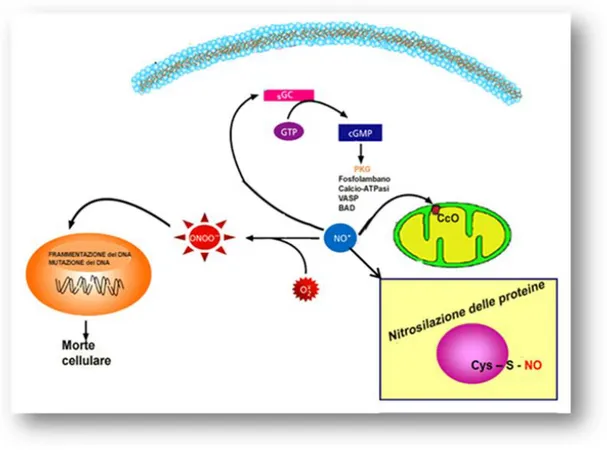
I meccanismi molecolari attraverso cui il NO esercita le sue funzioni possono essere così riassunti (Fig. 1):
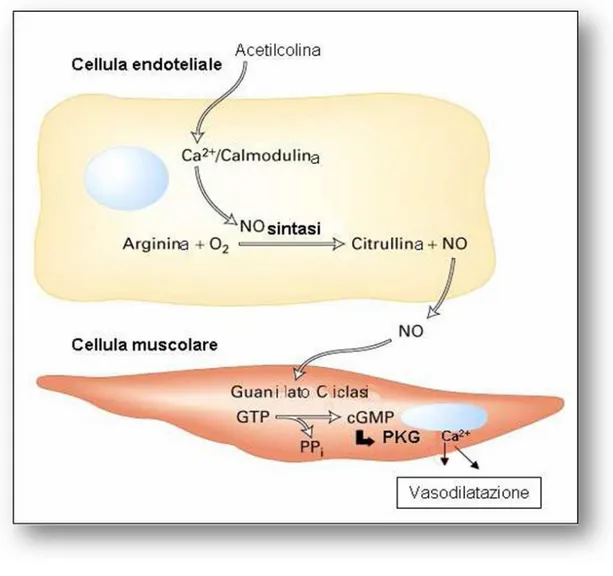
Attivazione della guanilato ciclasi: Il NO attraverso la nitrosilazione
del gruppo eme della guanilato ciclasi solubile (sGC) ne stimola l’attività, regolando così una varietà di importanti risposte fisiologiche, tra cui la proliferazione e il differenziamento di numerosi tipi cellulari e il rilassamento della muscolatura vasale. La sGC è una proteina eterodimerica costituita da una subunità α e una β, contenente un gruppo eme in grado di legarsi al NO, formando un complesso Fe-NO (Fig. 2). La formazione di tale complesso induce un cambiamento conformazionale dell’enzima tale da attivarlo e portare all’accumulo del guanosin-monofosfato ciclico (cGMP), un noto secondo messaggero in grado di stimolare la protein-chinasi G (PKG) ed i
17
Figura 1. Meccanismi molecolari di segnalazione del NO. Il NO
regola l’attività di alcune emoproteine come la guanilato ciclasi solubile (sGC) e la citocromo c ossidasi (CcO). Interagisce con
l’anione superossido (O2
-) generando perossinitriti (OONO-), reazione
molto importante nell’induzione del danno e della morte cellulare. Nitrosila i residui di cisteina di varie proteine bersaglio regolandone l’attività.
18
Figura 2. Rappresentazione schematica della struttura della guanilato ciclasi solubile. Il legame del NO con lo ione Fe2+ dell’eme determina una modificazione conformazionale dell’enzima che in tal modo si attiva e catalizza la sintesi di cGMP.
19
canali ionici cGMP-dipendenti, coinvolti nel rilassamento muscolare (Lincoln et al., 1994).
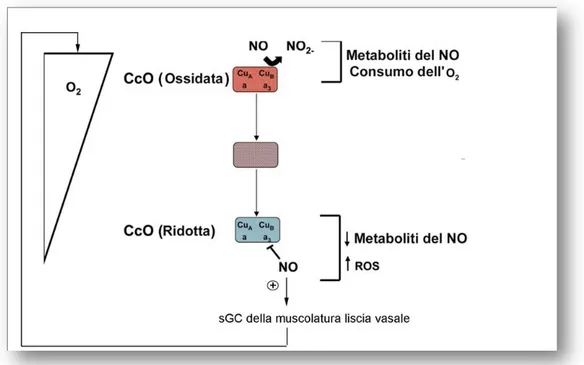
La modulazione dell’attività della sGC ha una importante
ripercussione sul consumo di ossigeno (O2) a livello dei mitocondri.
La citocromo c ossidasi (CcO o complesso IV nella catena di trasporto
degli elettroni mitocondriale) è il sito primario di consumo di O2
cellulare e, come tale, è fondamentale per la fosforilazione ossidativa e per la produzione di adenosina-trifosfato (ATP). Il NO modula l'attività della CcO in funzione della concentrazione intracellulare di
O2 e del conseguente stato redox dell’enzima (Fig. 3). Quando le
concentrazioni di O2 sono elevate, la CcO è prevalentemente in uno
stato ossidato, consuma O2 e metabolizza il NO convertendolo in
nitriti. Quando le concentrazioni di O2 diminuiscono, la CcO è in uno
stato ridotto, il NO non viene metabolizzato, quindi la sua concentrazione aumenta ed aumentono le specie reattive dell’ossigeno (ROS). Studi recenti hanno mostrato che, quando la concentrazione di
O2 è molto bassa, il NO attiva la guanilato ciclasi, la quale una volta
attiva induce vasodilatazione con conseguente apporto di O2 (Palacios
et al., 2007) (Fig. 3). Quindi il NO agisce come un regolatore della respirazione e in condizione di ipossia attiva meccanismi di protezione (Taylor and Moncada 2010).
-Modulazione dell’attività della citocromo ossidasi: Come appena
descritto, la CcO è in grado di modulare le concentrazioni intracellulari di NO. Viceversa, il NO è in grado di modulare l’attività della CcO. Infatti il messaggero gassoso lega questo enzima in modo reversibile, occupando lo stesso sito di legame riconosciuto dall’ossigeno. Tale legame inibisce la funzionalità della CcO
20
Figura 3. Modulazione dell'attività della citocromo c ossidasi. In
presenza di elevate [O2] la CcO, è prevalentemente in uno stato
ossidato, consuma O2 e metabolizza il NO convertendolo in nitriti.
Quando, la [O2]diminuisce, la CcO è in uno stato ridotto, il NO non
viene metabolizzato, quindi la sua concentrazione aumenta ed aumentono le ROS. Si veda il testo per ulteriori dettagli. (Modificata da Taylor and Moncada 2010).
21
determinando un rallentamento della respirazione (Clementi et al., 1999).
-Nitrazione ed S-Nitrosilazione: La nitrazione della tirosina è una
modificazione irreversible e spesso dannosa riscontrata in alcune malattie neurodegenerative come il morbo di Alzheimer, di Parkinson (Ebadi et al., 2005) ed in patologie cardiovascolari (Turko and Murad, 2002). In due studi interessanti è stata dimostrata un’azione fisiologica della nitrazione a livello della matrice extracellulare: la nitrazione della Tyr 388 della metalloproteinasi MMP-13 causa l’attivazione di questa proteasi, con conseguente degradazione di proteine della matrice extracellulare ed aumento della migrazione di cellule endoteliali e della guarigione da ferite (Lopez Rivera et al., 2005; Lizarbe et al., 2008).
Come già accennato, a differenza della nitrazione la nitrosilazione è una modifica reversibile, e notevole importanza è attribuita alla capacità del NO di nitrosilare il gruppo eme di svariate proteine; l’esempio meglio caratterizzato è quello della guanilato ciclasi che comporta l’attivazione enzimatica con conseguente aumento del cGMP. La regolazione dei livelli del cGMP è responsabile di molte delle risposte NO-dipendenti, tuttavia molti studi hanno focalizzato l’attenzione anche sulle modifiche post-traduzionali indotte dal NO mediante nitrosilazione dei residui cisteinici (S-nitrosilazione). Tramite tale reazione il NO regola l’attività di moltissime molecole: enzimi metabolici, proteine chinasi e fosfatasi, recettori e canali ionici di membrana, proteine del citoscheletro, fattori di trascrizione e molti altri (Hess et al., 2005).
22
-Azioni tossiche per interazione del NO con l’anione superossido
(O2
-): a causa dell’alta reattività del NO, molte delle reazioni in cui
esso è coinvolto dipendono dalla sua concentrazione locale, dallo stato ossidativo cellulare e dalla disponibilità delle molecole bersaglio. Il
NO può reagire rapidamente con l’O2
formando specie reattive
dell’ossido nitrico (RNS), fra i quali il perossinitrito (ONOO
-) (Moreno and Pryor, 1992; Pacher et al., 2007) ed il diossido di azoto
(NO2). Le RNS, allo stesso modo delle ROS, possono reagire con tutte
le biomolecole: il DNA, i gruppi tiolici degli aminoacidi ed i metalli portando all’ossidazione e alla nitrazione. Se prodotto ad elevate concentrazioni (dell’ordine di 1-3 µM) il NO, con i suoi derivati, può avere effetti deleteri per tutte le funzioni cellulari portando, per esempio, alla perdita di funzioni enzimatiche, all’alterazione dell’integrità di membrana ed a mutazioni del DNA (Fabbri et al., 2005).
1.2. Enzimi che sintetizzano il NO: nitrossido sintasi (NOS)
La famiglia delle ossido nitrico sintasi (NOS) è costituita da tre isoforme, clonate e purificate tra il 1991 e il 1994.
Le tre isoforme umane della NOS sono prodotte da tre geni distinti (Tabella 1); si distinguono sia per localizzazione intracellulare che per regolazione; inoltre presentano diverse proprietà catalitiche e diversa sensibilità agli inibitori. Sono comunemente chiamate (Alderton, 2001):
NOS-23
1), identificata per prima ed espressa nelle cellule del sistema nervoso centrale e periferico;
2) NOS inducibile (iNOS, nota anche come II o
NOS-2), è l’isoforma la cui espressione è inducibile in una vasta gamma di cellule e tessuti;
3) NOS endoteliale (eNOS, nota anche NOS-III o NOS-3),
identificata per la prima volta nelle cellule dell’endotelio vascolare, ma presente anche in molti altri tipi cellulari.
Isoforme Dimensione e struttura Localizzazione Numero di umane della gene cromosomiale amminoacidi NOS e dimensioni nNOS (NOS-1) 29 esoni, 28 introni, 12q24.2-12q24.3 1434 aa, 161 kDa 200 kpb del cromosoma 12
iNOS (NOS-2) 26 esoni, 25 introni, 17cen-q11.2 1153 aa, 131 kDa 37 kpb del cromosoma 17
eNOS (NOS-3) 26 esoni, 25 introni, 17cen-q11.2 1203 aa, 133 kDa 21-22 kpb del cromosoma 7
Tabella 1 (Alderton, 2001).
In passato queste isoforme sono state anche distinte sulla base della loro espressione costitutiva (nNOS, eNOS) o inducibile (iNOS) e
della loro calcio (Ca2+)-dipendenza (eNOS, nNOS) o (Ca2+
)-indipendenza (iNOS). Inoltre per ciascuna isoforma, sono state identificate anche delle varianti di splicing e non si può escludere che in futuro potranno essere identificati altri prodotti genici che estenderanno ulteriormente questa famiglia di proteine.
Oltre alle tre canoniche forme di NOS, esiste anche un'altra isoforma dell’enzima denominata NOS mitocondriale (mtNOS), presente all’interno dei mitocondri ed espressa costitutivamente (Ghafourifar and Richter, 1997; Ghafourifar and Cadenas, 2005; Kanai et al., 2001; Carreras and Poderoso, 2007). Recenti studi hanno
24
dimostrato che la mtNOS è l’isoforma nNOS che ha subito alcune modificazioni post-traduzionali quali la fosforilazione nel residuo di serina in posizione 1412 (Ser1412) e una miristoilazione insolita su un residuo interno della proteina (Carreras et al., 2007).
1.2.1. Struttura e meccanismo catalitico della NOS
Tutte le isoforme della NOS presentano una struttura costituita da
due domini. Il dominio ossigenasico all’NH2-terminale contiene i siti
di legame per l’eme, la tetraidrobiopterina (BH4) e l’L-arginina, ed è
connesso al dominio riduttasico, COOH-terminale, dalla regione deputata al riconoscimento del complesso calcio/calmodulina
(Ca2+/CaM). Il dominio riduttasico contiene i siti di legame per il
flavin adenina dinucleotide (FAD), il flavin mononucleotide (FMN) ed il nicotinammide adenina dinucleotide fosfato ridotto (NADPH).
Quando le concentrazioni di Ca2+ intracellulari aumentano, il Ca2+ si
lega alla CaM formando il complesso Ca2+/CaM che interagisce con il
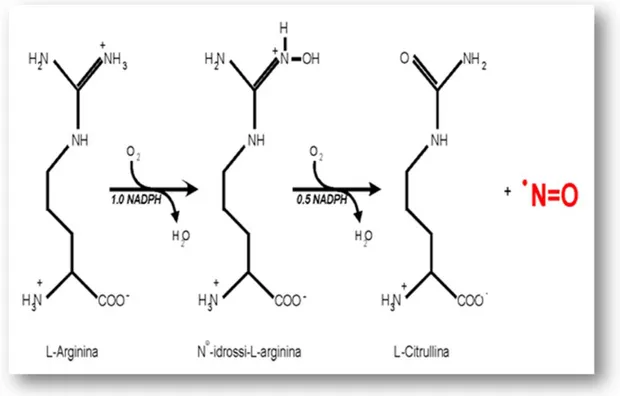
domino riduttasico della NOS. In seguito a tale legame, i due domini dell’enzima interagiscono e si allineano. Quindi gli elettroni, donati dal NADPH al domino riduttasico mediante la via FAD-FMN, passano al domino ossigenasico ed interagiscono con l’eme e il
cofattore BH4 per catalizzare la reazione che genera citrullina e NO a
partire dalla L-arginina (Fig. 4 e 5).
Le NOS sono enzimi comunemente considerati dimerici ma non è ben chiaro se la dimerizzazione oltre a rendere stabile la struttura sia necessaria per la loro attività (Xie et al., 1996; Li et al., 1999;
25
Figura 4. Rappresentazione della regolazione della NOS mediante il complesso Ca2+/CaM. Quando le concentrazioni di Ca2+
intracellulari aumentano, il Ca2+ si lega alla CaM formando il
complesso Ca2+/CaM che interagisce con il domino riduttasico della
NOS. In seguito a tale legame, i due domini dell’enzima interagiscono e si allineano. Quindi gli elettroni, donati dal NADPH al domino riduttasico, mediante la via FAD-FMN passano al domino
ossigenasico ed interagiscono con l’eme e il cofattore BH4 per
catalizzare la reazione che genera citrullina e NO a partire dalla L
27
Figura 5. Schema della reazione di ossidazione catalizzata dalle NOS. La NOS usa O2 che viene ridotto dagli elettroni forniti dal
donatore NADPH e dal substrato. La sintesi di NO si articola in due successive ossidazioni. Nel primo passaggio si ha la formazione dell’intermedio stabile N-idrossi-L-arginina attraverso l’ossidazione di 1.0 equivalente di NADPH; nel secondo passaggio viene prodotto L-citrullina e NO attraverso l’utilizzo di 0.5 equivalenti di NADPH.
28
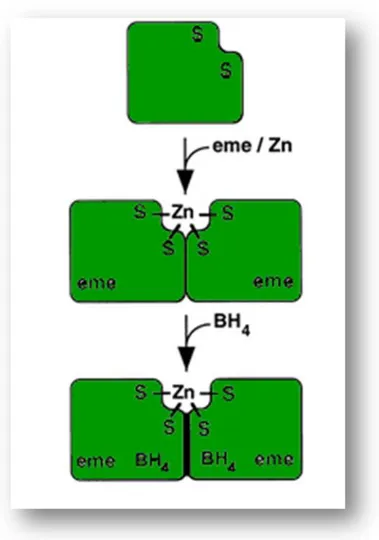
Hemmens et al., 2000). Alcuni autori hanno concluso che il trasferimento degli elettroni avvenga dal dominio riduttasico di un monomero al dominio ossigenasico del partner e che questo trasferimento trasversale sia necessario per l’attività (Erwin et al., 2006). Altri studi, invece non confermano tale ipotesi (Klatt et al., 1995; Hemmens et al., 2000). L’interfaccia di associazione di due monomeri coinvolge il dominio ossigenasico ed include il sito di
legame per la BH4: questa favorisce la formazione della tasca del sito
attivo contenente l’eme e l’L-arginina, ed include due residui di cisteina (Cisteina 94 e Cisteina 99) per monomero che sono
responsabili del legame di uno ione zinco (Zn2+) ai due monomeri
stessi (Fig. 6).
La reazione di ossidazione della NOS, inoltre, può generare O2
-invece che NO quando l’accettore finale degli elettroni non è
l’arginina ma l’O2. Tale processo, noto come “reazione non
accoppiata” della NOS, è mediato dal gruppo eme del dominio ossigenasico (Stroes et al., 1998) e dipende dalle concentrazioni del
substrato arginina e dal cofattore BH4 (Vasquez-Vivar et al., 1998;
Wever et al., 1997). In presenza di entrambi la NOS produce NO; quando la concentrazione di uno dei due elementi è relativamente
bassa, l’enzima genera l’anione O2
29
Figura 6. Dimerizzazione della NOS. I due monomeri che
costituiscono l’enzima, presentano due residui di Cisteina (S) in
posizione 94 e 99. Lo ione Zn2+ si lega ai residui di cisteina favorendo
30 1.3. La NOS endoteliale (eNOS) 1.3.1. Generalità
La eNOS, espressa soprattutto nelle cellule endoteliali, nei miociti e nelle piastrine, è essenziale per la regolazione del tono vascolare (Ignarro, 1989). Essa gioca un ruolo chiave nella regolazione della fisiologia cardiovascolare (Dudzinski et al., 2006), nell’aggregazione piastrinica (Radomski et al., 1987), nell’adesione delle piastrine e dei leucociti all’endotelio (Kubes et al., 1991; Radomski et al., 1987), nella proliferazione delle cellule muscolari (Garg and Hassid, 1989), nell’apoptosi delle cellule endoteliali (Hoffmann et al., 2001), nell’angiogenesi (Papapetropoulos et al., 1997) e nell’infiammazione acuta durante lo stravaso (Cirino et al., 2003). E’ stato documentato che la eNOS è espressa anche in altri tipi cellulari tra cui i mastociti, le cellule dell’epitelio renale, gli eritrociti ed i leucociti T e B (de Frutos et al., 2001; Lowenstein and Michel, 2006).
Dal momento che la produzione del NO deve essere attentamente controllata per rispondere ai diversi stimoli fisio-patologici, la eNOS è un enzima che è soggetto ad una serie di meccanismi di controllo che ne regolano finemente l’attività (Dudzinski and Michel, 2007).
31
1.3.2. Gene della eNOS: regolazioni trascrizionali e post trascrizionali
Il gene umano della eNOS contiene 26 esoni e 25 introni ed è localizzato sul cromosoma 7. La lunghezza dell’intera sequenza di lettura è 3609 bp, codificante per una proteina di circa 133 kDa.
Studi in vivo hanno dimostrato che la produzione della eNOS è fondamentale per la normale fisiologia endoteliale e che la sua espressione è soggetta a svariati meccanismi di controllo che comprendono la regolazione della trascrizione del gene eNOS ed alcune modificazioni post-trascrizionali (Searles, 2006).
In cellule endoteliali in coltura è stato dimostrato che stimoli quali lo shear stress, il fattore di crescita trasformante-β1 (TGF- 1) (Saura et al., 2002), la ciclosporina A (Lopez-Ongil et al., 1996), la lisofosfatidilcolina (Cieslik et al., 1998), l’acido linoleico ossidato, le statine (Hernàndez-Perera et al., 1998; Sen-Banerjee et al., 2005) e le ROS (Zhen et al., 2008) aumentano i livelli di mRNA della eNOS. Al contrario, il fattore di necrosi tumorale Tumor Necrosis Factor alpha (TNF-α) (Anderson et al., 2004), l’ipossia (Arnet et al., 1996), i lipopolisaccaridi, la trombina e le lipoproteine a bassa densità ossidate (LDL) (Liao et al., 1995) possono ridurre tali livelli.
Lo shear stress è uno dei più importanti fattori di regolazione della trascrizione dell’mRNA della eNOS: cellule in coltura (Uematsu et al., 1995, Topper et al., 1996; Weber et al., 2005) o animali da laboratorio (Sessa et al., 1994; Nadaud et al., 1996; Mattsson et al., 1997) sottoposti a stimoli che riproducono stress da stiramento presentano i livelli di tale mRNA aumentati. Alcuni autori (Davis et al., 2001) hanno proposto che tale aumento potrebbe essere attribuito a
32
due meccanismi distinti: un’attivazione transitoria della trascrizione del gene della eNOS seguita da una prolungata stabilizzazione dell’mRNA. E stato dimostrato che lo shear stress induce il legame del fattore trascrizionale Nuclear Factor-Kappa B (NF-kB) al promotore del gene della eNOS con conseguente attivazione della trascrizione, aumento dei livelli della proteina eNOS e successivo aumento della produzione di NO (Grumbach et al., 2005). Secondo questi autori, inoltre, il NO finisce per nitrosilare la subunità p50 di NF-kB inattivandolo e quindi chiudendo un’ansa di feedback negativo con blocco della via di regolazione indotta dallo shear stress.
Nelle condizioni in cui è stato osservato un aumento dell’mRNA della eNOS è stato rivelato anche un aumento della produzione di NO di circa due-tre volte. Anche se questo livello di modulazione può sembrare modesto, bisogna sottolineare che piccole variazioni dei livelli del NO possono determinare effetti fisiologici significativi. Per esempio, nel caso del rilassamento vascolare, la risposta al NO è dose dipendente; incrementi minuscoli delle concentrazioni del NO possono provocare grandi cambiamenti del tono vascolare (Myers et al., 1990; Sellke et al., 1990).
Oltre alla regolazione a livello trascrizionale, anche i controlli
post-trascrizionali sono importanti; questo perché l’emivita
dell'mRNA della eNOS è lunga, (10-35 h), e la sintesi della proteina può avvenire anche dopo che la trascrizione del gene è repressa. Le modifiche post-trascrizionali sono in gran parte mediate da elementi cis-acting RNA che si localizzano nelle regioni 5' e 3' non tradotte (UnTranslated Regions) dell’mRNA (5' e 3'-UTRs) (Grzybowska et al., 2001; Pesole et al., 2001).
33
1.3.3. Regolazione dell’attività della eNOS.
Una volta sintetizzata, l’eNOS è soggetta regolazione per
modificazioni post-traduzionali quali l’acilazione, la fosforilazione e
la nitrosilazione (Fleming and Busse, 2003). La sua localizzazione sub-cellulare e l’attività biologica sono regolate anche mediante l’associazione reversibile con numerose proteine regolatrici quali la CaM, la caveolina-1, NOSIP, NOSTRIN, Hsp90 ed altre.
I vari meccanismi di controllo non si escludono a vicenda, bensì sono strettamente correlati nel mantenere una corretta organizzazione spaziale e temporale del signalling della eNOS.
1.3.3.1. Miristoilazione e palmitoilazione
Delle tre isoforme la eNOS è l’unica a subire la N-miristoilazione e la S-palmitoilazione, modificazioni covalenti che ne favoriscono l’attività enzimatica promuovendo la sua localizzazione al complesso del Golgi e alla membrana plasmatica (Dudzinski and Michel, 2007).
La eNOS, a differenza delle altre due isoforme, presenta
all'NH2-terminale una sequenza consenso per la miristoilazione
(Pollock et al., 1992; Liu and Sessa, 1994). La miristoilazione è una modificazione irreversibile; è un processo co-traduzionale catalizzato dall’enzima N-miristoil transferasi (NMT), che trasferisce il miristato dal miristoil-CoA al peptide nascente (Resh, 1999). Il peptide, per
poter essere miristoilato, deve avere all’NH2-terminale, una precisa
34
posizione 2 è assolutamente obbligatoria, la serina o la treonina in posizione 6 facilitano la miristoilazione, ma la loro presenza non è
strettamente richiesta. Dopo rimozione della metionina NH2-terminale
da parte di una amminopeptidasi, l’acido miristico viene legato mediante legame ammidico al gruppo α-amminico della glicina. Una mutazione nella sequenza di miristoilazione (glicina ad alanina in posizione 2: G2A) rende l'eNOS solubile, dimostrando che la miristoilazione è necessaria per la sua associazione alle membrane (Busconi and Michel, 1993; Liu and Sessa, 1994).
La eNOS, oltre ad essere miristoilata, lega un altro acido grasso, l'acido palmitico. I siti che vanno incontro al processo di palmitoilazione, tramite legame tioestere, sono due residui di cisteina in posizione 15 e 26 (Liu et al., 1995); tale modificazione, post-traduzionale e reversibile, avviene solo se l’enzima è associato alla membrana (Liu et al., 1995; Robinson et al., 1995). Per chiarire l’importanza delle modificazioni lipidiche in relazione alla sua localizzazione cellulare, sono state costruite delle proteine di fusione eNOS-Green Fluorescent Protein (GFP), ed è stata studiata la loro localizzazione tramite microscopia a fluorescenza (Liu et al., 1997;
Sowa et al., 1999). La eNOS non miristoilata (mutante myr-), risulta
anche non palmitoilata e presenta una distribuzione citosolica diffusa, per cui la miristoilazione è necessaria per la palmitoilazione. Si ritiene, quindi che la miristoilazione sia richiesta per un’associazione transitoria della eNOS alle membrane e che la successiva palmitoilazione renda tale associazione stabile e duratura. Inoltre, chimere costituite dal mutante G2A non miristoilabile legato ad un dominio transmembrana (glicoproteina CD8), possono subire la
35
palmitoilazione in quanto legate alla membrana, suggerendo che è la localizzazione alla membrana, piuttosto che la miristoilazione a supportare la palmitoilazione (Prabhakar et al., 2000).
Secondo un gruppo di ricercatori, la quantità della eNOS presente sulla membrana plasmatica dipende da continui cicli di palmitoilazione-depalmitoilazione che corrispondono ad una continua traslocazione dell’enzima tra il Golgi e la membrana plasmtica (Garcia-Cardena et al., 1996). Un simile ciclo di acilazione è stato descritto per l'oncogene Ras (Rocks et al., 2005). Sia per la eNOS che per Ras, questo ciclo comprende due tipi di modificazioni, ovvero un’acilazione irreversibile (la miristoilazione della eNOS e la farnesilazione di Ras), e la palmitoilazione reversibile. Agonisti come la bradichinina o la ionomicina attivano la eNOS promuovendo la depalmitoilazione e causando la traslocazione della eNOS dalla membrana plasmatica al citosol (Michel et al., 1997). Il rapido turnover del palmitico legato alla eNOS (45min), rispetto al miristico (18h), suggerisce che il ciclo pamitoilazione-depalmitoilazione possa regolare la quantità di enzima presente sulla membrana plasmatica (Garcia-Cardena et al., 1996).
Mediante esperimenti di knockdown dell’enzima
palmitoiltransferasi (DHHC21) mediante short interfering RNA (siRNA), è stato possibile ottenere una ridotta palmitoilazione della eNOS, con conseguente modifica della sua localizzazione e riduzione del rilascio di NO dopo stimolazione degli agonisti (Fernandez-Hernando et al., 2006). Questo studio dimostra ancora una volta come la localizzazione subcellulare della eNOS sia strettamente correlata con la sua attività.
36 1.3.3.2. Fosforilazione
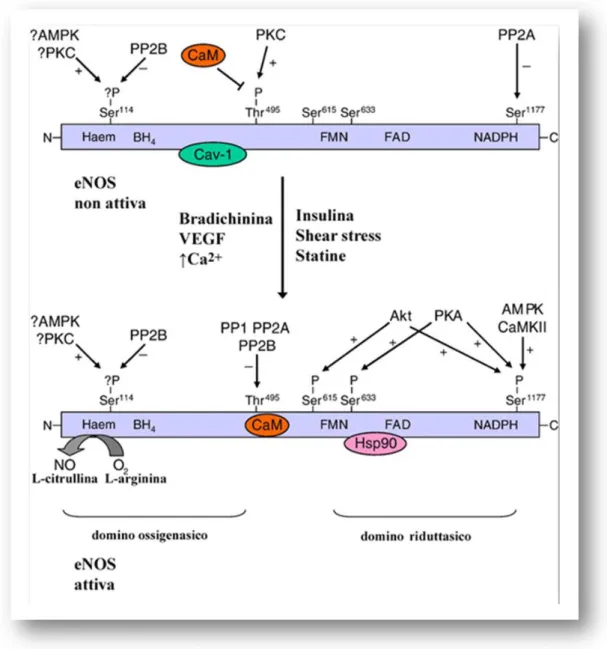
La fosforilazione della eNOS, mediata da proteine chinasi di varie vie di segnalazione, è un processo fondamentale nella regolazione della sua attività. Esso si verifica in risposta ad un’ampia gamma di stimoli quali: fattori meccanici (shear stress), umorali [fattore di crescita endoteliale vascolare (VEGF), bradichinina, insulina, estrogeni, sfingosina 1-fosfato (S1P)] e farmacologici (statine) (Montagnani et al., 2001; Harris et al., 2001; Igarashi et al., 2001; Boo et al., 2002a e b; Harris et al., 2004; Guo et al., 2005) (Fig. 7).
Nell’enzima sono stati identificati sei siti specifici che possono subire fosforilazione (Fig. 7): Tyr-81, 114, Thr-495, 615, Ser-633 e Ser-1177 (nella sequenza umana, equivalenti a Tyr-83, Ser-116, Thr-497, Ser-617, Ser-635 e Ser-1179 nella sequenza bovina) (Fleming e Busse, 2003; Sessa, 2004; Fulton et al., 2005). La fosforilazione di questi siti aumenta (Ser-1177, Ser-633 e Ser-615) o diminuisce (Thr-495 e Ser-611) l'attività dell'enzima.
Il sito di fosforilazione della eNOS più importante, e meglio caratterizzato, è quello della Ser-1177, sito che si trova nel dominio riduttasico a livello del COOH-terminale (Fig. 7). Il più importante stimolo fisiologico per la fosforilazione di tale sito è lo shear stress. Lo shear stress, così come alcuni ormoni e fattori di crescita [il VEGF, il fattore di crescita insulino-simile 1 (IGF-1), le statine e gli estrogeni], attivano la fosforilazione in questo sito dereprimendo il loop autoinibitorio presente al dominio COOH-terminale dell’enzima,
riducendo la dissociazione del complesso Ca2+/CaM dalla eNOS e
37
Figura 7. Regolazione dell’attività della eNOS mediata da chinasi e fosfatasi. L’attività della eNOS è coordinata da eventi di
fosforilazione e defosforilazione, mediati da numerose protein chinasi e fosfatasi. Con il simbolo + è indicata la fosforilazione mediata dalle chinasi, con il simbolo - è indicata la defosforilazione mediata dalle
fosfatasi. La figura illustra anche come eventi di
fosforilazione/defosforilazione sono coordinati con il legame di proteine regolatorie attivanti (CaM e Hsp90) ed inattivanti
38
dominio ossigenasico (Dimmeler et al., 1999; Fulton et al., 1999; Michell et al., 1999; McCabe et al., 2000; Bauer et al., 2003). La mutazione della Ser-1177 con un’alanina (S1177A) impedisce la fosforilazione e di conseguenza la produzione basale e quella indotta del NO; al contrario, la sostituzione della serina con un aspartato (S1177D) genera un enzima costitutivamente attivo (Bauer et al, 2003).
Numerose proteine chinasi, tra cui Akt (Protein chinasi B), la proteina chinasi A (PKA), la proteina chinasi AMP-dipendente
(AMPK), la protein chinasi Ca2+/CaM-dipendente (CaMKII), la
protein-chinasi cGMP-dipendente (PKG), e fosfatasi, tra cui la protein fosfatasi 2 (PP2A), attivate da diversi stimoli, sono implicate nella regolazione della fosforilazione in Ser-1177 (Fig. 7) (Chen et al., 1999; Fulton et al., 1999; Butt et al., 2000; Michell et al., 2001; Morrow et al., 2003; Levine et al., 2007; Okayasu et al., 2008; García et al., 2008).
Da uno studio del 2002 (Gonzalez et al., 2002) è emerso che la fosforilazione della eNOS in Ser-1177 è influenzata dalla sua localizzazione alla membrana. E’ stata infatti confrontata, mediante immunoprecipitazione, la forma wild-type dell’enzima con mutanti
palm-/eNOS e myr-/eNOS; si è osservato che la fosforilazione avviene
normalmente nel wild-type, risulta parzialmente ridotta nel mutante
palm-/eNOS ed è quasi assente in myr-/eNOS, sia in condizioni basali
che in seguito ad attivazione dell’enzima da parte di VEGF o S1P. Tali mutanti possono essere rifosforilati se ad essi viene legato un dominio transmembrana come la glicoproteina CD8, che consente una corretta rilocalizzazione alle membrane. Considerando questi dati
39
risulta evidente che per la fosforilazione della Ser-1177 non è fondamentale l’acilazione, bensì il targeting alle membrane. Giustiniani et al., (2009) hanno inoltre osservato che per la fosforilazione in Ser-1177 sembra essere necessaria la stabilità dei microtubuli, nonché l’acetilazione della tubulina.
Il significato fisiologico della fosforilazione indotta in altri siti (come in Ser-633 e Ser-615) della eNOS è meno conosciuto. La fosforilazione in Ser-633 è mediata dalla PKA ed aumenta la produzione del NO (Fig. 7) (Michell et al., 2002; Boo et al., 2003a). Diversi agonisti della eNOS che determinano la fosforilazione della Ser-1177, inducono anche la fosforilazione in Ser-633. Con alcuni di tali stimoli quali il VEGF, lo shear stress, ed un analogo del cGMP l’8-bromo-cAMP (8-Br-cAMP) si è osservato che i livelli di fosforilazione in Ser-633 sono più bassi di quelli in Ser-1177. Inoltre la fosforilazione in Ser-633 non richiede un aumento delle
concentrazioni di Ca2+ intracellulare (Boo et al., 2003a). Tali dati
suggeriscono che questa fosforilazione è importante nel mantenere l’attività della eNOS dopo un’iniziale attivazione dovuta al flusso di
Ca2+ e/o fosforilazione in Ser-1177 (Mount et al., 2007).
La fosforilazione in Ser-615 è regolata da Akt (Fig. 7). I dati presenti in letteratura sulla fosforilazione di tale sito sono molto contrastanti. Michell et al. (2002) hanno riportato che la mutazione di tale serina con un aspartato (S615D, che genera una forma che si comporta come quella fosforilata) aumenta l’affinità della eNOS per il
complesso Ca2+/CaM ma non stimola ulteriormente l’enzima già
attivo. Bauer et al., (2003) hanno invece osservato che questa mutazione (S615D) aumenta l’attività enzimatica in saggi in vitro,
40
mentre la mutazione con un’alanina (S615A, che genera una forma di eNOS costitutivamente defosforilata) genera una forma meno attiva nei saggi in vitro. Tuttavia, ambedue queste mutazioni risultano in un maggior rilascio di NO sia basale che stimolato in vivo. Gli autori suggeriscono che l’effetto stimolatorio della mutazione S615A in vivo sia dovuta ad effetti indiretti, comportanti sia l’interazione della eNOS con altre proteine sia la fosforilazione in altri siti. Recentemente Tran et al. (2009) hanno dimostrato che la contemporanea fosforilazione in Ser-615 e in Ser-1177 aumenta notevolmente l’attività della eNOS in
presenza di concentrazioni basali di Ca2+, mentre una sola di queste
fosforilazioni aumenta di poco l’attivita dell’enzima e richiede alte [Ca2+].
Anche la fosforilazione in Tyr-81, indotta da Src chinasi, ha un’azione stimolatoria sull’enzima. Questa fosforilazione contribuisce ad un aumento dell'attività della eNOS, o influenzando l’attivazione
dell’enzima Ca2+
-mediata, o alterando le interazioni proteina-proteina, oppure modificando la localizzazione dell'enzima (Fulton et al., 2005, 2008).
Il sito Thr-495, localizzato all'interno della sequenza dove si
lega il complesso Ca2+/CaM, a differenza di quanto riportato per i
residui indicati finora, quando fosforilato riduce l’attività catalitica
della eNOS, interferendo con il legame della Ca2+/CaM all’enzima
(Harris et al., 2001; Michell et al., 2001; Fleming et al., 2001; Matsubara et al., 2003). La defosforilazione della Thr-495, indotta da agonisti della sintesi del NO come la bradichinina, VEGF e ionomicina è mediata da protein fosfatasi 1 (PP1), fosfatasi 2 (PP2A) e dalla calcineurina (PP2B) (Michel et al., 2001; Greif et al., 2002)
41
(Fig. 7). In cellule endoteliali, è stato osservato che la defosforilazione di tale residuo e la contemporanea fosforilazione in Ser-1177, sono essenziali per l’attività della eNOS (Harris et al., 2001; Michell et al., 2001; Fleming et al., 2001).
Le relazioni tra Ser-1177 e Thr-495 sono state investigate utilizzando dei mutanti (Lin et al., 2003): mutando la Thr-495 in alanina (T495A) per generare una forma costituivamente defosforilata della eNOS alla posizione 495 è stato osservato un aumento dell’attività enzimatica nei relativi lisati cellulari; tuttavia la produzione di NO in vivo era ridotta. Se alla prima mutazione si aggiunge quella della Ser-1177 in acido aspartico (S1177D), che da sola renderebbe l’enzima costitutivamente attivo, è stata osservata un’attività estremamente incrementata, con maggiore produzione di
O2
rispetto alla forma wild-type. Questo studio potrebbe chiarire il ruolo della Thr-495 quale importante sito di regolazione della “reazione non accoppiata” della eNOS, ma non spiega perché agonisti della eNOS come la bradichinina, che causano la defosforilazione in tale sito (Harris et al., 2001; Fleming et al., 2001), stimolano la sintesi
del NO piuttosto che del O2
-. Quindi saranno necessari ulteriori studi per chiarire se la defosforilazione in Thr-495 causa prevalentemente
un aumento della sintesi del NO o del O2
(Mount et al., 2007). In accordo con un’azione inibitoria di fosfo-Thr-495, Yoon et al. (2010) hanno recentemente osservato che in cellule endoteliali, Human Umbilical Vein Endothelial Cells (HUVEC), l’invecchiamento della cellula riduce la produzione di NO, i livelli della eNOS ed inibisce l’attività dell’enzima favorendo la fosforilazione in Thr-495 piuttosto che la fosforilazione in Ser-1177.
42
Il sito di fosforilazione in Ser-116, è l’unico sito individuato nel domino ossigenasico della eNOS (Fig. 7). Sebbene alcuni studi condotti su cellule endoteliali in coltura hanno mostrato che la fosforilazione di questo residuo contribuisce all’attivazione della eNOS in seguito a shear stress (Gallis et al., 1999), ad esposizione alle lipoproteine ad alta densità (HDL) ed all’apolipoproteina AI (Drew et al, 2004), altri autori hanno riportato che tale sito è fosforilato in condizioni basali e che la sua defosforilazione determina una incremento dell’attività della eNOS (Kou et al., 2002). Li et al. (2007) utilizzando mutanti fosfomimetici (S116D) e mutanti non fosforilabili (S116A) hanno confermato che la fosforilazione in Ser116 è inibitoria piuttosto che stimolatoria, e ciò sembra essere dovuto non ad un diretto effetto sull’attività catalitica della eNOS, ma piuttosto ad un’aumentata associazione dell’enzima con la caveolina-1.
Recentemente, è stato osservato che la fosforilazione in questo sito, dovuta alla chinasi S-ciclina-dipendente 5 (Cdk5), induce l’inattivazione della eNOS e quindi una minore produzione di NO con conseguente diminuzione dei danni dovuti alla nitrazione di diverse proteine (Lee et al., 2010).
1.3.3.3. S-nitrosilazione
La S-nitrosilazione è una reazione che consiste nell’attacco covalente di gruppi nitrosilici (NO-derivati) ai gruppi tiolici degli amminoacidi cisteina presenti nelle proteine (si veda la sezione 1.1).
43
La S-nitrosilazione/denitrosilazione è ormai riconosciuta come un meccanismo di fondamentale importanza nella regolazione delle vie di
trasduzione del segnale pari ai processi di
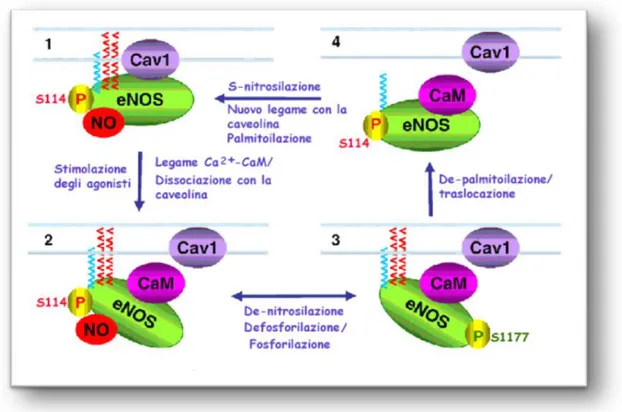
fosforilazione/defosforilazione (Fig. 8). Nelle cellule endoteliali a riposo la eNOS è inibita mediante S-nitrosilazione a livello delle Cys-94 e Cys-99 che, come detto nella sezione relativa alla struttura della eNOS, sono localizzate all’interfaccia di associazione tra due monomeri (Erwin et al., 2005).
Il meccanismo attraverso cui la S-nitrosilazione inibisce la
eNOS non è ancora stato chiarito. Dal momento che lo ione Zn2+ viene
rilasciato dal cluster tetratiolato dopo S-nitrosilazione (Ravi et al., 2004), è stato suggerito che il meccanismo di inibizione potrebbe essere dovuto alla conseguente dissociazione degli omodimeri in monomeri inattivi, tra i quali non può avvenire il trasferimento degli elettroni (Erwin et al., 2006; Dudzinski et al., 2006). Altri studi hanno invece suggerito che la S-nitrosilazione della eNOS possa influenzare il legame del substrato o di cofattori all’enzima stesso (Erwin et al., 2005).
Agonisti quali il VEGF e l'insulina promuovono la denitrosilazione della eNOS e la sua fosforilazione in Ser-1177, attivandola. Sebbene la denitrosilazione avvenga simultaneamente alla fosforilazione in Ser-1177, tali modifiche non sembrano essere interdipendenti (Erwin et al., 2005). In corrispondenza della diminuzione della fosforilazione in Ser-1177 e al ritorno dell’attività dell'enzima allo stato basale, la eNOS è nuovamente S-nitrosilata (Musicki et al., 2009) (Fig. 8).
44
Figura 8. Rapporto dinamico tra la nitrosilazione e le principali modificazioni post-traduzionali della eNOS. Questa figura mostra le
relazioni tra i processi di regolazione dell’attività della eNOS più frequenti e importanti: la nitrosilazione, l’acilazione, la fosforilazione e l’associazione con la cav-1. Pannello 1: Mostra la eNOS a riposo, miristoilata e palmitoilata (catene blu e rosse, rispettivamente), ancorata alla membrana ed inibita mediante: la nitrosilazione (indicata con NO gruppi nitrosilici legati ai gruppi tiolici delle 94 e Cys-99); il legame con la caveolina-1 (cav-1) (come riportato nella sezione relativa all’interazione con la cav-1); la fosforilazione inibitoria in Ser-114. Pannelli 2 e 3: Mostrano come gli agonisti promuovono il
legame del complesso Ca2+/CaM all’eNOS e la dissociazione dalla
cav-1 (pannello 3) la de-nitrosilazione e la contemporanea defosforilazione in Ser-114/fosforilazione in Ser-1177. Pannello 4: La prolungata stimolazione degli agonisti induce la de-palmitoilazione con conseguente traslocazione della eNOS dalla membrana al citosol. La eNOS nuovamente palmitoilata, ritorna alla membrana dove va incontro alla nitrosilazione, alla fosforilazione in Ser-114 e al legame con la cav-1. (Modificata da Dudzinski et al., 2007).
45
Sembra che sia la eNOS a generare il NO necessario per la propria S-nitrosilazione (Erwin et al., 2005) e che la localizzazione intracellulare dell’enzima stesso influenzi tale reazione. In particolare, in esperimenti in cui sono stati usati dei mutanti non miristoilabili
(myr--eNOS) è stato osservato che la S-nitrosilazione è abolita, mentre
un mutante myr- fuso al dominio transmembrana della glicoproteina
CD8 (CD8-myr--eNOS) è iper-nitrosilato (Erwin et al., 2006). Il
motivo per cui la S-nitrosilazione richiede la localizzazione dell’enzima alla membrana potrebbe dipendere dalla diversa chimica degli ambienti cellulari che possono essere a favore o sfavore di questa modifica (Hess et al., 2005). È stato ipotizzato che l'ambiente idrofobico delle membrane facilitino la reazione del NO con l’ossigeno gassoso necessaria per la formazione di molecole
nitrosilanti, quali N2O3 (Erwin et al., 2006; Liu et al., 1998). Al
contrario, la de-nitrosilazione nel citosol potrebbe essere facilitata dall’ambiente riducente, dalla più bassa concentrazione di NO e dalla
rapida idrolisi di N2O3 in ambiente acquoso (Erwin et al., 2006).
1.3.3.4. Acetilazione
Recentemente è stato scoperto che i residui di lisina (Lys 496) e 506 della eNOS, che si trovano nel dominio che lega la CaM, possono essere acetilati e che questa acetilazione riduce il legame tra eNOS e CaM (Mattagajasingh et al., 2007). Il significato biologico di questa modificazione è discussa nella sezione 1.4.1.
46 1.3.3.5. Interazioni proteine-proteine
Caveolina-1. Le caveoline (proteine palmitoilate di 20-24 Kda),
formano l’impalcatura strutturale delle caveolae, causando l’invaginazione di microdomini della membrana plasmatica, i cosiddetti rafts, ricchi di colesterolo e sfingolipidi (Galbiati et al., 2001). Le caveoline sono comunemente classificate in: caveolina-1 (cav-1) presente in un’ampia varietà di cellule, compreso l’endotelio; 2 (cav-2) espressa principalmente negli adipociti; caveolina-3 (cav-caveolina-3) contenuta nei muscoli striati, incluso il miocardio (Rothberg et al., 1992).
Le caveolae sono particolarmente abbondanti nelle cellule endoteliali; grazie alle loro proprietà dinamiche, partecipano alla transcitosi, alla permeabilità vascolare ed al controllo del tono vascolare (Frank et al., 2003).
La membrana plasmatica non è, tuttavia, l’unico sito di localizzazione della cav-1; la si trova anche nel complesso del Golgi (Kurzchalia et al., 1992; Smart et al., 1994; Luetterforst et al., 1999), negli endosomi (Pol et al., 1999), nei corpi lipidici (Pol et al., 2001), sulle membrane mitocondriali e in vescicole di trasporto coinvolte nel traffico della via secretoria (Li et al., 2001). Si ritiene che cav-1 leghi il colesterolo prodotto nel reticolo endoplasmatico e lo trasporti alla membrana plasmatica, passando per il Golgi; è inoltre implicata in una via di endocitosi indipendente dalla clatrina (Hansen and Nichols, 2009) e in vie di trasduzione del segnale (Anderson et al., 1998).
Attraverso esperimenti di co-immunoprecipitazione, di pull-down e two-hybrid (Ju et al., 1997), è stato dimostrato che la eNOS interagisce, attraverso una sequenza di legame consenso (amminoacidi
47
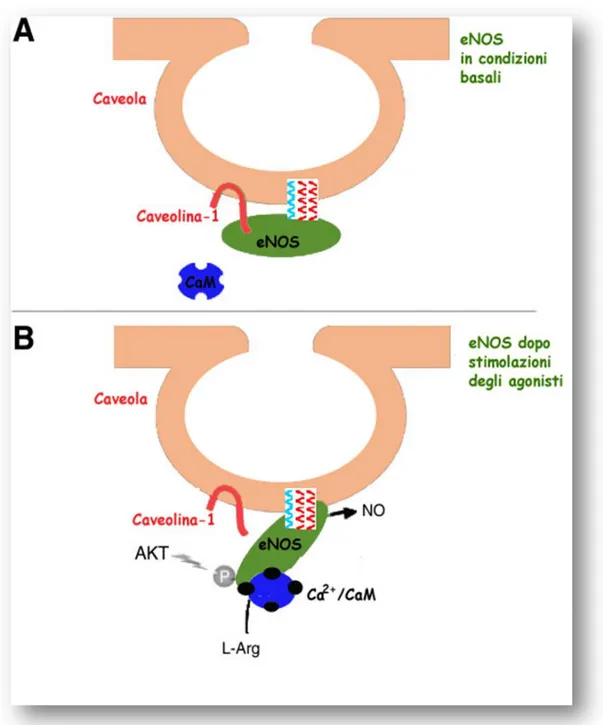
350-358), direttamente con la cav-1 legandosi al cosiddetto caveolin scaffolding domain (amminoacidi 82-101) (Michel et al., 1997). Tale associazione riduce fortemente l’attività basale della eNOS perché interferisce con il flusso di elettroni che passano dal dominio riduttasico a quello ossigenasico (Ju et al., 1997; Ghosh et al., 1998). E’ stato dimostrato che l'interazione inibitoria della eNOS con la
cav-1, può essere antagonizzata dal complesso Ca2+/CaM (Michel et al.,
1997; Feron et al., 1998): questa serie di esperimenti hanno portato a
proporre l’esistenza di un ciclo di regolazione
nell’attivazione/inattivazione dell’enzima, dipendente dal complesso
Ca2+/CaM e dalla cav-1 (Fig. 9). Diversi agonisti come lo shear stress,
la bradichinina, gli estrogeni o l’aumento del flusso vascolare,
aumentano le concentrazioni intracellulari di Ca2+, promuovendo il
legame del complesso Ca2+/CaM all’eNOS e la sua dissociazione dalla
cav-1, con conseguente attivazione della stessa. Quando i livelli
intracellulari di Ca2+ ritornano a livelli basali, il ciclo è invertito, la
CaM si dissocia e la caveolina si riassocia con la eNOS ormai inattiva (Feron et al., 1998).
Livelli elevati di LDL in pazienti ipercolesterolemici aumentano l'espressione della cav-1 e la formazione del complesso cav-1/eNOS limitando sia il rilascio del NO basale che quello stimolato dagli agonisti (Feron et al., 1999 e 2001). Questa risposta patologica può spiegare in parte la disfunzione endoteliale presente nei pazienti con ipercolesterolemia.
Bucci et al., (2000) mediante studi in vivo hanno osservato che l’interazione della cav-1 con la eNOS riduce la permeabilità vascolare e la formazione di edema tissutale. Il blocco della permeabilità
48
Figura 9. Regolazione della eNOS mediata dall’interazione con la caveolina-1 e dal complesso Ca2+/CaM. Pannello A. In condizioni
basali l’attività della eNOS è inibita dal legame con la cav-1. Pannello B. Diversi agonisti come lo shear stress, la bradichinina, gli estrogeni
promuovono il legame del complesso Ca2+/CaM alla eNOS, la sua
dissociazione dalla cav-1 e fosforilazioni stimolatorie come quella mediata da Akt, con conseguente attivazione dell’enzima e produzione di NO.
49
vascolare dovuto all’associazione cav-1/eNOS è simile a quello
osservato nei topi knockout per la eNOS (Fukumura et al., 2001). Inoltre studi su topi knockout (Drab et al., 2001; Razani et al., 2001) e animali transgenici per la cav-1 (Bauer et al., 2005), hanno dimostrato l’importanza della regolazione negativa della cav-1 sulla eNOS per l’intero organismo. I topi knockout per il gene della cav-1 (cav-1 -/-) sono vitali e fertili ma privi di caveolae nella maggior parte dei distretti tissutali (polmone, tessuto adiposo, rene, cuore), a conferma del ruolo essenziale della cav-1 nella biogenesi di tali strutture. Gli stessi topi non rispondono al trattamento con il vasocostrittore fenilefrina. Dal momento che la vasocostrizione viene ripristinata somministrando Nω-Nitro-L-Arginina-Metil-Estere (L-NAME), un classico inibitore della eNOS, si è dedotto che, alla base della mancata risposta vasocostrittrice, ci sia un’aumentata attività della eNOS. Al contrario, risulta incrementata la vasodilatazione indotta dall’acetilcolina ACh (un fenomeno NO-dipendente) confermando l’azione inibitoria della cav-1 sulla eNOS.
La risposta vasodilatativa ACh-dipendente è stata studiata anche in topi transgenici per la cav-1 (che sovraesprimono tale proteina). Come previsto la vasodilatazione risulta ridotta, in quanto l’attività catalitica della eNOS è inibita dalla cav-1. Da questa serie di lavori emerge che a livello endoteliale il ruolo principale della cav-1 sia quello di regolare le funzioni del letto vascolare attraverso l’interazione con la eNOS.
Considerata l’importanza dell'attività della eNOS e della conseguente produzione del NO nel determinare una risposta
50
fornire un nuovo approccio terapeutico per la regolazione della
produzione del NO, e per attenuare gli effetti pro-infiammatori dovuti ad un’eccessiva produzione del NO.
Mentre è ormai nota l’interazione tra la cav-1 e la eNOS, meno chiari sono i dati in letteratura circa “il luogo” di interazione tra le due proteine. Diversi lavori riportano che nella membrana plasmatica la eNOS si localizzi nelle caveolae (Garcia-Cardena et al., 1996; Shaul et al., 1996; Rizzo et al., 1998; Sowa et al., 2001). Gran parte di questi studi sono basati su una particolare tecnica di frazionamento che isola proteine insolubili in soluzioni fredde di detergenti non denaturanti (come il Triton-X-100). La equazione tra insolubilità in Triton e localizzazione alle caveolae è oggi messa in discussione da diversi ricercatori nel campo (Parton and Simons, 2007). Si deve anche aggiungere che entrambe le proteine sono state trovate anche in altri compartimenti intracellulari (Kurzchalia et al., 1992; Sessa et al., 1995; Li et al., 2001), per cui si potrebbe pensare che la loro interazione possa avvenire in uno di tali compartimenti [anche se uno studio di Govers et al. (2002) suggerisce che il complesso del Golgi non sia un sito di interazione tra le due proteine].
In un studio di microscopia confocale ad alta risoluzione sulla
localizzazione della eNOS, è stata riportata una scarsa
colocalizzazione dell’enzima con la cav-1 sulla membrana plasmatica
di cellule HeLa indotte ad esprimere la eNOS e di cellule HUVEC; in
particolare in cellule in migrazione, è stata osservata una netta segregazione tra le due proteine, con la eNOS localizzata al margine avanzante di queste cellule e la cav-1 concentrata nella parte posteriore (Bulotta et al., 2006). Anche in cellule confluenti, non
51
migranti, il grado di colocalizzazione tra eNOS e cav-1 era scarso. Questi risultati indicano che la maggior parte della eNOS presente sulla membrana plasmatica non è presente in caveolae; tuttavia, il sito di interazione tra l’enzima e la cav-1 rimane ancora da scoprire.
Recentemente è stato osservato che in cellule endoteliali di aorta bovina (BAECs) coltivate in assenza di siero si ha lo spostamento del complesso eNOS/cav-1 dalla membrana plasmatica e dal Golgi a siti intracellulari. In seguito al trattamento con insulina il complesso ritorna alla membrana plasmatica e questa traslocazione è accompagnata dalla riduzione della produzione di NO. Il knockdown della cav-1 e l’inibizione della palmitoilazione della eNOS e della cav-1 determinano, in presenza di insulina, un aumento dei livelli di NO. Da ciò emerge che il complesso eNOS/cav-1, nonché l’associazione dello stesso alla membrana plasmatica, rivestano un ruolo determinante nel modulare l’attività dell’enzima indotta dall’insulina (Wang et al., 2009).
Heat shock protein 90 (Hsp90). Hsp90 fa parte del gruppo delle
“Heat Shock Proteins”, abbondanti proteine citosoliche che agiscono da chaperon regolando il ripiegamento, la maturazione, l’attivazione e il traffico di proteine e intervenendo anche nella regolazione di proteine coinvolte nella trasduzione del segnale (Pratt, 1997).
L’interazione tra eNOS e Hsp90 è stata rilevata con esperimenti di co-immunoprecipitazione ed è stato dimostrato che questa associazione incrementa l’attivazione della eNOS in risposta a svariati
52
stimoli, quali VEGF e shear stress (García-Cardeña et al., 1998). In cellule endoteliali a riposo, Hsp90 sembra essere associata in minime quantità alla eNOS. In cellule stimolate da agonisti (VEGF, estrogeni, istamina e shear stress) vi è un aumento dell'associazione tra le due proteine correlata con una maggiore produzione del NO (Garcia-Cardena et al., 1998). A conferma del ruolo positivo di Hsp90, la introduzione di un dominante negativo di Hsp90 in cellule endoteliali determina il blocco dell’associazione tra Hsp90, eNOS ed Akt in seguito a trattamento con VEGF, con conseguente defosforilazione di Akt (Ser-473), defosforilazione della eNOS (Ser-1177) e diminuzione del rilascio di NO (Miao et al., 2008). Il controllo positivo attuato da Hsp90 sulla eNOS è stato dimostrato anche in vivo (Shah et al., 1999). Recentemente, Yoon et al. (2010) hanno riportato che, in cellule HUVEC invecchiate, aumenta l’associazione della eNOS con la cav-1 rispetto a quella dell’enzima con Hsp90 e Akt.
Il meccanismo d’azione di Hsp90 non è ancora del tutto compreso. È stato suggerito che lo chaperon agisca da modulatore allosterico della eNOS, inducendo un cambiamento conformazionale dell’enzima, portando ad un aumento della sua attività oppure ad una stabilizzazione del suo legame con la calmodulina; in alternativa, hsp90 potrebbe agire da proteina di ancoraggio (scaffold) per il reclutamento all’enzima di altre proteine regolatrici quali chinasi e fosfatasi (Fulton et al., 2001; Fontana et al., 2002). L’attivazione comporta la dissociazione CaM-dipendente della eNOS dalla cav-1, il successivo reclutamento della eNOS e di Akt ad Hsp90 e la fosforilazione (Akt-mediata) dell’enzima stesso (Garcia-Cardena et