Scuola Dottorale in Biologia - Dottorato di Ricerca in Biologia
Applicata alla Salute dell’Uomo
XXV Ciclo
TELOMERES AND GENOME STABILITY IN
IRRADIATED MAMMALIAN CELLS: EFFECT OF
RADIATION QUALITY, DOSE RANGE, AND
MITOCHONDRIAL FUNCTIONALITY STATE
TELOMERI E STABILITÀ GENOMICA IN CELLULE
DI MAMMIFERO IRRADIATE: EFFETTO DEL TIPO
DI RADIAZIONE, RANGE DI DOSE E DELLA
CONDIZIONE FUNZIONALE DEI MITOCONDRI
Dottorando/PhD student:
dr. Dino Nieri
Docenti guida/Tutors:
Prof. Antonio Antoccia Dott.ssa Antonella Sgura
Coordinatore/Coordinator:
prof. Paolo Visca
INDEX
Riassunto
... vSummary
... viii1.
Introduction
... 11.1. IRs and Linear Energy transfer ... 1
1.2. Photon’s interactions with living matter ... 2
1.3. Charged particles’ interactions with living matter ... 3
1.4. Types of DNA radio-induced damage ... 4
1.5. Clusters of damage ... 6
1.6. Relative Biological Effectiveness of IR on the living matter ... 6
1.7. Chromosome DSBs repair ... 7
1.8. Chromosomal aberrations, genesis, nature and fate ... 8
1.9. Telomeres ...10
1.10. Consequences of telomere shortening: senescence, apoptosis and genomic instability ...11
1.11. Telomeric damage-related chromosomal instability ...13
1.12. Telomere-related radio-sensitivity ...13
1.13. Mitochondrial oxidative functions ...15
1.14. ROS homeostasis: physiol. release and detoxification ....16
1.15. Mitochondrial dysfunctions and ROS homeostasis imbalance ...17
1.16. Low-dose range and non-linear effects ...18
1.17. HRS and IRR ...19
1.18. Adaptive response ...21
1.19. Genomic instability ...21
1.20. Bystander effect ...21
1.21.
Considerations on linearity failure ...222.
Aim of the study
...232.1. Cytogenetic effects of low doses on human fibroblasts ...23
2.2. Radio-protective significance of the telomere ...23
2.3. Contribution of inner ROS activity to the radio-induced damage .24 3.
Results
...253.1. Induction of structural chromosomal aberrations in the low-dose range ...25
3.2. Telomeric length modulation in the low-dose range ...29
3.3. Telomere-related genomic instability in the low-dose range: anaphase bridge formation with telomeric involvement ...30
3.4. Radio protective effect of the telomere ...31
3.4.1. Telomere length and telomere loss analysis in the TK6 whole population ...31
3.4.2. Telomere length and telomere loss in TK6 clones survived to 4Gy of X-rays ...33
3.4.3. Telomerase activity after irradiation ...34
3.4.4. Selection of TK6 clones with short and long telomeres ..34
3.4.5. Radio-sensitivity in short and long telomere clones ...36
3.4.6. Telomere loss but not telomere length correlates with radio-sensitivity ...37
3.5. Role of the mitochondrion in response to IRs ...38
3.5.1. mtDNA depletion via ddC-treatment ...38
3.5.2. Electron microscopy in mitochondrial morphology ...41
3.5.3. Mitochondrial membrane potential analysis ...42
3.5.4. Evaluation of internal ROS homeostasis imbalance ...43
3.5.5. DSB induction and repair kinetics ...44
3.5.6. Telomere length analysis by q-FISH ...46
3.5.7. Chromosomal aberration analysis by m-FISH ...46
4.
Discussion
...484.1. Chromosomal radio-induced damage ...48
4.2. Radiobiological meaning of the telomere ...49
4.2.1. Telomeric damage induced by low doses ...49
4.2.2. Radio-protective effect of the telomere at radiotherapy doses ...50
4.3. Role of mitochondrial dysfunction in response to IRs ...52
5.
Conclusions
...575.1. Cytogenetic effect of low doses on human primary fibroblasts ....57
5.2. Radio-protective significance of the telomere ...57
5.3. Contribution of the inner ROS activity to the rad-induc. damage .57 Appendix A - Materials and methods ...58
Appendix B – Symbols and abbreviated terms ...67
v
Riassunto
Il bersaglio più importante delle radiazioni ionizzanti (IR) nelle cellule eucariotiche è il corredo cromosomico. La radiosensibilità delle cellule ciclanti dipende generalmente dalla capacità di riparazione del danno e da un insieme di fattori promuoventi il mantenimento dell’ integrità genomica. Lo scopo di questo lavoro, riguardante diversi tipi di radiazione ed il loro effetto su colture cellulari, è molteplice: è uno studio sul danno citogenetico da basse dosi su fibroblasti umani normali (i), sull’ipotetico ruolo radiosensibilizzante delle disfunzioni dell’omeostasi telomerica su cellule linfoblastoidi umane (ii), e in ultimo (iii) è rivolto a far luce sul ruolo delle specie reattive dell’ossigeno (ROS) citosoliche e del loro contributo all’induzione di danno da radiazioni su fibroblasti umani normali, appositamente trattati farmacologicamente.
(i) In accordo con il cosiddetto modello lineare senza soglia (LNTM), a lungo si è ritenuto che la risposta di un sistema biologico alle IR fosse proporzionale alla dose, anche quando minima. In verità si tratta di un approccio pratico e prudente dovuto alla penuria di dati su basso range di dose. Basato su effetti deterministici rilevati a dosi più alte, il LNTM è una linea guida di riferimento per la previsione di rischio nelle situazione più diverse.
La percezione è cambiata con l’emergere di un insieme di fenomeni non lineari (§ 1.16) come l’ipersensibilità/radioresistenza indotta, effetto bystander e risposta adattativa. Benché a oggi il saggio clonogenico sia il test di più largo uso per tali verifiche, in questo lavoro, si sono usate tecniche citogenetiche per ricercare comportamenti non lineari su endpoint come il danno cromosomico, la modulazione della lunghezza telomerica e l’instabilità genomica radio-indotta in dipendenza dalla dose e dal tipo di radiazione (alto o basso LET).
In accordo al LNTM le curve di regressione dei grafici dose-risposta dovrebbero essere strettamente monotone anche per basse dosi, mentre in caso di non linearità si dovrebbero osservare insoliti picchi di risposta o andamenti piatti.
Abbiamo misurato il danno radio-indotto in due modi: come induzione di rotture e scambi cromosomici, con una tecnica di colorazione dell’intero cariogramma (§ 3.1), e come induzione di ponti di cromatina in anafase (§ 3.3), con tecniche tradizionali. Come risultato, sostanziali deviazioni dalla linearità non sono state dimostrate. Il modello alla base è puramente
vi probabilistico: la quantità e la complessità del danno indotto dipende dalla dose, dall’ efficacia biologica relativa (RBE) della radiazione (fig 14, 15, 18) e dal contenuto in DNA del bersaglio (fig 16), senza ipersensibilità o effetti soglia (§ 4.1, 4.2.1).
(ii) Venendo alla questione dell’effetto delle IR sul telomero, si ricordi che questi è un importante fattore di stabilità del genoma: evita la fusione cromosomica e che le estremità cromosomiche siano processate come rotture a doppio filamento (DSB). Ad alte dosi di IR è noto un effetto di modulazione sulla lunghezza, sia come allungamento che accorciamento, a seconda del tipo di radiazione. Dai nostri dati invece non si evincono effetti simili a basse dosi con nessuna delle IR testate (§ 3.2). In più, dalla osservazione dei ponti anafasici, il telomero non sembra partecipare all’instabilità genomica innescata dalle IR (§ 3.3 fig 18). Su questi risultati si è autorizzati a supporre una mancanza di linearità della risposta alle basse dosi (§ 4.2.1).
In definitiva basse dosi di IR inducono instabilità genomica, ma non perturbando l’omeostasi dei telomeri malgrado il loro noto ruolo nella destabilizzazione/stabilizzazione del corredo cromosomico.
Questo ci ha spinto a valutare l’importanza del telomero nella radioresistenza a dosi di raggi X di ordine radioterapico.
Come il telomero funzionale è un fattore di stabilizzazione del genoma, le IR sono per definizione agenti destabilizzanti, capaci di inattivare la cellula attraverso danni al genoma. Qualsivoglia cellula ha poi una caratteristica lunghezza telomerica e una frequenza spontanea di perdita di telomeri (telomere loss). Su queste premesse è ragionevole aspettarsi, in caso di disfunzione telomerica, un aumento di radiosensibilità.
Dal momento che all’interno della stessa linea cellulare la lunghezza telomerica è dispersa su una media diversa da cellula a cellula, si può supporre che la stabilità genomica conferita dal telomero sia diversa da cellula a cellula (§ 3.4.1), dal che potrebbe ipoteticamente derivare una diversa radioresistenza.
Abbiamo testato questa supposizione su diverse linee linfoblastoidi monoclonali isolando vari cloni, coltivandoli indipendentemente e caratterizzandoli per funzionalità telomerica e radioresistenza.
Ne è risultato che i telomeri, se anche corti, non rendono radiosensibile la linea, mentre una frequenza di telomere loss oltre una data soglia dà questo effetto (§ 3.4.6 fig 25), cosicché questa disfunzione telomerica può pesare
vii sulla radiosensibilità differenziale di cloni originati dalla stessa popolazione (§ 4.2.2) e si mostra come potenziale marcatore di radiosensibilità.
(iii) Dal momento che il mitocondrio è una sorgente fisiologica di ROS e ne è il principale responsabile dell’omeostasi, si può supporre un suo ruolo, pur indiretto, nei meccanismi di induzione di danno da radiazioni.
A questo proposito si ricordi che il DNA è danneggiato sia dalla radiolisi diretta che tramite le specie reattive derivanti dalla radiolisi dell’acqua. Lontano dall’essere chiaro, questo ruolo è oggetto di svariate congetture: sembra ad esempio che mitocondri irradiati siano in grado di stabilire un rilascio di ROS a lungo termine, oppure il metabolismo mitocondriale è sospettato di amplificare l’attività chimica dei ROS generati dalle IR (§ 1.15). L’aumento di attività dei ROS causato da disfunzioni mitocondriali è stato messo in relazione ad anomalie cromosomiche, instabilità genomica e danno telomerico. Parimenti, la sovrapproduzione di ROS originantesi dalla stessa disfunzione, può paradossalmente essere intesa come protettiva per la stimolazione di contromisure come nel caso della cosiddetta risposta adattativa (§ 1.18).
Allo scopo di definire il ruolo dei ROS mitocondriali nella risposta alle IR, abbiamo farmacologicamente messo a punto una linea di fibroblasti con una degenerazione mitocondriale, mostrante un aumento basale della concentrazione di ROS causata dalla perturbazione della loro omeostasi cellulare.
Scopo di questo modello, in cui i mitocondri diventano diffusori di ROS (fig 29), è riportare, qualsivoglia risposta differenziale alle IR, rispetto al controllo, per verificare in ultima analisi se i ROS endogeni agiscono additivamente o in sinergia con il radio-danno esogeno.
Il trattamento farmacologico è risultato in un incremento di DSB spontanei (§ 3.5.5) e di frammentazione cromosomica (§ 3.5.7), mentre non si riscontra alcun danno al telomero (§ 3.5.6 fig 30, 31). In seguito all’ irraggiamento, abbiamo osservato che la produzione di danno citogenetico corrisponde alla somma del danno radio-indotto e del danno basale causato dalla disfunzione mitocondriale senza effetti sinergici. In conclusione, limitatamente ai nostri dati, il mitocondrio non riveste un duolo determinante nell’induzione di danno da radiazioni (§ 4.3).
viii
Summary
The most important ionizing radiation (IR) target in eukaryotic cells is the chromosomal asset. The radio-sensitivity of cycling cells, is generally dependent on their damage repair ability and a set of factors which promotes the maintenance of genome integrity.
The aim of this work, dealing with several types of IRs and their effects on cycling cell cultures, is manifold: it is a study on the cytogenetic damage of low doses on normal human fibroblasts (i), it is a study addressed to investigate any hypothetic radiation sensitizer role of the dysfunctions of the telomere homeostasis on lymphoblastoid human cells (ii), and finally (iii) it is addressed to highlight the role of the cytosolic reactive oxygen species (ROS) activity and its contribution to the cytogenetic radio-induced damage on normal human fibroblasts pharmacologically treated to this purpose. (i) For a long time the response of a biological system to IR exposure has been supposed proportional to the dose delivery, even the low-dose range, according to the so-called Linear No Threshold Model. Indeed it is a practical and conservative approach due to the general lack of data in that dose range. Based on deterministic data obtained at higher doses, it is suitable as guideline for the risk assessment in a variety of different situations.
Things have changed since a set of non-linear trends (§ 1.16) emerged, such as the low-dose hyper-radio-sensitivity/induced-radio-resistance, the bystander effect, and the adaptive response. So far, the cell survival is the most used endpoint to test the occurrence of such non-linear phenomena, in this work instead, cytogenetic skills have used in order to verify hypothetic non-linear behaviors of endpoints such as the chromosomal damage, the telomere length modulation, and the induced genome instability, in dependence on the dose and the type of IR (low- and high- LET).
According to the LNTM the fitted functions of the dose-response charts should be strictly monotonic even in the low-dose range, whilst in event of non-linear behaviors, abnormal peaks or flat trends should be reported. We measured the radio-induced damage in two ways: as induction of chromosome exchanges and breakages, through a whole karyotype painting technique (§ 3.1), and as induction of chromatine bridges in anaphase (§ 3.3), through traditional staining. About the induction of such aberrations, the
ix outcome is that no substantial deviation from linearity has been demonstrated. The underlying model is mere probabilistic: the yield and complexity of damage depends only on the dose delivery, the Relative Biological Effectiveness (RBE) of the radiation (fig 14, 15, 18) and the DNA content of the target (fig 16) without hypersensitivity or threshold effects (§ 4.1, 4.2.1).
(ii) Coming to the effect of IR exposure on the telomere, to be reminded that it is an important genome stabilizing factor: it avoids the chromosomal termini to be processed as Double Strand Breaks (DSBs), and the chromosome end-end fusion. An effect of IRs on the modulation of telomere length, in terms both of elongation and shortening depending on the radiation type used, has been reported for high doses. Instead, on the basis of our data, such a modulation is missing in the low-dose range (§ 3.2) for all the radiation types tested. Moreover, the telomere does not significantly participate in the genomic instability triggered on by IRs as revealed by the anaphase bridges analysis (§3.3 fig 18). On the basis of this outcome relative to the low-dose range, speculations on a linearity failure are allowed (§ 4.2.1).
Ultimately, low doses of IRs induce chromosome instability, but not via the telomere homeostasis perturbation, despite its well-known role played in the chromosome asset destabilization/stabilization. Therefore, we have been persuaded to weight the telomere role in the radio-resistance at doses of X-rays of therapeutic order of magnitude.
As the functional telomere is one genome stabilizing factor, IRs are by definition destabilizing agents, able to induce cell inactivation through the genome injury. Moreover, a cell is characterized by a mean telomere length, and a mean spontaneous lack of some telomeres (basal telomere loss frequency). On the aforementioned assumptions, an enhanced radio-sensitivity rationally is expected in case of telomere homeostasis dysfunction. As amid the same cell line the telomere length is spread around a mean value different from cell to cell, then the telomere-related genome stability is supposed to differ from cell to cell (§ 3.4.1), and by hypothesis it may result in a different inherent radio-resistance.
We tested this speculation on several monoclonal lymphoblastoid lines: several clones have been isolated, independently cultured and characterized for the telomere functionality and for the radio-resistance. The outcome is that the short telomeres do not confer radio-sensitivity to the line, while a frequency of telomere loss beyond a threshold does (§ 3.4.6 fig 25), thus the
x telomere dysfunction may be involved in the differential radio-sensitivity of clones obtained from the same population (§ 4.2.2), and shows potential as radio-sensitivity marker.
(iii) The mitochondrion is a physiological source of ROS and the main player of their homeostasis, therefore it is supposed to have some indirect involvement in the radio-damage mechanisms. To be reminded in fact that DNA is injured both by direct radiolysis and by means of reactive species released by the water radiolysis.
Far to be clear, this role is object of several speculations: for example, irradiated mitochondria are supposed to establish a long-term ROS release, moreover, the mitochondrial metabolism is supposed to amplify the activity of ROS generated by IRs (§ 1.15).
The increased ROS activity, generated by mitochondrial impairment, has been related to chromosomal abnormalities, genomic instability, and telomere damage. On the other hand, ROS overproduction, due to the same impairment, paradoxically may be seen as cytoprotective, through engaging countermeasures, as in the so-called adaptive response (§ 1.18).
To address the role of mitochondria-related ROS activity in the response to IRs, we pharmacologically set a fibroblast line upset in the mitochondrial functionality, that showed an increase of the basal ROS activity due to ROS homeostasis imbalance (§ 3.5.1). This model, in which mitochondria are turned into ROS diffusers (fig 29), is aimed to report, through the analysis of cytogenetic endpoints, any differential response to the IRs in comparison to a control line, to ultimately verify whether the increased endogenous ROS activity acts additively or synergistically with the exogenous radio-damage. The pharmacological treatment resulted in a basal increased frequency of DSB (§ 3.5.5) and spontaneous chromosomal fragmentation (§ 3.5.7), conversely no damage on the telomere was detected (§ 3.5.6 fig 30, 31). After irradiation, we observed that the cytogenetic damage yield was the sum of the radio-induced and the basal ROS-induced damage via mitochondrial impairment, without synergistic effect.
In conclusion, relatively to our data the mitochondrion is not an important player in the damage induction via IRs (§ 4.3).
1
1. Introduction
This work deals with the effect of ionizing radiations (IRs) on cultured cell lines, explored through cytogenetic endpoints and clonogenic assay. The development of new molecular probes in the last decade largely improved the resolving power of the modern cytogenetic techniques, both allowing accurate quantitative analysis, and extending the staining to the whole karyotype at once. To carry out the scheduled experimental panel both electromagnetic radiations (X-rays), proton (H+) and α-particle (4He2+) beams were used.
1.1. IRs and Linear Energy Transfer
Ionizing radiations are energy propagations, through electromagnetic waves or particle flux, that carry enough energy to kick out an electron from an hit atom without thermal ionization.
The borderline between ionizing and non-ionizing radiations is wide. One reliable landmark is around 10 eV energy, close to the ionization energy of hydrogen or oxygen (14 eV), or 12.6 eV, the ionization energy of the water. Beyond this threshold, radiations are supposed to be effective to damage biological biomolecules such as DNA.
Mainly, both electromagnetic radiations and charged particles injury the living matter through the inelastic collisions of the secondary electrons (i.e. δ-electrons) produced at the primary interaction with the medium. The photon transfers all its own energy in one event of interaction with matter (§ 1.2), while one charged particle do it all along its trajectory, a bit at time, through several physical collisions (§ 1.3). The outcome is a difference in the spatial distribution of the energy deposition, and consequently the spatial arrangement of the ionizations: spread and uniform for the electromagnetic radiations, clustered in tracks for the particulate type. The ensemble of the interactions of the radiation with the medium is the proper track (Fano, 1970). This set is stochastic but characteristic as a fingerprint of the upstream radiation, in terms of length, width, and new and unstable species that can move and react in their environment.
The International Commission of Radiation Units & Measurements (ICRU, 2011), defines the adsorbed dose in units of Gray (1Gy = 1 J/Kg) according to formula 1.1 (ICRU, 2011):
D [Gy] = ̅/m (1.1) where ̅ is the mean energy transferred by radiation to the m mass.
2 In general, the sequence of occurrences concerning the irradiation of living matter can be summarized in one physical-chemical and one biological stage. In the first step, the energy is deposited on an atom and an electron is kicked out (i.e. the secondary electron or δ-electron); δ-electron is scattered and rapidly loses energy producing further ionizations until it reaches the thermal equilibrium. In simple terms, secondary electrons are the principal damage effectors. Afterwards, the new-born species diffuse and react both each other, and with the solvent.
Usually IRs are characterized by the Linear Energy Transfer parameter (LET). This represents the linear density of energy (E) deposition along the path (x) through the medium. LET depends on the type of the radiation as well as on the material traversed, and is expressed according to formula 1.2 (ICRU, 1970):
LET [KeV/µm] = dE/dx (1.2) Electromagnetic ionizing radiations (i.e. X- and γ-rays ), cannot be described by LET because photons are absorbed in a single process, and don’t produce linear tracks as charged particles do. Therefore those are simply termed sparsely ionizing or low-LET radiations.
About the subsequent biological cascade of events, in this work we investigated at the level of cellular consequences, although the biological effects may be at different level:
1. Cellular effects, lasting up days or weeks
2. Organismal effects, lasting up to the whole lifespan 3. Transgenerational effects, no predictable expiration
1.2. Photons’ interactions with living matter
The X- and γ- rays flux into the medium undergoes an exponential decay depending on the energy of the incident photons, the arresting power of the medium and the depth of penetration. They randomly interact with the matter in one of those three ways: the Photoelectric Effect (PE), the Compton Effect (CE), and Pair Production (PP). In PE, the photon annihilates onto an atom, and an electron is ejected provided with a kinetic energy equal to the photon energy minus the bound energy of the electron removed. In CE case, a fraction of the photon energy is transferred to an atomic electron, and the reduced-energy photon is scattered at angles up to 180°. In PP case, the photon energy is needed to be higher than 1.022 MeV; in such a case it annihilates and one electron (β-) and positron (β+) are
3 produced, provided with a kinetic energy equal to the photon energy minus 1.022 MeV.
In water-based environments, such as the cells, the main photons’ injury mechanism is by mean of the reactivity of the water radiolysis compounds. In fig 1a is presented the time scale for the sequence of events triggered on by the energy deposition. Water radicals (H2O∙) and water ions (H2O+ + e-)
(i.e. primary radiolysis products) are generated by the interaction of the IR with the water molecules in one of the aforementioned modes. The physicochemical process ends in 10-14s when those compounds are decomposed into their primary products: ·H + ∙OH, H2 + O·, and ∙OH +
H3O+, while the δ-electron continues to ionize the water as far as its energy
growths lower than 12.6 eV (§1.1) until it is solved (10-12 sec). A set of compounds derives from the primary radiolysis products (fig 1b) and this reaches the chemical equilibrium in 10-7 sec through chemical diffusion. It is composed both of radicals, and non-radical molecules.
Figure 1: a) Time scale of radiolysis of water. b) Spur reactions in water; e-aq represents the electron solvated in water (modified, Gregory et al., 2002).
In absence of solutes the radical unstable compounds react each other and water is reformed. Conversely, in the water-based solutions, such as cytosol, the chemical changes of the solute reflect their reactivity towards the reductants ·H and eaq- (i.e. solvated electron) and the oxidant ∙OH. Moreover
in oxygenated solutions (such as in physiological conditions) eaq- reacts with
O2 to form superoxide anion O2∙-, one further highly reactive specie (§ 1.14). 1.3. Charged particles’ interactions with living matter
Particulate radiations (H+ or heavier accelerated ions), are classified as densely ionizing or high-LET radiations for they transfer their own kinetic energy to the hit matter, through several direct collisions along their straight trajectory (i.e. direct damage), in contrast both with the scattered path of the
4 electrons, and the random interaction of the photons. Paretzke (1987) summarized in few statements the general process of a charged particle penetrating into the matter: (a) the mass is responsible for the straightness of the trajectory; (b) the fraction of interactions produced directly by the ion (i.e. collisions) compared to those produced by secondary electrons decreases with the ion energy; (c) the higher is the energy of the ion, the sparser is the track (LET decreases); (d) the distance of the interactions decreases with the square of the effective charge of the ion (Zeff2).
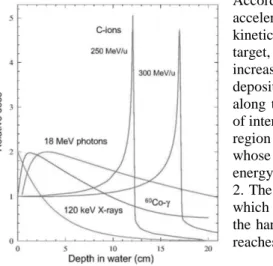
According to these statements, the accelerated charged particles lose kinetic energy penetrating into the target, so that they are forced to increase the frequency of energy deposition (i.e. direct collisions) along the path and to reach a peak of interaction at a defined depth, the region of the so-called Bragg peak, whose depth is determined by the energy of the beam as showed in fig 2. The Bragg depth is the region in which is the ionization density (i.e. the harmful power of the radiation) reaches the top.
Figure 2: a) For low-LET radiations (18 MeV photons, Co-γ and X-rays) the dose decreases exponentially with the depth of penetration. For the high-LET radiations instead (C-ions) the maximum dose is deposited at the Bragg peak at the end of the track (courtesy Weber U). The dose delivered downstream of the Bragg peak is due to the fragmentation of the primary ions into lighter “fireballs”, which are allowed to run forward beyond the peak depth (Sihver et al., 1998).
1.4. Types of DNA radio-induced damage
DNA is the cell target that far apart realizes the most important consequences on cell viability when harmed. IRs can injury DNA both by indirect (§ 1.2) and direct damage (§ 1.3) prevalent for low- and high-LET radiations respectively.
In the latter case the radiation ionizes and breaks directly the DNA molecule by direct interaction, whilst in the first case the ionization occurs by mean of reaction of the DNA with water radiolysis compounds released in solution, that are able to diffuse for nanometers from the origin point (Krämer and Kraft, 1994).
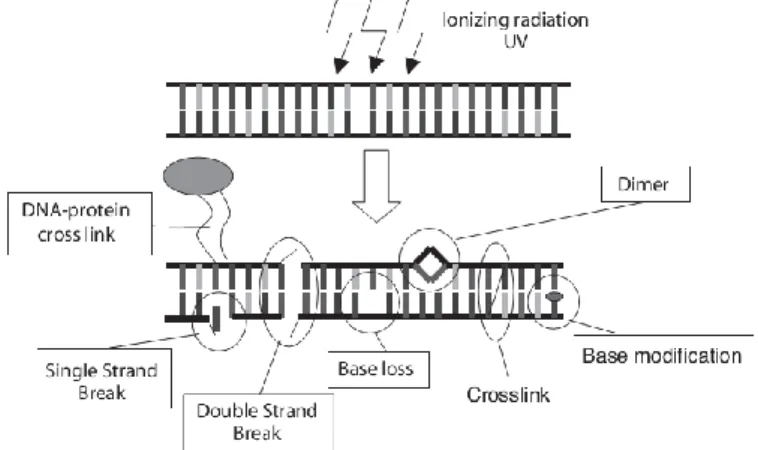
5 In fig 3 is schematized the plethora of heterogeneous damages due to IRs interaction that undergoes the double-stranded DNA. The level of combination and the proximity of the lesions determinate the severity of the injury, in fact, since the information on both strands of the DNA molecule is complementary, all injuries only on one side of the double strand can potentially be repaired by using the information on the intact complementary strand. In fact single-strand breaks (SSBs) are considered less severe than double-strand breaks (DSBs), for the integrity of chromosome is preserved. In fact DSBs are considered as the critical event for the induction of lethal lesions since they are responsible for the structural chromosomal aberrations (§ 1.8): even only one of those may be enough to inactivate the cell and to upset the whole chromosomal asset.
Figure 3: Types of radiation-induced DNA damage (Scholz, 2003).
In tab 1 are approximately summarized the damage types yield for 1 Gy of low-LET radiation. The most frequent types are base lesions, sugar modifications, apurinic / apyrimidinc sites, cross-links, SSBs and DSBs.
Table 1: Approximate yields of DNA damage per Gy per cell (Sholz, 2003)
SSB 1000
DSB 30-40
DNA-protein crosslinks 50 Complex damage (SSB + base lesion) 60
6
1.5. Clusters of damage
There is a straight relationship between the damage complexity and the LET of radiation. In general, the higher is the LET, the more clustered is the ionization distribution in the medium (§ 1.3), the more represented is the direct damage induction mode, the more complex are the lesions on the DNA duplex (Belli et al., 1996) and their reparability is hampered (Okayasu, 2012). In fig 4 several track structures, representative of radiations with increasing LET, are shown: the contribution of the direct damage radiations increases from γ-rays to higher LET. Their increasing density accounts for the increased DSBs yield in dependence of the LET, because of the increased probability of break of both DNA strands in two close position (Krämer and Kraft, 1994).
Figure 4: Schematic representation of sparsely (γ-rays) and densely (ion beams) IRs. LET increases from γ-rays to the top. The track density accounts for increasing complexity of the aberration induction. On the right for comparison is shown the DNA-solenoid (30 nm in diameter) (based on Paretzke, 1987 and Krämer and Kraft, 1994).
1.6. Relative Biological Effectiveness of IR on the living matter
The main consequence of the different yield of DSBs in dependence of the LET, is the fact that the same energy amount delivered in one living target by different radiation types can give rather different effects, in terms of deterministic effects, recovery capabilities, and downstream stochastic consequences. This fact imposes the definition of a parameter that allows the dose adsorbed to be normalized on its biological effect. The Relative Biological Effectiveness parameter (RBE) was issued by ICRU (ICRU, 1986) to fulfill this need, and was defined as the ratio of adsorbed dose of photon radiation to the adsorbed dose of the radiation used, to get a fixed biological effect according to formula 1.3:
7 Ultimately, the complexity of induced lesions accounts for the RBE of the considered radiation type.
From RBE derives the equivalent dose (H) concept, whose unit is the Sievert (Sv) according to formula 1.4:
H [Sv] = RBE D [Gy] (1.4) RBE has been determined for a large set of radiation qualities on a large set
of radiobiological endpoints, and despite the initial purpose, this parameter is not extendable and generalizable in every condition: it depends on the cell type, on the biological endpoint investigated, and in particular on the absorbed dose: in fact it generally decreases with the dose for the radio-sensitivity to the photons increases with the dose up to a plateau. Thus ICRU issued the so-called quality factor (wR) as a weighting factor lumping
several RBE values got from different endpoints, effectively applied in case of mixed radiation field. In such a case the final adsorbed dose is defined according to formula 1.5 where R is the radiation type:
[ ] ∑ [ ] (1.5)
1.7. Chromosome DSBs repair
Normal human cells contain about 3 x 109 base pairs (bp) in 46 chromosomes consisting of 22 pairs of autosomes and one pair of sex chromosomes, so that the normal male karyotype is 46 XY and the female one is 46 XX.
In case of DSB the unpaired free strands are too much short to stabilize the spontaneous rejoining. In such a case, the broken ends become “sticky” and the misrepair is frequent due to the random facing of sticky ends belonging to different chromosomes.
In DSB occurrence, there are two major enzymatic types of DSB repair modes: the homologous recombination repair (HRR) and the non-homologous end-joining (NHEJ). It is generally believed that NHEJ plays a more important role than HRR in mitotically replicating cells. NHEJ is more important during G1 phase (Takata et al., 1998), it uses little to no
sequence homology in a process, considered error prone, accomplished by the DNA ligase IV/XRCC4 (Lobrich and Jeggo, 2005). HRR instead seems to play a more prominent role during meiosis and when sister chromatids are available during the late S and G2 phases (Haber, 2000). The HRR is a
very complex process and it is allowed when homologous sequences, in the form of sister chromatid or homologous chromosome or DNA repeats, are
8 available to a “strand invasion” aimed to recover the lost genetic information of the injured DNA duplex (Valerie and Povirk 2003).
1.8. Chromosomal aberrations, genesis, nature and fate
Chromosomal aberrations can be sorted into two main groups: numerical (i.e aneuploidies) and structural. A structural aberration occurs in general when the normal structure of a particular chromosome is missing. It can involve both only one and several chromosomes. Instead, the numerical aberration derives from the loss or gain of one or more whole chromosomes. Structural aberrations can be produced in every moment of the cell cycle, whilst numerical disorders are usually produced during the cellular division (both mitosis and meiosis) for incorrect segregation in anaphase (Albertini et al, 2000).
Structural chromosomal aberrations (hereafter chromosomal aberration
tout-court) derive from misrepaired or unrepaired DSBs (Rothkamm and,
Löbrich, 2002).
DSBs can occur in G0/G1 phase, when one single chromatid per
chromosome is present. In case of misrepair or repair failure, the lesion will be replicated during the S-phase determining a CSA (chromosome aberration), in which the sister chromatids have a break in the same position, well detectable in metaphase by microscopic observation (fig 5). In contrast, one damage occurred in S or G2 phase results in one CDA
(chromatid aberration) (Natarajan 2002). Though in different manner (i.e. CDA or CSA), the DSB emerges regardless of whether the cell has undertaken or not the phase, and therefore IRs are classified as
S-independent agents.
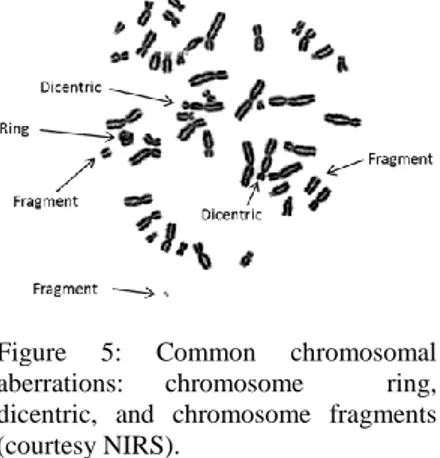
In general, a chromosome with more than one functional centromere in a cycling cell, is considered an unstable aberration because it results
in an anaphase bridge with
consequent mitotic failure and probable cell death (fig 6a). The acentric fragments too are unstable elements, as they cannot interact with the microtubules, thus they are generally lost in the anaphase. The loss via acentric fragments release of
important parts of genetic
information may lead to cell death
Figure 5: Common chromosomal aberrations: chromosome ring, dicentric, and chromosome fragments (courtesy NIRS).
9 for the loss of important metabolic functions. Furthermore, the cells with lasting unrepaired centric fragment can undergo apoptosis (fig 6b) (d’Adda di Fagagna et al., 2003). Whatever is the mechanism, the death of the aberrant cell or the loss of the aberrant element, a clearance of unstable aberrations is established.
Figure 6: Cell death for DNA lesions: reproductive cell death (a) for mitotic failure with chromatine fragment release and anaphase bridge due to a dicentric chromosome (DAPI and telomere staining), (b) apoptotic cell (top), and control (bottom) (Hoecsht and Acridine Orange).
Stable aberrations instead, such as balanced translocations or inversions, do not affect the total amount of genetic information and do not pose mechanical problems to the chromosomal segregation at anaphase, therefore these are transmitted to daughter cells. For their persistence throughout the
lifespan of the cells, such aberrations are even more threatening than short-living unstable aberrations, and are allowed to initiate carcinogenesis in case of gain a selective growth advantage (Jeggo 2009).
In this respect, the development of multicolor in situ hybridization FISH (m-FISH), that allows to differentiate all homologue pairs, has greatly improved the ability to identify stable chromosome aberrations (fig 7) which particular attention to the complex aberrations. Those are defined as chromosome aberrations involving three or more breaks in two or more chromosomes (Savage and Simpson, 1994). Complex aberrations are expected to occur more frequently after exposure to high-LET densely ionizing radiations, compared to low-LET sparsely ionizing radiations (§ 1.5), in that energy deposition patterns produced by densely ionizing radiation produces highly localized multiple DNA damage at the chromosomal level (Brenner and Ward 1992). The biological outcome of qualitatively different lesions is dependent on the quality of radiations as well described by RBE (§ 1.6).
Figure 7: Representative m-FISH image of one simple translocation between the chromosome 4 (green) and 9 (brown).
10
1.9. Telomeres
The preservation of integrity of the chromosomal complement is a crucial need for cells and organisms; likewise it is known for long time that chromosomal termini need a special protection to avoid the accidental “erosion”, enzymatic or not, of genetic information. Telomere literally means terminal monomer and as such it constitutes the cap of the linear chromosomes of eukaryotic cells, and fulfills the need for the termini’s protection.
This capping function consists in the avoidance that the ends are recognized as DSBs and so processed (Felder et al., 2003), leading to chromosomal fusions and dramatic consequences on cell viability.
The telomeres of all the vertebrates, share the same tandemly repeated TTAGGG sequence, the length instead can be very different: in the humans a normal telomere length is between 5 and 15 Kbps while for instance in the mouse is several times higher (Marion et al., 2009).
The telomere is arranged in the so-called telomere-loop (T-loop), the most part of it is double-stranded, whilst the single-stranded -3’ terminal part consists of about 150 nucleotides (Blackburn, 1991).
This single-stranded short region is crucial for functionality: it is free to fold back and to bind an unfolded string of the tandemly repeated region forming the structure termed displacement loop (D-loop) which stabilize the whole T-loop (fig 8) (de Lange 2005). This “triple stranded” region is supposed to be the primordial mechanism of protection of linear chromosomes ends. Two kinds of protein complexes are associated to form the so-called shelterin component (de Lange, 2002). One group (TRF1 group) regulates the telomere length, and the other (TRF2) fulfill a protection function and it is composed of proteins involved in the normal DNA repair process. TRF protein cluster binds the double-stranded DNA while POT1 the single-stranded DNA (fig 8). For the stabilization of T-loop a link between POT1 and the double-stranded part is needed and it is provided by TIN2 and TPP1 (not shown in figure) (reviwed in Blasco, 2005).
11
Figure 8: The T-loop structure. The 3′ overhang is strand-invaded into the adjacent duplex telomeric repeat array, forming the displacement-loop (D-loop). Telomere-associated proteins: the TRF1-headed groups fulfill the length control function, the TRF2-headed fulfill the telomere capping function (Blasco, 2005).
1.10. Consequences of telomere shortening: senescence, apoptosis and genomic instability
In every replication of linear chromosomes about 50-100 nucleotides are lost because of the intrinsic inability of the DNA-polymerase to polymerize DNA in 3’5’direction (i.e. end replication problem, Harley et al., 1991). This fact results in the telomere shortening, which causes the reproductive limit of the normal cells and senescence, and only through the expression of the telomerase can be prevented (Cong et al., 2002).
Telomerase is the main compensating enzyme which adds DNA telomeric sequences (i.e. TTAGGG in vertebrates) to the 3' end of DNA strands to overcome the end replication problem. This enzyme is typically active in the late S-phase (Greider and Blackburn 1985) and it consists of two subunits: the catalytic (TERT or TRT), and the RNA template core. To be noted that, though with exceptions, in general the more one cell is far from the stem condition, the less this enzyme is expressed, in fact it is not active in somatic cells.
The senescent cells lie stuck in G0 phase in the so-called mortality stage 1
(M1); the initiation of the senescence could be a protective mechanism to avoid the consequences of any unstable chromosomal complement
12 potentially leading to tumorigenesis (Wright et al., 1989). However, even in the senescent cell the least telomere presence is needed for the cell viability (Verdun and Karlseder, 2007): in fact in case of excessive telomere impairment the cell activates the proper countermeasures up to the apoptotic pathway.
The monitoring task of telomeric reliability is basically charged to the systems ATM-p53 and pRb activated by DNA double-strand breaks (Hemann et al., 2001). In case of telomeric dysfunction pRb inhibits transcription factors of the E2F family that participate in the progress into S phase. Likewise p53 activates p21 that inhibits the cyclin-CDK2 or -CDK1 complexes, and thus it arrests the cell-cycle progression in G1 and
eventually triggers the apoptosis.
During the senescence, when the p53/pRb control fails, and the cell continues to cycle, telomeres are allowed to indefinitely shorten until the cell undergoes a crisis state for the genome instability related to telomere impairment (Felder et al., 2003). Genomic instability is a general term to describe the processes that can increase the rate of mutation of a population, enabling cells to develop new and aggressive phenotypes, and involves whole chromosomes or large portions of them that are gained, lost or rearranged (Rebuzzini et al., 2009).
When the sequence is too short or virtually absent (i.e. telomere loss), the telomere cannot be arranged as a T-loop (§ 1.9). In such a condition the chromosome end is recognized as DSB and processed through the pathway available (i.e. HRR or NHEJ) (§ 1.7) leading to the genesis of abnormal chromosomes with more than one functional centromere (dicentric). Such an occurrence leads to an anaphase bridge with direct involvement of the telomere resulting in the reproductive death (§ 1.8), or to the so-called break-fusion-break cycle for the cells that survive the anaphase bridge occurrence (Ojima et al., 2004). Such an event perpetuates and enhances the genome instability in the subsequent cell generations and establishes a high mutation rate. Though this crisis phase is characterized by a very high cell mortality, the high mutation rate related to the genomic instability can produce a random reactivation of telomere lengthening through telomerase gene activation or the so-called alternative lengthening of telomere (ALT) (Londoño et al., 2004), thus the cell can
overcome the crisis and be immortalized
(Shay and Woodring, 2005) with a manifest tumorigenesis risk.
In conclusions telomeres are intimately linked to the maintenance of chromosomal stability and the consequences
13 of damaged telomeres have short, as well as long term effects, as during multistep carcinogenesis. (Murnane and Sabatier 2004)
1.11. Telomeric damage-related chromosomal instability
Ultimately, the telomere length is ruled by the competition of lengthening, and shortening factors, and there are evidence that several chemical or physical agents could alter this refined homeostasis.
Among these, IRs can harm the telomere with a direct break of the telomeric DNA (Bolzan and Bianchi 2006), or through the oxidation to 8-oxodG of the deoxyguanosine of the sequence TTAGGG (von Zglinicki, 2000), moreover it should be considered that telomere is repaired less efficiently than the rest of the genome (Opresko et al., 2005).
On the other hand the exposure to IRs is able to trigger cellular responses that promote the telomere maintenance and even the elongation.
For instance, high-doses of either high-LET protons are able to significantly increase the length of telomere in human primary fibroblasts shortly after exposure, while such an effect was not achieved after exposure to low-LET X-rays, on the contrary accompanied by telomere erosion (Berardinelli et al., 2010). This fact allows to speculate that the complexity of a subclass of lesions introduced into DNA by high-LET radiations, differs at qualitative level from those lesions produced by low-LET radiations in terms of cytogenetic outcome.
On such premises, and on the premises of the detrimental effects of the telomere impairment (§ 1.10), it is worth to pay attention on the possibility of occurrence of genomic instability driven by the radio-induced alteration of the telomeric homeostasis.
1.12. Telomere-related radio-sensitivity
A major factor in the failure of radiotherapy is inherent or induced cellular Radio-resistance. This is a characteristic of many different tumor types which is also retained in cultured cells. Radio-resistance could develop in cancer cells by several possible mechanisms. Among the possibilities, differences in the amount of initial DNA damage related to chromatin conformation and capacity to repair, DNA DSBs generally have been considered the most obvious mechanisms.
Other factors, such as apoptosis failure and altered expression of genes taking part in the regulation of cell cycle checkpoints, have been proposed as bases for radio-resistance (Skinner et al., 2012). In addition, a possible relationship between radio-sensitivity/resistance and telomere dysfunctions /telomere length has been reported (Fumagalli et al 2012,) leading to the
14 possible use of telomere length as a marker for chromosomal radio-sensitivity. Moreover, the telomere loss (§ 1.10) has been associated with genomic instability (Hemann et al., 2001) supporting the notion that it plays a central role in cancer transformation, especially during the crisis phase (Muraki et al., 2012).
Even cancer cells commonly show telomere loss despite the expression of telomerase. This lower rate of telomere loss is tolerated by the cell, and can therefore result in chromosome rearrangements that can continue to occur throughout the lifetime of the tumor (Muraki et al., 2012). In addition to chromosomal instability generated as a direct response to excessive telomere shortening, it should be pointed out that such telomeric modification alters the kinetics of DNA damage response in irradiated human fibroblasts, thus contributing also indirectly to genomic instability (Drissi et al., 2011). Interestingly, primary cells carrying mutations in genes devoted to DNA repair as DNA-PKcs, Ku70, Ku80, NBS1, MRE11, RAD50,
ATM, FA, BRCA1 and WRN display accelerated telomere shortening (Cabuy
et al., 2005), and increased sensitivity to irradiation. Overall, these studies indicate that an inverse relationship between telomere length and the degree of individual sensitivity to IRs exists in primary cells with an absent or insufficient level of telomerase (Castella et al 2007).
However, it should be pointed out that such a relationship is still a matter of debate, in that controversial data are available in the literature. A 7-8 fold reduction in telomere length was observed in radiosensitive murine lymphoma cells L5178Y-S compared with the radio-resistant parental cells L5178Y, possessing 7 kb and 48 kb, respectively (McIlrath et al., 2001). McIlrath and coworkers (2001) reported an inverse correlation between telomere length and radiation-induced cytogenetic damage in a survey carried out in lymphocytes drawn from 24 breast cancer patients and 5 normal individuals. On the contrary, the analysis of Large Cell Lymphomas (LCLs) established from 33 cancer patients who suffered significant tissue damage in the irradiated area (radiosensitive patients), showed that a subset of them had abnormally long telomeres, even longer than telomeres observed in 18 LCLs from individual with normal tissue response after radiotherapy (Sprung et al., 2008). Recently, Zongaro and coworkers immortalizing human fibroblast clones by ectopic expression of telomerase, showed that in the presence of an active enzyme, even short telomeres do not cause an increase in radio-sensitivity (Zongaro et al., 2008).
Overall, these conflicting results show that further investigation is needed to strengthen the link between telomere length and radio-sensitivity. In addition, it should be pointed out that in the studies above reported, based
15 on Telomere Restriction Fragment (TRF), q-FISH or f-FISH analysis, principally the mean telomere length was investigated, whereas the role of telomere loss was almost totally neglected.
1.13. Mitochondrial oxidative functions
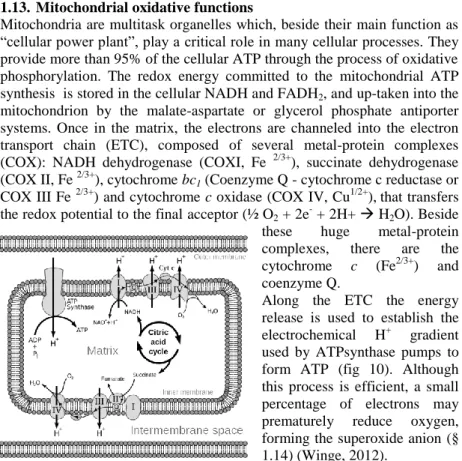
Mitochondria are multitask organelles which, beside their main function as “cellular power plant”, play a critical role in many cellular processes. They provide more than 95% of the cellular ATP through the process of oxidative phosphorylation. The redox energy committed to the mitochondrial ATP synthesis is stored in the cellular NADH and FADH2, and up-taken into the
mitochondrion by the malate-aspartate or glycerol phosphate antiporter systems. Once in the matrix, the electrons are channeled into the electron transport chain (ETC), composed of several metal-protein complexes (COX): NADH dehydrogenase (COXI, Fe 2/3+), succinate dehydrogenase (COX II, Fe 2/3+), cytochrome bc1 (Coenzyme Q - cytochrome c reductase or
COX III Fe 2/3+) and cytochrome c oxidase (COX IV, Cu1/2+),that transfers the redox potential to the final acceptor (½ O2 + 2e- + 2H+ H2O). Beside
these huge metal-protein
complexes, there are the
cytochrome c (Fe2/3+) and coenzyme Q.
Along the ETC the energy release is used to establish the electrochemical H+ gradient used by ATPsynthase pumps to form ATP (fig 10). Although this process is efficient, a small percentage of electrons may prematurely reduce oxygen, forming the superoxide anion (§ 1.14) (Winge, 2012).
The subunits of the most part of the ETC proteins are encoded both by nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). This dual genetic
Figure 10: Electron transport chain and involved protein complexes. The reducing power (e-) is provided by the citric acid cycle and addressed to oxygen. Some electrons are released along the streaming and contribute to the accidental ROS generation. The overall process: NADH → COX I → Q → COX III → cytochrome
c → COX IV → O2 ↑ FADH → COX II
16 control is responsible for 13 mtDNA-encoded proteins and 74 nDNA- encoded proteins (Kulkarni et al., 2009). Mutations in either mtDNA or nDNA leading to respiratory chain and ATPase deficits are now known to have profound adverse effects (§ 1.15).
1.14. ROS homeostasis: physiological release and detoxification
ROS is a family of unstable compounds, toxic for all the organisms, that usually react with the biological polymers (Hoeijmakers, 2009). Generally, a millimolar concentration of ROS leads to alteration of the cell homeostasis, driving it to the apoptosis or necrosis (Jacobson, 1996). The primary specie of this class of compounds is the superoxide anion O2∙−.
In water it exists in dynamic equilibrium, both as such (O2∙ −, 99.7%), and as
hydroperoxyl radical (∙HO2, 0.3%). The hydrogen peroxide (H2O2)
originates via its dismutation, with a stoichiometry close to 2, and it finally decomposes into two hydroxyl (OH-) according to reactions in 1.6:
2O2∙ − + 2H2O → O2 + H2O2 + 2OH− → O2 + 2(∙OH) + 2OH- (1.6)
ROS are also physiologically produced by NADPH oxidase in activated neutrophil granulocytes or macrophages and released by respiratory burst for use in oxygen-dependent killing mechanisms of invading pathogens. Moreover, ROS are released by exogenous factors such as UV light and IRs via the aforementioned water radiolysis (Lehnert and Iyer, 2002) (§ 1.2). Besides these particular occurrences, mitochondria are the major intracellular source of superoxide anion due to the small fraction of oxygen that is not fully reduced to water by the ETC (§ 1.13). There are two steps in the respiratory chain where these reactions mainly occur: COX I and COXIII (reviewed by Turrens, 2003) (fig 10), hence mitochondria are provided of an Mn-Superoxide Dismutase enzyme (SOD) inducible in a variety of conditions including hyperoxia and radiation exposures, or as a response to cytokines. In the cytoplasm also, this enzyme class is constitutively present, is extremely efficient, and catalyzes the neutralization of superoxide nearly as fast as this diffuses.
H2O2 in mitochondria it is mainly removed by glutathione peroxidase, while
in the cytoplasm mainly by catalase enzyme.
However, mitochondria are particularly vulnerable to oxidative damage because they are constantly exposed to high levels of ROS, and despite these enzymatic countermeasures, mtDNA bears a high base-oxidation rate compared to the nDNA (104-fold) (Sohal and Sohal, 1991). The mitochondrial ROS production has been related with several process, such
17 as aging, apoptosis, alcoholism-related tissue damage. Under such conditions, the detoxifying defenses are overwhelmed, causing oxidative stress and eventually damaging other compartments, up to the cell death.
1.15. Mitochondrial dysfunctions and ROS homeostasis imbalance
Mitochondrial dysfunctions, arising either by genetic mutations or exogenous factors, lead to decrease ATP synthesis and to an increased ROS production, as a result of inefficiencies in ETC (Kulkarni et al, 2009). Cellular redox imbalance is likely to affect all macromolecules, including nucleic acids, proteins, carbohydrates, and lipids. This leads to pronounced DNA damage, mostly SSB and base damage (Pollycove and Feinendegen, 2003), to spontaneous chromosomal aberrations (Samper et al., 2003), and a variety of cellular responses, including cell-cycle arrests, senescence, apoptosis, and possibly cancer.
In particular, telomeres are preferential targets of acute oxidative damage for ROS are able to induce 8-oxodG from triplet GGG present in human telomeric sequence TTAGGG (von Zglinicki, 2000), and it is repaired less efficiently compared to the rest of the genome (Opresko et al., 2005) (§ 1.11). Besides targeting a plethora of molecules, oxidative stress triggers on signaling cascades leading to cell death, by either apoptosis, or necrosis, or autophagy as well (Guachalla and Rudolph; 2010). Therefore, due to their central role in cellular homeostasis, impairment of mitochondrial functions may turn highly detrimental for cell survival. Furthermore, mitochondrial dysfunction may exacerbate the detrimental effects of genotoxic agents or paradoxically, may bring beneficial cellular responses to stressful agents. Actually, ROS overproduction, resulting from mitochondrial dysfunctions, may be seen as cytoprotective, in that a chronic stress, that precedes the genotoxic stimulus, may engage a regulatory component, as occurs in the so-called adaptive response (§ 1.18) (Lehnert and Iyer, 2002).
To address the role of mitochondria in the response to DNA damaging-agents, and in particular to IRs, both mitochondrial mutant cells and mtDNA-chemically depleted normal and tumor cells have been used (Kulkarni et al 2009). In particular it has been speculated that mitochondria, beside the major ROS production due to short-lived primary ionization events, may act as source for extranuclear amplification mechanism of secondary ROS after irradiation (Leach et al., 2001).
It has been proposed that radiations are able to activate a reversible form of membrane transition, propagating the signal from one mitochondrion to the next, with a consequent ROS release. This mitochondrial-generated ROS (m-ROS) persist at late times (>15 min) after irradiation with both low-
18 and high-LET radiations (Leach et al., 2001), or even weeks later in the progeny of irradiated cells.
The long-term increase of ROS intracellular level as generated by mitochondrial alterations, has been closely related to chromosomal abnormalities and nuclear genomic instability (Kim et al., 2006). In addition, this mitochondria-dependent amplification mechanism would account for the results concerning the bystander effect in cytoplasm-irradiated cells (§ 1.20) (Tartier at al., 2007).
To note that all the above mentioned studies have been performed either in transformed lymphoblastoid or in tumor cells, and mutations in mtDNA occur rather frequently in human tumors, leading to altered mitochondrial functions and association with overproduction of ROS. (Chatterjee et al., 2012). Instead, no investigations dealt so far with the effect on mtDNA deprivation and response to IRs in primary cell cultures.
1.16. Low-dose range and non-linear effects
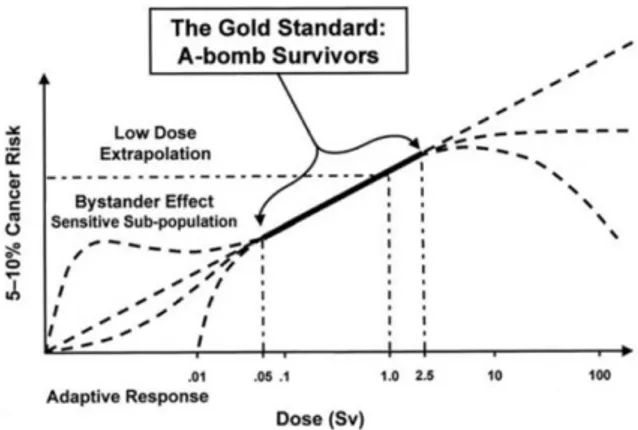
The cancer risk assessment due to IR exposures has been mainly based on the follow-up of the A-bomb and nuclear accident survivors (Sakata, 2012; Saenko et al 2012). The large amount of available data approximately covers the range between 0.5 and 2.5 Gy, and provides a satisfying linear dependence of the risk on the adsorbed dose. Conversely, out of this range, several theoretical models have been proposed, largely to be tested yet. As epidemiological studies are insufficient to elucidate the shape of the dose-response relationship at low doses, a Linear No Threshold Model (LNTM) has been applied to assess the risks resulting from exposure to IRs
(
Suzuki K and Yamashita, 2012).The LNTM is based on a conservative approach and on the a priori hypothesis that the response of biological systems is uniformly dose-dependent for low as for high doses. Indeed, the basic assumption that sustains the LNTM in the last years was questioned for the unfolding of clues and evidences of unexpected non-linear trends (fig 11). The product of dose by wR as well as RBE (§ 1.6) is supposed to be satisfactory to predict
the effect of one irradiation (ICRU 1986), but in extreme situation, as in the low-dose range, equation 1.4 fails and is inadequate to account for unexpected outcomes such as hyper sensitivity/increased radio-resistance (HRS/IRR, § 1.17), adaptive response (§ 1.18), genomic instability (§ 1.19), bystander effect (§ 1.20).
Even not yet clarified, the non-linear behavior of the biosystems is an issue composed of biological facts (and as such hard to investigate), and physical facts, which lie behind the biological outcome.
19 The heterogeneous deposition of dose in the low-dose range (Feinendegen, 2005) might be one of the underlying physical reasons, while the cytoplasmic pairing via gap-junctions and the diffusion of reactive species (e.g. ROS and RNS) released via irradiation are supposed to be important factors, in fact the addition of reactive species scavengers fades the phenomenon in the opinion of Kashino and coworkers (2007). On the other hand, the reactive species diffusion might stimulate the up-regulation of DNA surveillance and repair activities, resulting in the adaptive response (Matsumoto et al., 2007).
Figure 11: Risk assessments in function of the equivalent dose (§ 1.6): possible deviations from linearity in the extrapolations of high and low-dose range (courtesy INFN).
1.17. HRS and IRR
Hyper radio-sensitivity (HRS) can be defined as effect for which cells undergo a larger damage per unit dose after a single exposure in the low- dose range, but show an increased radio-resistance (IRR), to larger doses (fig 12a) (Marple et al., 2004).
The mainstream approach to investigate such an effect is the clonogenic assay, which is used to verify the ability of an irradiated cell population to continue the replication forming colonies from single cell, while few reports, and restricted to classical non-molecular cytogenetic techniques, are available on the induction of chromosomal damage in vitro for human (Nasonova et al., 2006) and rodent cells (Tsoulou et al., 2001)
HRS is demonstrated to be quite common in mammalian cells, and so far it is the core of the non-linearity issue in the low-dose range. It is generally
20 agreed that a threshold dose around 20 cGy, triggers on a set of responses which makes the cell more radio-resistant (fig 12b).
One general explanation could be that for the organism it should be better to let a little cells amount to die than initiate the DNA repair cascade, error and carcinogenesis prone.
The result is the self-clearance of the tissue from the dangerous elements, provided that the persistence of some unrepaired DSBs does not increase the risk of mutation more than the repair process. To be noted that HRS/IRR is known to be a predominant in G2 phase cells (Short et al., 2003), and it
might be a response exclusive of cells in G2 because the G2/M checkpoint is
strongly suggested to be the critical point in which HRS establishes.
Briefly, DSBs cause the activation of ATM and this results in the recognition of the strand breaks by damage recognition proteins. Ultimately, only if the damage is beyond a threshold the G2-phase specific checkpoint is
activated and promotes repair and cell survival (fig 12b) (Marple et al. 2004). Definitely the checkpoint “ineffectiveness” may be the key that underlies the increased mortality within 0.5 Gy.
Figure 12: a) Cell survival at increasing doses of X-rays (V79 hamster fibroblasts). The inactivation peak at low doses shows that the inactivation per unit dose is higher for low doses than for high doses (modified, Marples and Joiner, 2000). b) Over the damage threshold the cell activates the DSBs countermeasures resulting in the G2 -phase-specific checkpoint activation which promotes repair and cell survival (Marple et al. 2004).
21
1.18. Adaptive response
Usually, when a cell culture is exposed to a very low dose (i.e. priming dose) before a larger exposure (i.e. challenge dose), the response in terms of chromosomal aberrations is lower than in case of only challenge dose, and this occurrence it termed adaptive response (AR).
The retrospective review of Wolff (1995) showed that cells from different individuals respond differently to the primer dose, making evident that the genetic background masters the AR. Afterwards the investigation was also extended in vivo and for several endpoints, finding out that primer doses appear to reduce, below the non-irradiated controls, the spontaneous frequency of biological changes, such as the transformation rate (Redpath et al., 2001) and chromosome damage (Hooker et al., 2002). Moreover, Schollnberger and coworkers (2002) observed a decreased spontaneous tumorigenesis in cancer-prone animal models after low-dose exposures. Further confirmations come from cellular and molecular studies (Elmore et al., 2008), suggesting that the risk for late effects of exposures is decreased by triggering biological mechanisms of repair and DNA stability surveillance after least priming doses.
1.19. Genomic Instability
Rather recently, beside the well-known effects of IRs, such as DSBs, cell killing and so on, it emerged that the exposure might establish an event cascade resulting in a delayed increased mutation rate in the irradiated cells progeny. Those cells are prone to lose the control on their genomic complement and this genomic instability is supposed to be the cause behind the increased radio-sensitivity of the subsequent generations.
These phenomena were observed both for high-LET and low-LET irradiation in several lines (Kadhim et al., 2006).
Such a fact was observed even for the progeny of non-directly hit subpopulations in contact with the hit cells through gap-junctions, or by sharing the culture medium as for the bystander effect (§ 1.20) (Moore et al., 2005).
1.20. Bystander effect
In case of non-homogeneous irradiation of a cell population, the non-hit subpopulation can react to the injury as the hit population does, though by a different order of magnitude of consequences, so that the cells without energy deposition undergo biological changes generally termed “bystander effects”.
22 Mothersill and Seymour (2001) first focused on the release of soluble factors in the medium by the hit cells, able to induce reaction after-exposition-like in the bystander cells (§ 1.15). Beside this mechanism, the tight cell-cell pairing has been proposed as the mean through which the response might broadcast in the neighborhood for the cytoplasmic continuity, via gap junctions, of the hit and bystander cell.
Such an effect has been demonstrated both in vitro and in vivo in several cell lines, both normal and tumor, with a large variety of experimental approaches.
The target that switches on the cell activation to initiate the “bystander cascade”, seems to be the nucleus, however the kind of response seems to be “all-or-none” type, and independent from the dose delivered into the cell, provided above the activation threshold (Schettino et al., 2005).
The consequences of such damage propagation, may result in the extension of genomic instability to large regions of tissue, even if the hit portion is relatively little (Mancuso et al., 2012).
Since in particular α-particle are known to be able to trigger on the bystander effect, this fact assumes some further importance for the Rn222 (α-particle source) exposure risk assessment.
1.21. Considerations on linearity failure
At present, it is impossible to attribute the occurrence of the reported non-lineal responses to one specific and unique cause, and even to determinate with certainty whether the indirect or direct (§ 1.4) radio-damage mechanism is responsible for the production of such effects. Nevertheless some considerations are possible.
The reactive species release via irradiation is supposed to be one important factor, and the theoretical perspective is twofold: on the one hand those might be the set of supposed soluble molecule responsible for the diffusion of the irradiation signal in the bystander effect (Kashino et al., 2007); on the other hand, the reactive species diffusion might stimulate the up-regulation of DNA surveillance and repair activities, resulting in the adaptive response (Matsumoto et al., 2007).To this regard must be kept in mind that the mitochondrial compartment is the main ROS source in the cell due to electron leak along the respiratory chain (§ 1.14), and that it surely plays a role concerning the non-nuclear irradiation. This target seems to be involved in the irradiation signal transmission and adaptive response, uprising via reactive species release (Tartier et al., 2007).