III Facolt`
a di Ingegneria
Corso di Laurea in Ingegneria delle Telecomunicazioni
Tesi di Laurea
Studio delle propriet`
a di sequenze
di DNA
Relatore:
prof. Roberto Garello
Candidato:
Diego Alberto
Dedico questa tesi a genitori, parenti e amici che mi sono sempre stati vicino e che mi hanno espresso il loro affetto, seppur in forme differenti.
Un pensiero particolare va a Gabriella, spero di essere per lei ci`o che lei `e per
me, e a Diana che mi ha sopportato per tutto questo tempo.
Desidero infine ringraziare Roberto Garello, amico e professore (cronologicamente
non nell’ordine), che mi ha aiutato in questo lavoro e che forse mi stima pi`u del
dovuto.
L’individuo diventa la realizzazione
di un programma dettato dall’eredit`a
Ringraziamenti I
1 Introduzione 1
1.1 Genomic Signal Processing . . . 1
1.2 Progetto Genoma . . . 3
1.3 Introduzione alla genetica . . . 10
2 Lineare Dipendenza 22 2.1 Introduzione . . . 22
2.2 Esempio di applicazione: Yeast FLO9(fungo dello lievito) . . . 24
2.3 Organismi esaminati . . . 29 2.3.1 Saccharomyces Cerevisiae . . . 34 2.3.2 Caenorhabditis Elegans . . . 40 2.3.3 Magnaporthe Grisea . . . 46 2.3.4 Arabidopsis Thaliana . . . 51 2.3.5 Homo Sapiens . . . 56
2.4 Conclusioni e tempo di elaborazione . . . 61
2.5 Listato . . . 65
3 Entropia 68 3.1 Introduzione . . . 68
3.2 Esempio di applicazione: Viola Clauseniana . . . 69
3.3 Organismi esaminati . . . 75 3.3.1 Saccharomyces Cerevisiae . . . 76 3.3.2 Caenorhabditis Elegans . . . 82 3.3.3 Magnaporthe Grisea . . . 87 3.3.4 Arabidopsis Thaliana . . . 92 3.3.5 Homo Sapiens . . . 98 3.4 Conclusioni . . . 104 3.5 Listato . . . 105
4.2 Esempio di applicazione: Viola Clauseniana . . . 109 4.3 Organismi esaminati . . . 118 4.3.1 Saccharomyces Cerevisiae . . . 119 4.3.2 Caenorhabditis Elegans . . . 125 4.3.3 Magnaporthe Grisea . . . 130 4.3.4 Arabidopsis Thaliana . . . 135 4.3.5 Homo Sapiens . . . 140 4.4 Conclusioni . . . 146 4.5 Listato . . . 147
5 Numero medio di Lempel-Ziv 154 5.1 Introduzione . . . 154
5.2 Esempio di applicazione: Viola Clauseniana . . . 156
5.3 Organismi esaminati . . . 166 5.3.1 Saccharomyces Cerevisiae . . . 167 5.3.2 Caenorhabditis Elegans . . . 172 5.3.3 Magnaporthe Grisea . . . 177 5.3.4 Arabidopsis Thaliana . . . 182 5.3.5 Homo Sapiens . . . 187 5.4 Conclusioni . . . 192 5.5 Listato . . . 193 6 Analisi Spettrale (PSD) 201 6.1 Introduzione . . . 201
6.2 Esempio di applicazione: Viola Clauseniana . . . 203
6.3 Organismi esaminati . . . 204 6.3.1 Saccharomyces Cerevisiae . . . 205 6.3.2 Caenorhabditis Elegans . . . 210 6.3.3 Magnaporthe Grisea . . . 215 6.3.4 Arabidopsis Thaliana . . . 220 6.3.5 Homo Sapiens . . . 225 6.4 Conclusioni . . . 230 6.5 Listato . . . 232
A Come scaricare da Internet le sequenze di DNA 234 B Bibliografia 243 Bibliografia . . . 243
Introduzione
1.1
Genomic Signal Processing
E’ risaputo che molte delle scoperte scientifiche e tecnologiche che avverranno nel corso del XXI secolo dipenderanno in larga scala dall’essere in grado di processare
e interpretare un’enorme quantit`a di dati.
In questi ultimi anni si `e resa necessaria una capacit`a di gestione e catalogazione
sempre maggiore che l’uomo non `e pi`u riuscito a garantire: di qui l’importanza
del data processing, una nuova frontiera, un nuovo approccio informatico che permette di trattare studi ed esperimenti che generano grandi moli di dati.
Nel campo della genetica, ad esempio, settore di ricerca molto vasto e inter-disciplinare che va dalla medicina alla biologia, dall’ingegneria all’agricoltura, sta nascendo un nuovo filone atto a gestire e sondare il libro della vita: il Genomic Signal Processing(GSP), il quale deriva dal Digital Signal Processing (DSP) . Si tratta di una disciplina ingegneristica che studia il DNA come un segnale qualsiasi: il segnale genomico.
Considerando il ruolo fondamentale, giocato nella genetica, dai segnali di
tras-crizione, `e naturale che la teoria del Signal Processing debba essere utilizzata per
ottenere una comprensione sia della parte strutturale che di quella funzionale del fenomeno in questione.
L’obiettivo del GSP `e quello di integrare la teoria e i metodi del SP con una
globale comprensione della genomica funzionale e con particolare enfasi sulle re-golazioni genomiche; in particolar modo abbraccia svariate tecniche di espressione come la scoperta, la predizione, la classificazione, il controllo e la modellizzazione statistica e dinamica delle reti di geni (gene networks).
costituisce un modello strutturale basato su analisi e sintesi che sta alla base del rigore matematico ingegneristico [B.2].
Esempi di tecniche mutuate dall’Ingegneria delle Telecomunicazioni sono :
• Digital Signal Processing (sequence spectral analysis, filtering, prediction, image processing);
• Information theory(entropy, source coding, channel coding); • Networks theory.
Nei capitoli che seguono ci occupiamo dell’analisi di sequenze di DNA mediante tec-niche di Teoria dell’Informazione come misurazioni di entropia, numeri medi calco-lati con gli algoritmi di Huffman e Lempel-Ziv applicati a sottosequenze individuate con il metodo della sliding window. Ricerchiamo sequenze ridondanti e periodiche ed infine eseguiamo un’analisi spettrale (Power Spectral Density) delle porzioni di
DNA considerate. L’obiettivo `e individuare le caratteristiche del segnale genetico,
come ad esempio la delimitazione tra aree codificanti (che servono per costruire le proteine) e non codificanti.
Nei due paragrafi che seguono vengono introdotti il Progetto Genoma ed i concetti di base della genetica per fare un po’ di luce in questo nuovo campo di applicazione dell’ingegneria.
1.2
Progetto Genoma
Tra le scoperte scientifiche pi`u importanti, quella della decodifica dell’intero
geno-ma ugeno-mano `e forse quella che ci tocca pi`u nel profondo. Tale scoperta `e avvenuta
nell’aprile del 2000 ad opera della compagnia privata Celera Genomics (con il Geno-ma Project); per la priGeno-ma volta eravamo in grado di leggere il libro delle istruzioni, il progetto che ci ’disegna’ come ci vediamo e sentiamo. E’ forse tutto qui ? Il
lavo-ro era dunque finito ? Assolutamente no, perch´e una cosa `e leggere le tre miliardi
di basi azotate (circa 800 Bibbie messe insieme) che costituiscono il nostro codice
genetico, un’altra, molto pi`u complessa, `e comprenderne il significato, le relazioni
e la suddivisione in sottosequenze che determinano la costruzione di geni, proteine, anatomia e fisiologia di ogni nostro organo e apparato. E’ come essere riusciti a
risolvere un gigantesco puzzle (fig 1.1)1 formato da decine di milioni di pezzi, ma
dobbiamo ancora oggi comprenderne l’interazione e il ruolo [B.3].
Figura 1.1. Il DNA `e come un puzzle
La tecnica di decodifica adottata dalla Celera Genomics `e nota come
shotgun-ning ovvero ’spara nel mucchio ’: prima si isola il DNA dal nucleo, poi usando onde sonore si riduce ogni cromosoma in minutissimi frammenti, 60 milioni di pezzetti,
ognuno contenente da 2000 a 10000 molecole base. Ogni frammento `e quindi inserito
in una macchina robotizzata che lo decifra ad altissima velocit`a e qui viene il difficile
: una volta decodificato il frammento, il robot invia i risultati per posta
elettroni-ca ai supercomputer Celera (i pi`u potenti al mondo dopo quelli del Pentagono fig
1.2)2 che riassemblano ogni singolo pezzetto nei 46 cromosomi umani. Sarebbe come tagliuzzare la Treccani in striscioline di carta e da queste ricomporre i 36 volumi dell’enciclopedia.
Figura 1.2. Supercomputers nei Celera Labs
Per decifrare le sequenze delle quattro lettere (basi chimiche) che compongono il nostro patrimonio genetico, viene usato un metodo chiamato cromatogramma (o
metodo dei quattro colori fig 1.3)3: quando i frammenti di DNA vengono colpiti da
un laser la radiazione luminosa emessa viene captata e trasformata in una sequenza di picchi colorati che poi sono interpretati come basi dal computer.
Per comprendere in pieno l’informazione contenuta nel nostro codice genetico, bisogna riunire tre tipi di dati :
1. la sequenza delle basi del DNA;
2mensile Newton 06-2000
2. i dati che indicano quali geni sono effettivamente funzionanti, quando, in quali cellule e in quali malattie;
3. i dati che riguardano tutte le proteine prodotte nella cellula, le loro relative abbondanze nei vari tipi di cellule e nelle diverse malattie.
Invece di analizzare singolarmente i geni, come `e avvenuto finora, bisogner`a
ca-pire come l’azione di uno di essi dipenda da quella degli altri. Ci viene in aiuto
una tecnica detta gene array (matrice di geni fig 1.4)4 che permette di avere un
quadro generale di tutti i geni funzionanti nella cellula. Questa analisi, insieme
all’identificazione delle propriet`a e delle funzioni delle centinaia di migliaia di
pro-teine prodotte in un organismo, permetter`a di rispondere a domande fondamentali:
quali cambiamenti biochimici avvengono in una cellula nervosa quando arriva un neurotrasmettitore o quali sono le mutue relazioni tra proteine del sistema ormo-nale, immunitario o nervoso. Oppure ancora, capire tutti i geni coinvolti in una malattia. Inoltre particolarmente importanti sono i segmenti prima e dopo ogni gene: contengono le istruzioni su quando, per quanto tempo e dove far funzionare il
gene. Quest’informazione spaziale e temporale `e difficile da catturare semplicemente
leggendo la sequenza del genoma, occorreranno anni di esperimenti.
Un’importante applicazione di questo studio del Genoma sar`a la possibilit`a di
ca-pire l’origine genetica delle pi`u diverse malattie. Saranno sviluppati test diagnostici
che informeranno il singolo individuo a quali malattie `e geneticamente predisposto.
Inoltre la decifrazione del DNA di microrganismi come i batteri potr`a rivoluzionare
la ricerca medica. I batteri infettivi si evolvono continuamente mettendo in atto sotterfugi per contrastare le difese del nostro organismo e l’azione degli antibiotici,
che infatti perdono efficacia nel tempo. Decifrare il genoma dei batteri sar`a come
scoprire i loro futuri piani di attacco, ci`o consentir`a di creare nuove categorie di
farmaci, nuovi tipi di cura, ma anche di stabilire una nuova definizione delle malat-tie, basate sui difetti genetici che ne sono alla base, piuttosto che sui sintomi come avviene ora.
Tuttavia nella realt`a in tantissimi casi i geni vengono attivati da sostanze
chi-miche che provengono dall’ambiente. E’ esperienza comune che situazioni di stress possano abbassare le difese immunitarie e favorire raffreddori o febbre; sostanze can-cerogene possono provocare tumori. Malattie come l’asma dipendono non solo dalla reciproca interazione di molti geni, ma anche da come ciascun gene interagisce con l’ambiente. Piccoli eventi casuali che accadono durante lo sviluppo dell’organismo
danno luogo a notevoli variazioni nell’adulto. Perci`o in ultima analisi dovremmo
ac-cettare con gioia l’idea di non essere semplicemente macchine il cui funzionamento `e
precostituito dai geni poich`e non solo `e necessario ereditare un gene difettoso, ma `e
anche necessario che questo gene funzioni in un ambiente particolare perch´e l’intero
Figura 1.3. Cromatografia del batterio Neisseria Meningitidis (2 milioni di basi)
organismo contragga la malattia cui il gene era predisposto (fig 1.5)5. Dobbiamo
quindi essere cauti nel sostenere che alcune persone sono destinate ad ammalarsi
di una certa malattia. La genetica sta entrando sempre pi`u nella nostra vita
quo-tidiana, un po’ come vi sono entrati i computer, ed `e quindi possibile che si abusi
dell’informazione genetica per discriminazioni sociali, soprattutto nelle assunzioni e
nelle polizze assicurative. Al contrario, il progresso della ricerca mette sempre pi`u
Figura 1.4. Tecnica del gene array
in evidenza che ’ nessuno `e perfetto ’. Invece l’informazione contenuta nel DNA
rap-presenta una fantastica sorgente di informazioni sul passato dell’uomo: differenze nelle sequenze del genoma ci permettono di ricostruire le migrazioni delle popola-zioni umane. Per esempio, recenti analisi genetiche confermano la tradizione orale secondo la quale i Lemba, una popolazione che vive nell’Africa del Sud e che parla bantu, siano discendenti degli Ebrei che migrarono dal Medio Oriente 2700 anni fa.
Il timore generale, dopo la decifrazione del DNA da parte di una societ`a privata,
fu il seguente: in mano a chi finir`a lo schema biochimico dell’uomo? Bisogner`a
pa-gare per conoscerlo? E se verr`a brevettato, sar`a in un certo senso come brevettare la
vita? La Celera rese pubblica, gratuita e disponibile tutta la sequenza del genoma
umano, strada che abbandon`o quasi subito per intraprendere quella pi`u redditizia
dello studio dei microrganismi.
Brevettare i geni `e un bene o un male per il progresso medico ?
Non solo la Celera ma anche altre compagnie come Millenium Phermaceutical,
Figura 1.5. Mappa genetica delle malattie
di brevetto di sequenze di DNA. La loro intenzione `e di ottenere cospicui diritti da
ogni possibile farmaco o terapia medica che usi sequenze geniche che esse hanno sequenziato. Molti ricercatori sostengono per che tali brevetti impediranno il libero scambio di informazioni e faranno aumentare il costo dei farmaci. Senza dubbio
assisteremo a uno scontro, su cui `e ora difficile fare previsioni, tra l’economia di
mercato e i difensori dei diritti dell’uomo, quali soprattutto gli organismi pubblici internazionali. E’ questo il vero problema alla base dell’ingegneria genetica. Non certo il timore molto diffuso nell’opinione pubblica, ma molto meno reale, che i la-boratori diventino fabbriche per produrre mostri.
Le banche dati che contengono le sequenze di DNA umano e di altri orga-nismi viventi sono oggigiorno in massima parte accessibili gratuitamente su
In-ternet. Il sito principale al quale ci si riferir`a nei successivi capitoli `e
dell’NC-BI (National Center for Biotechnology Information)(fig 1.6) accessibile all’indirizzo
http://www.ncbi.nlm.nih.gov/, gestito dall’Istituto di Sanit`a degli Stati Uniti6.
Altri siti ai quali ci si pu`o riferire sono:
• www.tigr.org, The Institute for Genomic Research
• www.sanger.ac.uk, il Sanger Center `e il principale centro europeo dello studio
del genoma umano • www.ebi.ac.uk
• www.nhgri.nih.gov/ELSI, banca dati sugli aspetti etici, legali e le implicazioni sociali del Progetto Genoma
Figura 1.6. Map Viewer dell’NCBI
1.3
Introduzione alla genetica
La genetica `e quella parte della biologia che studia le modalit`a di trasmissione ed
espressione del genoma, ovvero dell’insieme dei geni contenuti in una cellula. Un
gene `e la porzione di Acido desossiribonucleico (DNA) contenente
l’informa-zione necessaria alla sintesi di una proteina. Le cellule possono essere procariote
(fig 1.8)7, nelle quali il patrimonio genetico `e disperso nel citoplasma ( `e la
strut-tura cellulare tipica degli organismi primordiali come i batteri), oppure eucariote
(fig 1.7)8, nelle quali il materiale genetico `e racchiuso all’interno di una struttura
chiamata nucleo ( `e tipica degli organismi complessi, uomo compreso).
Figura 1.7. Cellula Eucariota
7Le scienze biologiche H.Curtis, N.Barnes Zanichelli
Figura 1.8. Cellula Procariota
Nelle cellule eucariote esistono due tipi di acido nucleico. Il DNA `e contenuto
all’interno del nucleo ed `e costituito da due filamenti appaiati e spiralizzati secondo
la nota struttura a doppia elica identificata nel 1953 da J.Watson e F.Crick [B.4](fig 1.9, 1.13).
Ciascun filamento di DNA `e un polimero di nucleotidi (fig 1.10, 1.12), formati da uno zucchero pentavalente, o pentosio, denominato desossiribosio (fig 1.11), da
un gruppo fosforico e da una base azotata che pu`o essere Adenina (A), Citosina
(C),Guanina (G) o Timina (T) (fig 1.12)9.
Figura 1.10. Generico nucleotide
Figura 1.11. Desossiribosio
Figura 1.12. Basi azotate DNA
Ciascun nucleotide `e legato al successivo attraverso un legame fosfodiesterico ed
i due filamenti sono mantenuti appaiati da legami idrogeno che si stabiliscono fra ciascuna coppia di basi.
Per ragioni di natura chimica A si lega sempre a T e C sempre a G.
Figura 1.13. Doppia elica del DNA
L’Acido Ribonucleico (RNA) `e un polimero di nucleotidi, disposti su
fila-mento singolo, nei quali il pentosio `e il ribosio (fig 1.14) e la base azotata Uracile
(U) prende il posto di T (fig 1.15)10.
Figura 1.14. Ribosio
Figura 1.15. Basi azotate RNA
L’RNA `e presente sotto varie forme all’interno della cellula e ciascuna di esse
Come precedentemente accennato un gene `e la porzione di DNA contenente l’informazione necessaria alla sintesi di una proteina. Nel DNA umano sono stati identificati 30.000 geni in grado di codificare circa 100.000 proteine. Ma cosa
so-no queste proteine e perch´e sono cos`ı importanti? Le proteine sono anch’esse dei
polimeri le cui unit`a sono chiamate amminoacidi. In natura sono noti 20
am-minoacidi (fig 1.16)11, composti ciascuno da un gruppo amminico (basico) da un
gruppo carbossilico (acido) e da un radicale R che li caratterizza e li distingue.
Figura 1.16. I 20 amminoacidi
Ciascun amminoacido si unisce al seguente tramite un legame peptidico (fig 1.17)12.
Figura 1.17. Il legame peptidico
Le proteine si suddividono in strutturali ed enzimatiche. Le proteine strut-turali, come dice il nome, sono i mattoni alla base della costruzione di ciascuna struttura cellulare e, pertanto, dell’intero organismo; le proteine enzimatiche, o en-zimi, si occupano invece di regolare le reazioni chimiche cellulari e di controllare la
produzione delle altre molecole organiche. Ogni proteina `e caratterizzata da una
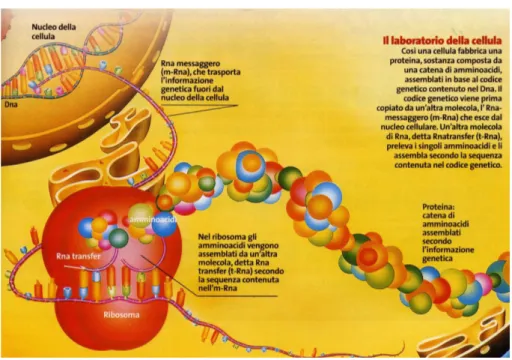
sequenza e da un numero preciso di amminoacidi che vengono determinati dall’es-pressione del messaggio genetico contenuto nel nucleo. Rimane da chiarire in che modo l’informazione genetica contenuta nel nucleo venga trasmessa nel citoplasma
e tradotta in polipeptidi. Il DNA `e una struttura troppo preziosa e delicata perch´e
venga lasciata fuoriuscire nel citoplasma, sede della sintesi proteica. Pertanto ne viene realizzata una copia da utilizzare come messaggero. La doppia elica del DNA viene despiralizzata ed i due filamenti separati. In corrispondenza del gene inter-essato viene costruita una nuova catena polinucleotidica complementare. La nuova catena formata da molecole di RNA ed chiamata RNA-messaggero trascritto
primario (fig 1.18)13.
12Biologia molecolare della cellula, J.Darnell Zanichelli
Figura 1.18. Trascrizione DNA in m-RNA
Tale struttura, rappresentando la copia fedele della catena di DNA, contiene un
eccesso di nucleotidi. Infatti ciascun gene `e formato da sequenze di nucleotidi dette
esoni, le quali contengono il messaggio di codifica per la proteina, e da sequenze denominate introni, le quali appaiono prive di informazione. Attraverso un processo
di maturazione chiamato splicing (fig 1.19)14l’RNA-messaggero trascritto primario
viene privato degli introni e le sequenze esoniche vengono unite a formare l’RNA-messaggero (m-RNA) vero e proprio.
Figura 1.19. Splicing
L’m-RNA fuoriesce nel citoplasma attraverso uno dei pori della membrana
nu-cleare e va ai ribosomi. Tali organelli citoplasmatici sono composti da due subunit`a
di RNA-ribosomiale (r-RNA) e sono la sede della sintesi proteica. L’m-RNA
si infila fra le due subunit`a del ribosoma, dove viene letto a sequenze di 3
ammi-noacidi. Ogni sequenza, chiamata codone (fig 1.20)15, corrisponde ad un preciso
amminoacido.
Figura 1.20. Codoni e rispettivi amminoacidi
Esistono un codone di avvio, rappresentato dalla sequenza di basi AUG, che innesca il processo di traduzione delle triplette in amminoacidi, e tre codoni di arresto ( o codoni non-senso), identificati nelle sequenze UAA, UGA,UAG, che indicano la fine della traduzione. Ogni volta che il ribosoma legge una tripletta compresa fra il codone di avvio ed il codone di arresto della traduzione, dal citoplasma giunge una molecola di RNA-transfert (t-RNA), che presenta la tripletta complementare o
anticodone. Alla molecola di t-RNA `e a sua volta legato un preciso amminoacido.
In questo modo, attraverso l’appaiamento fra codoni ed anticodoni, viene a formarsi, sulla superficie del ribosoma, una catena di amminoacidi. Questi ultimi interagiscono fra di loro formando legami peptidici e venendo a costituire la proteina di cui la
cellula aveva richiesto la sintesi (fig 1.2116, 1.22, 1.2317).
16Le scienze biologiche H.Curtis, N.Barnes Zanichelli
Figura 1.21. Costruzione di proteine
Lineare Dipendenza
2.1
Introduzione
L’analisi che ci si propone di fare in questa sezione prevede la ricerca della lineare dipendenza in matrici costruite con basi di DNA. Considerando una qualsiasi se-quenza genetica di lunghezza L, possiamo dividerla in frames di lunghezza N ed ogni N frames costruire una matrice NxN come in figura 2.1.
ctaggataga atggggtata attggacgcg cagacgtgat aaagctggtg atttgtcctg aagaagatga caaactggat gagatggcag tagttggagt
N=10
Frame 1
Frame 2
N
N
ctaggatagaatggggtaataattg gacgcgcagacgtgataaagctggt gatttgtcctgaagaaga……… ………ccgttggttccagtgatggtgg tcatctcagtagatgtagaggtgaa agtaccggtccatggctcggttgtaFigura 2.1. Suddivisione in frames
Si parte quindi dal primo frame per costruire la prima riga, con il secondo cos-truiamo la seconda e cos`ı via fino all’N-esimo. A questo punto non possiamo ancora
calcolare il rango perch´e la matrice quadrata `e composta da sole lettere (le basi
azotate), `e quindi necessario stabilire un labeling: il pi`u ovvio il seguente
a ==> 0
t ==> 1
c ==> 2
g ==> 3
Figura 2.2. labeling
Inoltre essendo l’alfabeto composto da soli quattro simboli passiamo ai campi finiti di Galois (GF4), otteniamo quindi una matrice di cui possiamo calcolare il
rango: se questo `e minore di N significa che esiste una qualche forma di correlazione,
la cui possibile spiegazione non `e di nostra competenza ma sar`a fornita da studi
biologici futuri. Per costruire la seconda matrice elimino la prima riga (ovvero il primo frame), sposto tutte le righe in su di una ed aggiungo come ultima riga l’ N+1esimo frame, abbiamo un’altra matrice NxN di cui possiamo calcolare il rango. Iterando cos`ı il procedimento analizziamo tutta la stringa e avremo L/N − N + 1 matrici NxN, dal numero di matrici con rango non pieno ricaviamo la percentuale di
matrici linearmente dipendenti con frame di lunghezza N. Ora `e sufficiente far variare
N: il valore minimo scelto `e 3 poich`e rappresenta la lunghezza di un amminoacido
che `e la pi`u semplice organizzazione di basi azotate; il valore massimo `e √L poich`e
per tale valore avremo una sola matrice NxN (= √L x √L ) che comprende tutta
la sequenza di basi azotate; superando tale valore avremmo matrici rettangolari la
cui dimensione massima sarebbe N ed il rango essendo alpi`u uguale alla dimensione
minima sarebbe necessariamente minore di N. Quando il rango di queste matrici
non `e massimo, `e generalmente inferiore a N di una o due unit`a, raramente `e ancora
minore; per questo motivo distingueremo solamente matrici a rango pieno da matrici a rango non pieno.
Questa `e la logica alla base dell’algoritmo implementato per svolgere questa
analisi, esso `e presentato alla fine di questo capitolo (§2.5). L’ambiente di lavoro `e
Matlab nella versione 6.5, preferibile al C in quanto pi`u flessibile alla trasformazione
2.2
Esempio di applicazione: Yeast FLO9(fungo
dello lievito)
Consideriamo come esempio una porzione di 1425 basi del DNA del fungo dello Lievito (Yeast FLO9):
c a g t t t c a t c a g t t g g t t g a c c g t t g g t t c c a g t g a t g g t g g t c a t c t c a g t a g a t g t a g a g g t g a a a g t a c c g g t c c a t g g c t c g g t t g t a g t t g t a a c c a a a c c t t c a c t g g t t g g a g t t c t g a t a a c a a t c a c g g t t t c g t c a g t t g g t t g a c c g t t a g t a c c g g t g a c g g t g g t c a t c t c a g t g g a t g t a g a g g t g a a a g t a c c a g t c c a t g g t t c a g t g g t g g t g c t g a t t a g a c c t t c a c t a g t t g g a g t t c t g a t g a c a a t g a c g g t t t c g t c a g t t g g a a c g c c g t t g g t a c c g g t g a c g g t g g t c a t t t c a g t g g a t g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t c t c a t c a g t t g g c a a a c c g t t g g t a c c g g t g a c g g t g g t g a t t t c a g t g g a t g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t t t c a t c a g t t g g c a a a c c g t t g g t a c c g g t g a c g g t g g t c a t t t c a g t g g a t g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t t a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t t t c a t c a g t t g g c a a a c c g t t g g t a c c g g t g a c g g t g g t c a t t t c a g t g g a t g t a g a g g t a a a a g t g c t g t t c c a t g g c t c a g t t g t a g t t a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t t t c a t c a g t t g g c a a a c c g t t g g t a c c g g t g a c g g t g g t c a t t t c a g t g g a t g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t c t c a t c a g t t g g c a a a c c a t t g g t a c c g g t g a c g g t g g t g a t t t c a g t g g a t g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t c t c a t c a g t t g g c a a a c c a t t g g t a c c g g t g a c t g t g g t c a a t t c g g t a g a a g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t c t c a t c a g t t g g c a a a c c a t t g g t a c c g g t g a c t g t g g t c a a t t c g g t a g a a g t a g a g g t a a a a g t g t c g t t c c a t g g c t g a g t t g t a g t c a t g g c a g t a g t g g c t g t t g t t g g t g t t c t g a t g a c a a t g a t g g t c t c a t c a g t t g g c a a a c c a t t g g t a c c g g t g a c t g t g g t c a a t t c g g t a g a a g t a g a g g t a a a a g t g c t g t;
la sequenza `e pubblica ed `e stata scaricata dal sito dell’NCBI: http://www.ncbi.nlm.nih.gov/
N n° TOT matr n° matr L.D. % 3 473 93 19,66 4 353 107 30,31 5 281 68 24,20 6 232 62 26,72 7 197 56 28,43 8 171 60 35,09 9 150 44 29,33 10 133 34 25,56 11 119 39 32,77 12 107 39 36,45 13 97 30 30,93 14 88 23 26,14 15 81 80 98,77 16 74 20 27,03 17 67 22 32,84 18 62 53 85,48 19 57 18 31,58 20 52 19 36,54 21 47 21 44,68 22 43 15 34,88 23 39 13 33,33 24 36 9 25,00 25 33 15 45,45 26 29 6 20,69 27 26 26 100,00 28 23 6 26,09 29 21 9 42,86 30 18 18 100,00 31 15 4 26,67 32 13 3 23,08 33 11 5 45,45 34 8 5 62,50 35 6 4 66,67 36 4 4 100,00 37 2 0 0,00 Figura 2.3. esempio
N varia tra 3 e √1425 = 37.7 quindi 37
e rappresentando tutto in un grafico:
Si possono osservare diversi picchi (almeno 5) in cui la percentuale di matrici
con righe o colonne linearmente dipendenti `e molto alta; tuttavia man mano che N
Figura 2.4. grafico esempio
un determinato periodo N di basi azotate. Sono quindi pi`u significativi quei picchi
che a parit`a di ordinata (% matr. L.D.) presentano un pi`u piccolo valore di ascissa
N perch´e sono in numero maggiore.
In questo caso N = 15 `e un buon candidato alla dimensione di un frame
periodi-co per ricercare un significato biologiperiodi-co a tale ridondanza e periodicit`a matematiche.
In questo capitolo, dedicato allo studio della Lineare Dipendenza, non cercheremo
di evidenziare le transizioni da regioni codificanti a non codificanti perch´e l’obiettivo
`e trovare sottosequenze (frames) ricorrenti.
Inoltre, per ottenere risultati in un tempo ragionevole (qualche ora di elabora-zione), esamineremo solo le aree geniche di ogni macro-sequenza scaricata da
differenza nell’applicare questo studio ad aree geniche, non geniche o regioni che le comprendono entrambe fino al valore di N minimo tra i 3 casi considerati.
0 10 20 30 40 50 60 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
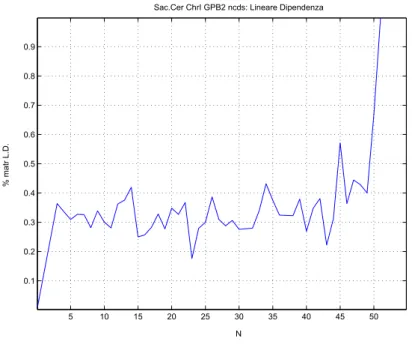
Sac.Cer ChrI GPB2 cds: Lineare Dipendenza
5 10 15 20 25 30 35 40 45 50 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 N % matr L.D.
Sac.Cer ChrI GPB2 ncds: Lineare Dipendenza
Figura 2.6. Area non codificante
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Sac.Cer ChrI GPB2 tot: Lineare Dipendenza
2.3
Organismi esaminati
Il fungo Saccharomyces Cerevisiae1 `e un importante modello per comprendere
i processi cellulari e molecolari in organismi eucarioti, lo studio del suo genoma `e
stato completato nel 1996 (Goffeau A. et al. ”Life with 6000 genes”, Science. 1996 Oct 25;274(5287):546, 563-7).
Pagina 1 di 1
02/08/2004 http://www.microbeworld.org/img/aboutmicro/gallery/gid_06_sacc.jpg
Figura 2.8. Saccharomyces Cerevisiae
Il Sanger Center e il Genome Sequencing Center alla Washington University
han-no completato lo studio della sequenza del gehan-noma del Caehan-norhabditis Elegans2
(un invertebrato) che consta di 100,274 Mb (milioni di basi azotate) organizzate
in 6 cromosomi ed in 19542 coding sequences. Questo organismo `e stato il primo
eucariota pluricellulare il cui genoma `e stato decodificato (Science, 1998).
Pagina 1 di 1
Figura 2.9. Caenorhabditis Elegans
1fotografia www.microbeworld.org
2fotografia www.nematode.net
Il Whitehead Institute/MIT Center for Genome Research (WICGR) e il Fungal Genomics Laboratory del North Carolina State University hanno collaborato per
sequenzializzare il genoma del Magnaporthe Grisea3 composto da circa 40 Mb
organizzate in 7 cromosomi. Il metodo utilizzato `e stato il ’whole genome shotgun’
(WGS). Questo organismo `e l’agente responsabile del danneggiamento delle piante
di riso: si calcola che ogni anno ne rovini una quantit`a tale da poter sfamare 60
milioni di persone. Oltre al riso, alcune varianti di Magnaporthe Grisea possono attaccare anche l’orzo, il grano e il miglio.
Pagina 1 di 1
Figura 2.10. Magnaporthe Grisea
3fotografia www.eolproject.org
L’ Arabidopsis Thaliana4`e una piccola pianta largamente usata come modello di organismo in botanica, fa parte della famiglia delle Brassicaceae (che include
anche il cavolo e il ravanello). Il suo genoma `e stato sequenzializzato mediante una
collaborazione internazionale chiamata Arabidopsis Genome Initiative (AGI) (The Arabidopsis Genome Initiative, 2000, Nature, 408:796-815) tra il TIGR, il MIPS e
il TAIR. Pagina 1 di 1
31/07/2004 http://www.erboristeriadulcamara.com/galleria_immagini/images/large/cardamineG.jpg
Figura 2.11. Arabidopsis Thaliana
Il completo genoma dell’ Homo Sapiens5, costituito da 3 miliardi di basi e
organizzato in 46 cromosomi, `e stato decodificato nell’aprile del 2000 ad opera della
Celera Genomics vedi capitolo §1.2 pag 3.
Pagina 1 di 1
Figura 2.12. Homo Sapiens
Per ognuno dei 5 organismi elencati considereremo 10 sequenze casuali del loro rispettivo genoma e le analizzeremo con i 5 algoritmi descritti nei capitoli §2, §3, §4, §5, §6.
4fotografia www.erboristeriadulcamara.com
5fotografia www.staticfriends.com
regno Chr Kbp gene contig start stop dim prec succes sovra sovra dim interv stop start start stop sovra NCDS CDS NCDS fungo I 230 GPB2 NC_001133 39260 41902 2642 24001 45900 36618 44544 7926 1 % 2642 5284 % 7926 III 316 BUD3 NC_001135 96280 101190 4910 90414 136870 91370 106100 14730 1 % 4910 9820 % 14730 IV 1531 DNF2 NC_001136 631276 636114 4838 520687 651120 626438 640952 14514 1 % 4838 9676 % 14514 V 576 ZRG8 NC_001137 218056 221286 3230 182277 347608 214826 224516 9690 1 % 3230 6460 % 9690 VI 270 RPO41 NC_001138 58781 62836 4055 42565 74425 54726 66891 12165 1 % 4055 8110 % 12165 VII 1090 SNT2 NC_001139 261651 265862 4211 225576 536059 257440 270073 12633 1 % 4211 8422 % 12633 VIII 562 SET1 NC_001140 346045 349287 3242 319816 402967 342803 352529 9726 1 % 3242 6484 % 9726 IX 439 NEO1 NC_001141 261436 264891 3455 154843 292632 257981 268346 10365 1 % 3455 6910 % 10365 X 745 BCK1 NC_001142 247171 251607 4436 217226 361165 242735 256043 13308 1 % 4436 8872 % 13308 XII 1078 FKS1 NC_001144 809997 815627 5630 776304 941480 804367 821257 16890 1 % 5630 11260 % 16890 00 0 invert I 15000 VAB10 NC_003279 11755812 11797512 41700 11565842 12886543 11714112 11839212 125100 1 % 41700 83400 % 125100 I 15000 UNC-89 NC_003279 4035894 4090428 54534 4031740 4321096 3981360 4144962 163602 1 % 54534 109068 % 163602 II 15000 2I41 NC_003280 7724824 7752244 27420 7649873 8488012 7697404 7779664 82260 1 % 27420 54840 % 82260 II 15000 PTP-3 NC_003280 10975442 11012156 36714 10787172 11050165 10938728 11048870 110142 1 % 36714 73428 % 110142 III 13000 DIG-1 NC_003281 6746243 6794697 48454 4643245 8279021 6697789 6843151 145362 1 % 48454 96908 % 145362 III 13000 TRA-1 NC_003281 11170715 11195028 24313 10677352 12031531 11146402 11219341 72939 1 % 24313 48626 % 72939 IV 17000 LIN-1 NC_003282 2275866 2300355 24489 2032997 2497821 2251377 2324844 73467 1 % 24489 48978 % 73467 IV 17000 ITR-1 NC_003282 7675163 7696913 21750 6004854 10324237 7653413 7718663 65250 1 % 21750 43500 % 65250 V 20000 5L325 NC_003283 10827618 10849015 21397 8441005 13155519 10806221 10849015 42794 1 % 21397 42794 % 42794 V 20000 SOS-1 NC_003283 4518409 4549084 30675 2799640 6467665 4487734 4579759 92025 1 % 30675 61350 % 92025 00 0 fungo I 9000 MG01468 NT_086089 1208172 1215878 7706 577447 1489503 1200466 1223584 23118 1 % 7706 15412 % 23118 II 7000 MG06822 NT_086168 699403 707075 7672 528088 altro contig 691731 714747 23016 1 % 7672 15344 % 23016 II 7000 MG05589 NT_086040 989482 997931 8449 195182 1192293 981033 1006380 25347 1 % 8449 16898 % 25347 III 9000 MG05998 NT_086146 754868 761307 6439 307179 1396529 748429 767746 19317 1 % 6439 12878 % 19317 IV 6000 MG04775 NT_086080 1597773 1604751 6978 1365858 1613884 1590795 1611729 20934 1 % 6978 13956 % 20934 IV 6000 MG04118 NT_086069 1331610 1339458 7848 1236560 2355457 1323762 1347306 23544 1 % 7848 15696 % 23544 V 2000 MG09784 NT_086096 350020 354811 4791 215524 1123981 345229 359602 14373 1 % 4791 9582 % 14373 V 2000 MG10072 NT_086101 1087176 1092647 5471 210264 1143664 1081705 1098118 16413 1 % 5471 10942 % 16413 VI 4000 MG00736 NT_086018 1556659 1562418 5759 1302410 2103129 1550900 1568177 17277 1 % 5759 11518 % 17277 VII 5000 MG02729 NT_086111 790356 796765 6409 744074 841263 783947 803174 19227 1 % 6409 12818 % 19227
regno Chr Kbp gene contig start stop dim prec succes sovra sovra dim interv stop start start stop sovra NCDS CDS NCDS pianta I 30000 AT1G67120 NC_003070 25073230 25099189 25959 24085571 25104578 25047271 25125148 77877 1 % 25959 51918 % 77877 I 30000 AT1G72270 NC_003070 27203395 27214784 11389 26504944 29815034 27192006 27226173 34167 1 % 11389 22778 % 34167 II 19000 AT2G25660 NC_003071 10923281 10934468 11187 7809312 10963380 10912094 10945655 33561 1 % 11187 22374 % 33561 II 19000 AT2G41220 NC_003071 17185012 17195829 10817 15476446 17390149 17174195 17206646 32451 1 % 10817 21634 % 32451 III 23000 AT3G10380 NC_003074 3219528 3228863 9335 3160186 3482155 3210193 3238198 28005 1 % 9335 18670 % 28005 III 23000 AT3G54280 NC_003074 20103339 20115133 11794 18711514 21854384 20091545 20126927 35382 1 % 11794 23588 % 35382 IV 18000 AT4G03300 NC_003075 1447372 1456750 9378 1174321 7044395 1437994 1466128 28134 1 % 9378 18756 % 28134 IV 18000 AT4G22970 NC_003075 12033448 12043583 10135 11623531 13558472 12023313 12053718 30405 1 % 10135 20270 % 30405 V 26000 AT5G15540 NC_003076 5047676 5057414 9738 4121430 5101191 5037938 5067152 29214 1 % 9738 19476 % 29214 V 26000 AT5G40480 NC_003076 16230589 16241210 10621 16212756 16359973 16219968 16251831 31863 1 % 10621 21242 % 31863 00 0 regno Chr Mbp gene contig GI dim interv mam I 246 FAF1 NC_000001 19528653 2610 III 199 GRM7 NC_000003 32528267 4113 V 181 FER NC_000005 4885230 2950 IX 136 RFX3 NC_000009 19743883 3018 X 135 ADK NC_000010 32484974 2018 XII 132 SOX5 NC_000012 30061560 4563 XIII 113 MYR8 NC_000013 42660130 7098 XV 100 NEO1 NC_000015 4505374 5297 XVII 81 NF1 NC_000017 4557792 8959 XX 63 ATRN NC_000020 21450860 8645 0 00 0 0 00 0 0 00 0 0 00 0 0 00 0
2.3.1
Saccharomyces Cerevisiae
0 10 20 30 40 50 60 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.Sac.Cer ChrI GPB2 cds: Lineare Dipendenza
Figura 2.13. Saccharomyces Cerevisiae chr I
Qui N=40 individua un insieme di matrici 40x40 in cui oltre il 50% `e linearmente
dipendente. Il picco che arriva all’80% non `e molto significativo a causa del limitato
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Sac.Cer ChrIII BUD3 cds: Lineare Dipendenza
Figura 2.14. Saccharomyces Cerevisiae chr III
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
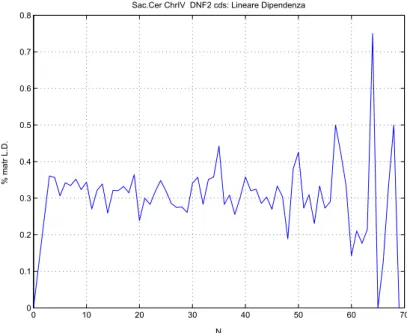
Sac.Cer ChrIV DNF2 cds: Lineare Dipendenza
0 10 20 30 40 50 60 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 N % matr L.D.
Sac.Cer ChrV ZRG8 cds: Lineare Dipendenza
Figura 2.16. Saccharomyces Cerevisiae chr V
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
Sac.Cer ChrVI RPO41 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
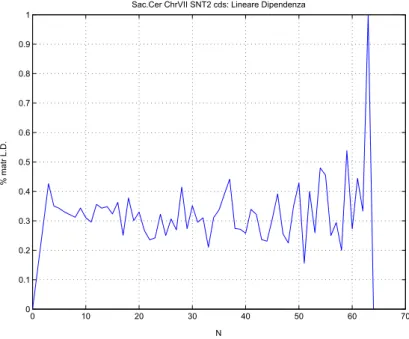
Sac.Cer ChrVII SNT2 cds: Lineare Dipendenza
Figura 2.18. Saccharomyces Cerevisiae chr VII
0 10 20 30 40 50 60 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 N % matr L.D.
Sac.Cer ChrVIII SET1 cds: Lineare Dipendenza
0 10 20 30 40 50 60 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 N % matr L.D.
Sac.Cer ChrIX NEO1 cds: Lineare Dipendenza
Figura 2.20. Saccharomyces Cerevisiae chr IX
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Sac.Cer ChrX BCK1 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D. Lineare Dipendenza Sacch.Cer chrXII FKS1: cds
2.3.2
Caenorhabditis Elegans
A causa dell’elevato tempo di elaborazione necessario per esaminare le sequenze codificanti di questo organismo (25-30 Kbp oltre le 15 ore ognuna), abbiamo dovuto operare una partizione e lavorare su sottosequenze di dimensione non superiore alle
9-10 Kbp. Inoltre se si evidenziasse una determinata periodicit`a elevata sulla sequenza
da 30 Kbp, non la perderemmo di certo analizzandone una porzione ridotta ad un
terzo poich`e sarebbe al limite riscalata.
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 N % matr L.D.
Caenorhabditis Elegans ChrI VAB10 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Caenorhabditis Elegans ChrI UNC-89 cds: Lineare Dipendenza
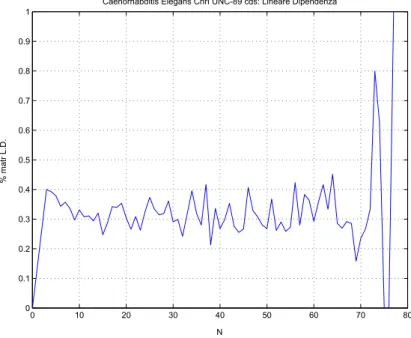
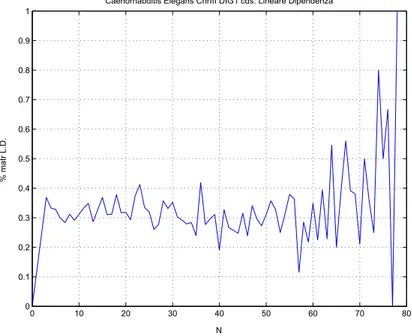
Figura 2.24. Caenorhabditis Elegans chr I
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
Caenorhabditis Elegans ChrII 2I41 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Caenorhabditis Elegans ChrII PTP3 cds: Lineare Dipendenza
Figura 2.26. Caenorhabditis Elegans chr II
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Caenorhabditis Elegans ChrIII DIG1 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Caenorhabditis Elegans ChrIII TRA1 cds: Lineare Dipendenza
Figura 2.28. Caenorhabditis Elegans chr III
Qui N=48 individua un insieme di matrici 48x48 in cui il 70% `e linearmente
dipendente. Tale valore `e il pi`u grande che abbiamo rilevato per il Caenorhabditis
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
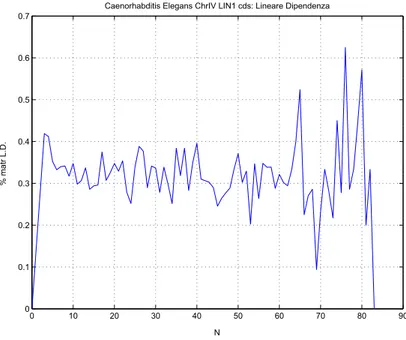
Caenorhabditis Elegans ChrIV LIN1 cds: Lineare Dipendenza
Figura 2.29. Caenorhabditis Elegans chr IV
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Caenorhabditis Elegans ChrIV ITR1 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Caenorhabditis Elegans ChrV 5L325 cds: Lineare Dipendenza
Figura 2.31. Caenorhabditis Elegans chr V
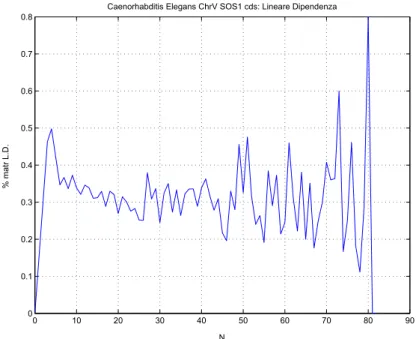
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
Caenorhabditis Elegans ChrV SOS1 cds: Lineare Dipendenza
2.3.3
Magnaporthe Grisea
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.Magnaporthe Grisea ChrI MG01468 cds: Lineare Dipendenza
Figura 2.33. Magnaporthe Grisea chr I
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
Magnaporthe Grisea ChrII MG06822 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Magnaporthe Grisea ChrII MG05589 cds: Lineare Dipendenza
Figura 2.35. Magnaporthe Grisea chr II
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
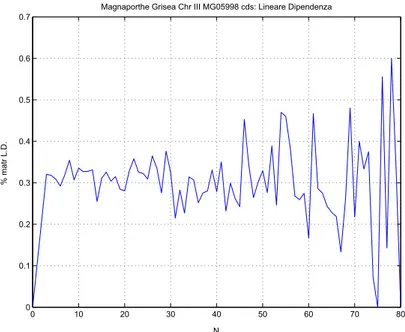
Magnaporthe Grisea Chr III MG05998 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
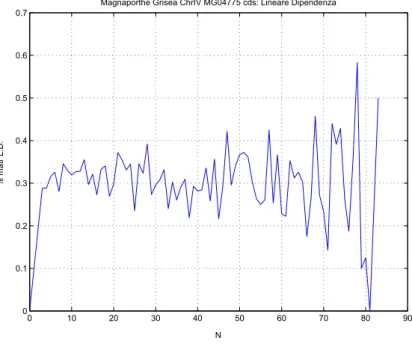
Magnaporthe Grisea ChrIV MG04775 cds: Lineare Dipendenza
Figura 2.37. Magnaporthe Grisea chr IV
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Magnaporthe Grisea ChrIV MG0411 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Magnaporthe Grisea ChrV MG09784 cds: Lineare Dipendenza
Figura 2.39. Magnaporthe Grisea chr V
0 10 20 30 40 50 60 70 80 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 N % matr L.D.
Magnaporthe Grisea ChrV MG10072 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Magnaporthe Grisea ChrVI MG00736 cds: Lineare Dipendenza
Figura 2.41. Magnaporthe Grisea chr VI
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Magnaporthe Grisea ChrVII MG02729 cds: Lineare Dipendenza
2.3.4
Arabidopsis Thaliana
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.Arabidopsis Thaliana ChrI AT1G67120 cds: Lineare Dipendenza
Figura 2.43. Arabidopsis Thaliana chr I
0 20 40 60 80 100 120 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 N % matr L.D.
Arabidopsis Thaliana ChrI AT1G72270 cds: Lineare Dipendenza
0 20 40 60 80 100 120 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Arabidopsis Thaliana ChrII AT2G25660 cds: Lineare Dipendenza
Figura 2.45. Arabidopsis Thaliana chr II
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Arabidopsis Thaliana ChrII AT2G41220 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Arabidopsis Thaliana ChrIII AT3G10380 cds: Lineare Dipendenza
Figura 2.47. Arabidopsis Thaliana chr III
0 20 40 60 80 100 120 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 N % matr L.D.
Arabidopsis Thaliana ChrIII AT3G54280 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 100 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 N % matr L.D.
Arabidopsis Thaliana ChrIV AT4G03300 cds: Lineare Dipendenza
Figura 2.49. Arabidopsis Thaliana chr IV
0 10 20 30 40 50 60 70 80 90 100 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 N % matr L.D.
Arabidopsis Thaliana ChrIV AT4G22970 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Arabidopsis Thaliana ChrV AT5G15540 cds: Lineare Dipendenza
Figura 2.51. Arabidopsis Thaliana chr V
0 20 40 60 80 100 120 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Arabidopsis Thaliana ChrV AT5G40480 cds: Lineare Dipendenza
2.3.5
Homo Sapiens
0 10 20 30 40 50 60 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.Homo Sapiens ChrI FAF1 cds: Lineare Dipendenza
Figura 2.53. Homo Sapiens chr I
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Homo Sapiens ChrIII GRM7 cds: Lineare Dipendenza
0 10 20 30 40 50 60 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Homo Sapiens ChrV FER cds: Lineare Dipendenza
Figura 2.55. Homo Sapiens chr V
0 10 20 30 40 50 60 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 N % matr L.D.
Homo Sapiens ChrIX RFX3 cds: Lineare Dipendenza
0 5 10 15 20 25 30 35 40 45 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Homo Sapiens ChrX ADK cds: Lineare Dipendenza
Figura 2.57. Homo Sapiens chr X
0 10 20 30 40 50 60 70 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 N % matr L.D.
Homo Sapiens ChrXII SOX5 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Homo Sapiens ChrXIII MYR8 cds: Lineare Dipendenza
Figura 2.59. Homo Sapiens chr XIII
0 10 20 30 40 50 60 70 80 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Homo Sapiens ChrXV NEO1 cds: Lineare Dipendenza
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Homo Sapiens ChrXVII NF1 cds: Lineare Dipendenza
Figura 2.61. Homo Sapiens chr XVII
0 10 20 30 40 50 60 70 80 90 100 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 N % matr L.D.
Homo Sapiens ChrXX ATRN cds: Lineare Dipendenza
2.4
Conclusioni e tempo di elaborazione
Complessivamente possiamo affermare che i risultati migliori li abbiamo ottenuti
con il Caenorhabditis Elegans dove i picchi significativi assoluti pi`u elevati hanno
raggiunto il 70%. Mentre i picchi significativi medi pi`u elevati sono stati quelli del
Magnaporthe Grisea.
Abbiamo provato a ricercare correlazioni anche su matrici costruite con frames di lunghezza N variabile prendendo un frame si e uno no e le percentuali di matrici linearmente dipendenti scendevano al di sotto del 10%. Siccome con il metodo tradi-zionale non si scende sotto un valore medio di circa 35-40% possiamo affermare che esiste una certa forma di correlazione. Risultati analoghi a questi ultimi (35-40%) sono stati trovati anche dal Georgia Institute of Technology [B.5].
5 10 15 20 25 30 35 40 45 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 N % matr L.D.
Homo Sapiens ChrY PR48: Lineare Dipendenza (N consecutivi)
2 – Lineare Dipendenza 0 5 10 15 20 25 30 35 40 45 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 N % matr L.D.
Homo Sapiens ChrY PR48: Lineare Dipendenza N (alterni)
Figura 2.64. Lineare Dipendenza frames alternati
Un ultimo tentativo prevedeva di cambiare il labeling: vedi figure 2.62 e 2.63, ma non abbiamo evidenziato particolari differenze dal metodo tradizionale.
a ==> 0
Î
a ==> 1000
t ==> 1 Î t ==> 0100
c ==> 2 Î c ==> 0010
g ==> 3 Î g ==> 0001
Lineare Dipendenza: Yeast FLO9 0,00 10,00 20,00 30,00 40,00 50,00 60,00 70,00 80,00 90,00 100,00 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 N % matri ci L. D. normale nuova
Tempo di elaborazione
Osservando il programma passo passo, si evidenzia un andamento non lineare del
tempo di elaborazione per ogni singolo valore di N (dove N `e la dimensione delle
matrici quadrate). Se rappresentiamo tutto in un grafico, notiamo che:
• quando N `e piccolo abbiamo molte matrici che vengono processate abbastanza
in fretta poich`e sono di dimensioni decisamente limitate;
• quando N `e grande (vicino al limite superiore √L), le matrici sono grandi ma
poco numerose e vengono elaborate in fretta;
• quando N `e vicino a √L/2 il tempo di elaborazione raggiunge il massimo
siccome abbiamo matrici abbastanza grandi e di dimensioni che cominciano a diventare considerevoli.
Nel grafico `e stato anche rappresentato il numero di matrici al variare di N
(ricor-diamo che segue la formula: L/N − N + 1 dove L `e la lunghezza della sequenza
considerata), inoltre questi valori sono stati normalizzati di un fattore 20 in modo da poter sovrapporre le curve. Nel nostro esempio la porzione di DNA considerata
appartiene all’Arabidopsis Thaliana ed il tempo di elaborazione complessivo `e stato
di 2h 21m 48s con un Pentium IV 2,4 GHz e 1024 MB di Ram.
10 20 30 40 50 60 70 80 90 100 0 20 40 60 80 100 120 140 160 N
nun tot matr norm/20
tempo [sec]
Tempo di elaborazione per ogni N
tempo [sec]
numero totale matrici / 20
(es: Arabidopsis Thaliana ChrV AT5G40480)
2.5
Listato
Listato del programma utilizzato per l’analisi della lineare dipendenza
%% questo programma richiede di inserire le stringa di basi azotate nella %% forma: ’base base base’(es: ’a c t’)nel vettore vect[ fine] lasciando il %% valore fine (=5) che serve per delimitare il vettore
%% Spezza la stringa in frames
%% di lunghezza N (N varia da 3 a sqrt(lunghezza stringa)), ogni N frames %% si pu costruire una matrice N x N e calcolarne il rango(trasformando la %% matrice in una matrice di Galois Gf4): se ci fosse
%% indipendenza statistica il rango dovrebbe sempre essere massimo = N;
%% questo programma calcola il numero totale di matrici N x N ed il numero di %% matrici N x N con rango non massimo quindi la percentuale che queste
%% rappresentano sul totale. Calcola inoltre il tempo macchina necessario %% a questo programma per dare il risultato. Inoltre alla fine emette un
%% segnale acustico per avvertire che il programma terminato
clear all close all
n_A=0; n_T=0; n_C=0; n_G=0; A=0; T=1; C=2; G=3;
a=0; t=1; c=2; g=3; fine = 5;
vect = [ fine];
time=cputime; %misuro il tempo macchina
i=1;
while vect(i) ~= fine % statistica di basi azotate %%
if vect(i)==0 % A n_A = n_A + 1; elseif vect(i)==1 % T n_T = n_T + 1; elseif vect(i)==2 % C n_C = n_C + 1;
elseif vect(i)==3 % G n_G = n_G + 1; end i = i + 1; end tot = n_A + n_T + n_C + n_G; %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%% ora faccio variare N, qui la lunghezza della stringa L = numero di basi
L = tot;
matr = 0; % inizializzo
riassunto = 0; % inizializzo
for N = 3 : 1 : sqrt(L) %% non ha senso andare oltre sqrt(L)
i = 1; j = 1; k=1; cnt = 0; cnt_r=0; while k < L matr(i,j)=vect(k); j=j+1; k=k+1; cnt=cnt+1; if cnt == N * i i=i+1; j=1; end if cnt==N*N %matr %%%%%%%%%%%%%%%%%%
matr_gf = gf(matr,2); %% per costruire matr di Galois
R = rank(matr_gf);
if R < N %% conto le matrici che non sono L.I.
end i=N; j=1;
cnt=(i-1)*N;
for z = 1 : 1 : N-1 % sposto le righe in su di 1
matr(z,:)=matr(z+1,:); end end end if cnt_r >= 0 riassunto(1,N) = N ; riassunto(2,N) = cnt_r;
n_tot_matr = ((L-rem(L,N))/N)-(N-1) ; % in totale ho questo numero
% di matrici di dimensione N x N
perc_matr_corr = cnt_r / n_tot_matr ;% percentuale matrici L.D. = correlate
riassunto(3,N) = n_tot_matr ;
riassunto(4,N) = perc_matr_corr ;
N % evidenzio mentre il programma gira
cnt_r n_tot_matr perc_matr_corr end
end %for
time=cputime-time %misuro il tempo macchina
s = ’ovvero’
x=riassunto(1,:); y=riassunto(4,:); figure plot(x,y) grid on
xlabel(’N’),ylabel(’% matr L.D.’),title(’Lineare Dipendenza’) x = rem(time,3600); y = rem(x,60);
ore = (time-x)/3600 minuti = (x-y)/60 secondi = y
Entropia
3.1
Introduzione
In questo capitolo cercheremo di distinguere le porzioni codificanti (CDS) da quelle non codificanti (NCDS) di una specifica sequenza di DNA valutando l’entropia con il metodo della Sliding Window. Supponiamo di dover analizzare una sequenza
geneti-ca di lunghezza L, il primo problema che dobbiamo affrontare `e il dimensionamento
della finestra di lunghezza N. L’idea `e quella di poter evidenziare le transizioni da
regioni geniche (CDS) a regioni non geniche (NCDS). Poich`e le regioni geniche non
superano il migliaio o poche migliaia di basi azotate, N non deve essere superiore
a 500 o 1000. Noi per comodit`a ci esprimeremo in potenze di 2, quindi 512,1024.
Quando parliamo di entropia ci riferiamo alla definizione data da Shannon [B.6], [B.7]:
H =X
i
pilog2pi
dove i = a,t,c,g (basi azotate) e p `e la probabilit`a di trovare una specifica base
azotata in una singola finestra calcolata come numero di basi ’t’ (ad esempio) su N (dimensione finestra). Avremo L-N+1 frames di lunghezza N e porremo in
rela-zione ogni frame con il rispettivo valore di entropia calcolato. Data la semplicit`a
dell’algoritmo anche questa volta abbiamo usato come ambiente di lavoro Matlab
3.2
Esempio di applicazione: Viola Clauseniana
Consideriamo il seguente esempio in cui abbiamo come sorgente una porzione di 198 basi del DNA della Viola Clauseniana (Eucariota):
a a c g t c g c c g c c a a c c c c a c c c c t a t g g g c g g g c a a t t g g g g g c g g a c t t t g g c c t c c c g t g c g c c t c g g c g c g c g g a t g g c c t a a a t t t c a g c t c c t g g g a a g g a t c g t c a c a a c a a g c g g t g g t t t t t t g a a c t a t g g a c c t c g g g t g t t g t c g t g c g g c c t c t c g g a a g g a a c g g a c c t g t t g t g c t t g c g c a c c ;
la sequenza `e pubblica ed `e stata scaricata dal sito Pubmed dell’NCBI:
http://www.ncbi.nlm.nih.gov/ otteniamo la seguente tabella :
# frame H bit 1 1,788 2 1,724 3 1,611 4 1,611 5 1,484 6 1,287 7 1,287 8 1,095 9 1,095 10 1,095 11 1,095 12 1,198 13 1,379 14 1,611 15 1,664 16 1,778 17 1,778 18 1,842 19 1,863 20 1,727 21 1,727 22 1,842 23 1,877 24 1,877 25 1,877 26 1,778 27 1,664 28 1,664 29 1,664 30 1,664 31 1,664 32 1,664 33 1,664 34 1,778 35 1,778 36 1,778 37 1,689 38 1,689 39 1,611 40 1,611 41 1,724 42 1,788 43 1,835 44 1,835 45 1,835
46 1,788 47 1,788 48 1,531 49 1,577 50 1,557 51 1,531 52 1,432 53 1,379 54 1,414 55 1,414 56 1,493 57 1,432 58 1,432 59 1,449 60 1,449 61 1,449 62 1,296 63 1,296 64 1,296 65 1,592 66 1,727 67 1,689 68 1,522 69 1,522 70 1,574 71 1,727 72 1,842 73 1,924 74 1,924 75 1,985 76 1,950 77 1,924 78 1,924 79 1,924 80 1,985 81 1,950 82 1,835 83 1,835 84 1,835 85 1,835 86 1,924 87 1,924 88 1,835 89 1,950 90 1,985 91 1,924
92 1,877 93 1,877 94 1,959 95 1,959 96 1,877 97 1,877 98 1,877 99 1,842 100 1,924 101 1,924 102 1,877 103 1,924 104 1,924 105 1,842 106 1,842 107 1,863 108 1,809 109 1,835 110 1,924 111 1,835 112 1,809 113 1,924 114 1,924 115 1,924 116 1,863 117 1,727 118 1,574 119 1,296 120 1,574 121 1,432 122 1,689 123 1,611 124 1,724 125 1,611 126 1,611 127 1,724 128 1,788 129 1,924 130 1,985 131 1,985 132 1,985 133 1,985 134 1,985 135 1,924 136 1,924 137 1,877
138 1,788 139 1,788 140 1,788 141 1,809 142 1,577 143 1,531 144 1,449 145 1,432 146 1,432 147 1,432 148 1,432 149 1,531 150 1,557 151 1,577 152 1,577 153 1,577 154 1,557 155 1,531 156 1,531 157 1,809 158 1,863 159 1,863 160 1,842 161 1,924 162 1,950 163 1,950 164 1,924 165 1,788 166 1,727 167 1,531 168 1,531 169 1,809 170 1,809 171 1,924 172 1,959 173 1,959 174 1,985 175 1,924 176 1,835 177 1,809 178 1,788 179 1,788 180 1,557 181 1,531 182 1,531 183 1,809 184 1,835 185 1,835
Il numero di frames (# frame) L-N+1 = 198-14+1 = 185.
Il grafico associato `e il seguente :
Figura 3.2. esempio entropia
Questa regione `e completamente non genica, non ci sono transizioni, inoltre
3.3
Organismi esaminati
Esamineremo 5 organismi vedi capitolo §2.3 pag 29. • Saccharomyces Cerevisiae
• Caenorhabditis Elegans • Magnaporthe Grisea • Arabidopsis Thaliana • Homo Sapiens
3.3.1
Saccharomyces Cerevisiae
0 1000 2000 3000 4000 5000 6000 7000 8000 1.85 1.9 1.95 2Sac.Cer ChrI GPB2 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
Figura 3.3. Saccharomyces Cerevisiae chr I
0 2000 4000 6000 8000 10000 12000 14000 1.9 1.91 1.92 1.93 1.94 1.95 1.96 1.97 1.98 1.99 2
Sac.Cer ChrIII BUD3 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
1024
CDS
0 2000 4000 6000 8000 10000 12000 14000 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Sac.Cer ChrIV DNF2 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
1024
CDS
Figura 3.5. Saccharomyces Cerevisiae chr IV
0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Sac.Cer ChrV ZRG8 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
Come possiamo notare l’entropia della CDS `e mediamente pi`u alta della regione NCDS. 0 2000 4000 6000 8000 10000 12000 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Sac.Cer ChrVI RPO41 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
Figura 3.7. Saccharomyces Cerevisiae chr VI
0 2000 4000 6000 8000 10000 12000 1.91 1.92 1.93 1.94 1.95 1.96 1.97 1.98
Sac.Cer ChrVII SNT2 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
1024
CDS
Figura 3.8. Saccharomyces Cerevisiae chr VII
0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 1.88 1.9 1.92 1.94 1.96 1.98 2
Sac.Cer ChrVIII SET1 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Sac.Cer ChrIX NEO1 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
Figura 3.10. Saccharomyces Cerevisiae chr IX
0 2000 4000 6000 8000 10000 12000 14000 1.92 1.93 1.94 1.95 1.96 1.97 1.98 1.99
Sac.Cer ChrX BCK1 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
512
CDS
0 2000 4000 6000 8000 10000 12000 14000 16000 18000 1.85
1.9 1.95
2 Entropia(SW) vs #frame con N fisso(da impostare)
# frame
H bit
512 bp S.Cer chrXII FKS1
CDS
3.3.2
Caenorhabditis Elegans
0 2 4 6 8 10 12 14 x 104 1.75 1.8 1.85 1.9 1.952 Entropia(SW) vs #frame con N fisso(da impostare)
# frame
H bit
1024 bp C.Elegans Chr1 vab10
CDS
Figura 3.13. Caenorhabditis Elegans chr I
0 2 4 6 8 10 12 14 16 18 x 104 1.7 1.75 1.8 1.85 1.9 1.95 2
Caenorhabditis Elegans ChrI UNC-89 tot: Entropia vs #frame con N fisso(da impostare)
# frame
H bit
1024
CDS
0 1 2 3 4 5 6 7 8 9 x 104 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Caenorhabditis Elegans ChrII 2I41 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
Figura 3.15. Caenorhabditis Elegans chr II
0 2 4 6 8 10 12 x 104 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98
2 Caenorhabditis Elegans ChrII PTP-3 tot: Entropia vs #frame con N fisso
# frame
H bit
CDS
1024
0 5 10 15 x 104 1.85 1.9 1.95 2
Caenorhabditis Elegans ChrIII DIG-1 tot: Entropia vs #frame con N fisso
# frame
H bit
2048
CDS
Figura 3.17. Caenorhabditis Elegans chr III
Come possiamo notare l’entropia della CDS `e mediamente pi`u alta della regione
NCDS. 0 1 2 3 4 5 6 7 8 x 104 1.7 1.75 1.8 1.85 1.9 1.95 2
Caenorhabditis Elegans ChrIII TRA-1 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
0 1 2 3 4 5 6 7 8 x 104 1.65 1.7 1.75 1.8 1.85 1.9 1.95 2
Caenorhabditis Elegans ChrIV LIN-1 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
Figura 3.19. Caenorhabditis Elegans chr IV
0 1 2 3 4 5 6 7 x 104 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Caenorhabditis Elegans ChrIV ITR-1 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 x 104 1.8 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Entropia vs #frame con N fisso(da impostare)
# frame
H bit
1024 bp C.Elegans Chr5 5L325
CDS
Figura 3.21. Caenorhabditis Elegans chr V
0 1 2 3 4 5 6 7 8 9 10 x 104 1.75 1.8 1.85 1.9 1.95
2 Caenorhabditis Elegans ChrV SOS-1 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
3.3.3
Magnaporthe Grisea
0 0.5 1 1.5 2 2.5 x 104 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2Magnaporthe Grisea ChrI MG01468 tot: Entropia vs #frame con N fisso
# frame
H bit
512
CDS
Figura 3.23. Magnaporthe Grisea chr I
0 0.5 1 1.5 2 2.5 x 104 0.6 0.8 1 1.2 1.4 1.6 1.8 2
Magnaporthe Grisea ChrII MG06822 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
0 0.5 1 1.5 2 2.5 x 104 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2
Magnaporthe Grisea ChrII MG05589 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
non decodif
Figura 3.25. Magnaporthe Grisea chr II
In figura 3.25 si osserva l’andamento di una sequenza non completamente
deco-dificata, le basi incognite sono state poste A (adenina) per comodit`a e l’Entropia va
quindi a zero. 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 104 1.96 1.965 1.97 1.975 1.98 1.985 1.99 1.995 2 2.005
Magnaporthe Grisea ChrIII MG05998 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
0 0.5 1 1.5 2 2.5 x 104 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98
2 Magnaporthe Grisea ChrIV MG04775 tot: Entropia vs #frame con N fisso
# frame
H bit
512
CDS
Figura 3.27. Magnaporthe Grisea chr IV
0 0.5 1 1.5 2 x 104 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2
Magnaporthe Grisea ChrIV MG04118 tot: Entropia vs #frame con N fisso
# frame
H bit
2048
CDS
0 5000 10000 15000 1.55 1.6 1.65 1.7 1.75 1.8 1.85 1.9 1.95 2
Magnaporthe Grisea ChrV MG09784 tot: Entropia vs #frame con N fisso
# frame
H bit
256
CDS
Figura 3.29. Magnaporthe Grisea chr V
In figura 3.30 si osserva l’andamento di una sequenza non completamente deco-dificata. 0 2000 4000 6000 8000 10000 12000 14000 16000 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2
Magnaporthe Grisea ChrV MG10072 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
0 2000 4000 6000 8000 10000 12000 14000 16000 1.97 1.975 1.98 1.985 1.99 1.995 2 2.005
Magnaporthe Grisea ChrVI MG00736 tot: Entropia vs #frame con N fisso
# frame
H bit
2048
CDS
Figura 3.31. Magnaporthe Grisea chr VI
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 104 1.88 1.9 1.92 1.94 1.96 1.98 2
Magnaporthe Grisea ChrVII MG02729 tot: Entropia vs #frame con N fisso
# frame
H bit
CDS
1024
3.3.4
Arabidopsis Thaliana
0 1 2 3 4 5 6 7 8 x 104 1.75 1.8 1.85 1.9 1.952 Arabidopsis Thaliana ChrI AT1G67120 cds: Entropia(SW) vs #frame con N fisso
# frame
H bit
1024 bp
CDS
Figura 3.33. Arabidopsis Thaliana chr I
0 0.5 1 1.5 2 2.5 3 3.5 x 104 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Arabidopsis Thaliana ChrI AT1G722700 tot: Entropia vs #frame con N fisso
# frame
H bit
2048
CDS
Figura 3.34. Arabidopsis Thaliana chr I
L’Entropia delle aree codificanti presenta un valore medio superiore
0 0.5 1 1.5 2 2.5 3 3.5 x 104 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98
Arabidopsis Thaliana ChrII AT2G25660 tot: Entropia vs #frame con N fisso
# frame
H bit
2048
CDS
0 0.5 1 1.5 2 2.5 3 3.5 x 104 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98
2 Arabidopsis Thaliana ChrII AT2G41220 tot: Entropia vs #frame con N fisso
# frame
H bit
CDS
1024
Figura 3.36. Arabidopsis Thaliana chr II
0 0.5 1 1.5 2 2.5 3 x 104 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Arabidopsis Thaliana ChrIII AT3G10380 tot: Entropia vs #frame con N fisso
# frame
H bit
CDS
1024
0 0.5 1 1.5 2 2.5 3 3.5 x 104 1.82 1.84 1.86 1.88 1.9 1.92 1.94 1.96 1.98 2
Arabidopsis Thaliana ChrIII AT3G54280 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS
Figura 3.38. Arabidopsis Thaliana chr III
0 0.5 1 1.5 2 2.5 3 x 104 1.75 1.8 1.85 1.9 1.95
2 Arabidopsis Thaliana ChrIV AT4G03300 tot: Entropia vs #frame con N fisso
# frame
H bit
1024
CDS