UNIVERSITÁ DI PISA
FACOLTÀ DI FARMACIA
TESI DI LAUREA IN FARMACIA
TITOLO
RITUXIMAB NEI PROTOCOLLI DI TRATTAMENTO PER LINFOMA NON-HODGKIN E RISCHIO DI LEUCOENCEFALOPATIA
MULTIFOCALE PROGRESSIVA
Relatore
Chiar.mo Prof. Corrado Blandizzi
Candidato Sabrina Montagnani
INDICE
PARTE GENERALE 3
1. Introduzione 4
2. Rituximab: efficacia e tollerabilità 6
2.1 Proprietà farmacologiche 7
2.2 Efficacia terapeutica 7
2.2.1 Linfoma non-Hodgkin indolente 7
2.2.2 Linfoma non-Hodgkin aggressivo 9
2.2.3 Leucemia linfocitica cronica 11
2.3 Tollerabilità 11
3. Leucoencefalopatia multifocale progressiva 13
3.1 Epidemiologia 14
3.2 Biologia 15
3.3 Patogenesi 19
3.4 Immunità umorale 20
3.5 Immunità cellulare 21
3.6 Segni clinici e sintomi 22
3.7 Diagnosi 25 3.8 Terapia 30 3.9 Prognosi 33 4. Rituximab e PML 34 PARTE SPERIMENTALE 38 5. Obiettivi 39 6. Metodi 40 7. Risultati 42
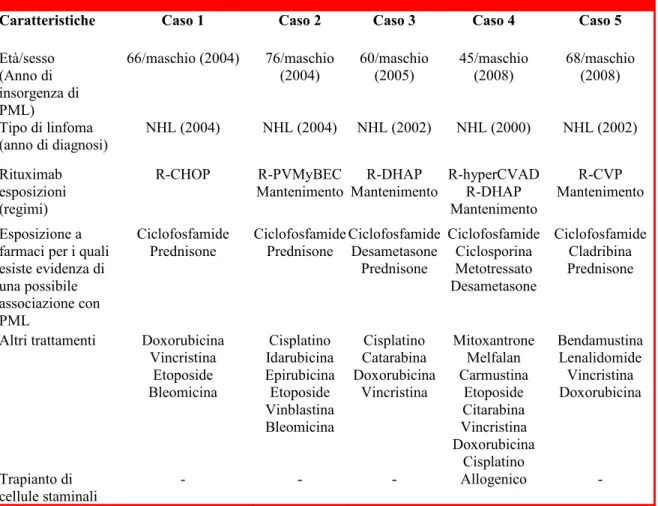
7.1 PML in pazienti trattati con rituximab presso l’Unità Operativa di Ematologia dell’Azienda Ospedaliera Universitaria Pisana
42 7.1.1 Case report 1 43 7.1.2 Case report 2 44 7.1.3 Case report 3 44 7.1.4 Case report 4 45 7.1.5 Case report 5 46
7. 2 Incidence rate di PML nei pazienti NHL trattati con rituximab e confronto con la letteratura
48
7.3 Rischio di PML nei pazienti NHL esposti a rituximab rispetto ai non esposti 50
9. Conclusioni 55
Bibliografia 56
Glossario 83
1. INTRODUZIONE
La leucoencefalopatia multifocale progressiva (progressive multifocal
leukoencepha-lopathy - PML) è l’unica patologia virale umana demielinizzante conosciuta [Weber and
Mayor, 1997]. La PML è causata dal poliomavirus JC (JCV). Il suo nome deriva dalle iniziali del primo paziente (John Cunningham) dal cui cervello è stato isolato il virus [Padgett et al., 1971]. La patologia fu descritta per la prima volta dal neuropatologo tedesco Hallervorden il quale non riuscì ad identificare la PML come entità nosologica distinta, ma la definì “inclassificabile” [Hallervorden, 1930]. Åström e coll. [Åstrom et al., 1958] coniarono il termine PML dopo l’identificazione di patologie croniche immuno-soppressive quali la malattia di Hodgkin, il linfoma non-Hodgkin, le leucemie linfatiche croniche e una possibile infezione virale. Cavanagh e coll. nel 1959 identificarono alcune inclusioni intranucleari come ulteriore evidenza di una causa virale della PML [Cavanagh et al., 1959]. Nel 1965, Zu Rhein e coll. [Zu Rhein et al., 1978] dimostrarono inclusioni intranucleari particellari simili al papovavirus. Sette anni dopo, Padgett all’Università del Wisconsin isolò con successo il virus JC dal cervello di un paziente [Padgett et al., 1971]. Nel 1984, Frisque e coll. [Frisque et al., 1984] pubblicarono la prima sequenza completa di RNA di questo virus, chiamato Mad-1. L’espandersi della pandemia di AIDS ha portato ad un incremento notevole dei casi di PML [Miller et al.,1982]. Ancora oggi, circa l’83% dei pazienti che sviluppa PML è affetta da AIDS [Eng et al., 2006]. Una percentuale compresa tra il 3% e il 5% degli individui HIV-positivi sviluppa PML [San-Andres et al., 2003]. L’infezione produttiva causata da JCV (il virus esce dalla fase di latenza e inizia a replicarsi e ad infettare le cellule) determina la comparsa di numerosi piccoli focolai di demielinizzazione che tendono a confluire. Le lesioni si sviluppano principalmente nella sostanza bianca, ma possono interessare anche la sostanza grigia [Jellinger et al., 2000].
Almeno un terzo dei casi mostra infiltrazioni di cellule rotonde negli spazi perivascolari che indicano una reazione immunitaria aspecifica [Jellinger et al., 2000, Neuenburg et al., 2002]. L’introduzione della terapia antiretrovirale ad elevata efficacia (highly active
antiretroviral therapy, HAART) ha determinato un cambiamento nel decorso clinico della
PML. Infatti in pazienti con AIDS e PML, dopo l’inizio della terapia HAART, si sviluppa una marcata risposta immunitaria denominata “sindrome infiammatoria da immunoricostituzione” (immune reconstitution inflammatory syndrome - IRIS) [Venkataramana et al., 2006, Hoffman et al., 2003]. Nella IRIS, le lesioni infiammatorie focali sono visualizzate alla risonanza magnetica (magnetic resonance imaging, MRI) come aree captanti gadolinio, mentre nella PML questo tipo di lesioni è stato osservato solo nel 15% dei casi [Venkataramana et al., 2006, Berger et al., 1998, Lima et al., 2005]. Queste lesioni possono colpire un intero emisfero cerebrale e appaiono molto intense dopo acquisizione dell’immagine in modalità T2. Raramente le lesioni sono diffuse. Le aree cerebrali più colpite sono le regioni paraventricolari e subcorticale dei lobi parietale-occipitale e frontale [Whiteman et al., 1993]. I segni clinici e i sintomi della PML sono molteplici e aspecifici. Mono o emiparesi predominano, seguiti da varie forme di afasia e disturbi di deambulazione e postura [Brooks and Walker,1984, Berenguer et al., 2003]. Non ci sono evidenze di una terapia efficace per la PML in pazienti affetti da AIDS o da altre patologie immunosoppressive [Roskopf et al., 2006].
Virtualmente tutti i farmaci dotati di attività immunosoppressiva possono innescare l’infezione produttiva da JCV. Nel 2005, la PML è diventata argomento d’interesse farmacologico, poiché è stata osservata in due pazienti con sclerosi multipla e in un paziente con morbo di Crohn trattati con l’anticorpo monoclonale natalizumab in grado di neutralizzare l’integrina a4/b1 [Sandborn et al., 2005-Kleinschmidt-Demasters and Tyler, 2005]. Il ruolo dei linfociti nellosviluppo della PML è sostenuto anche da almeno 25 casi di PML in pazienti trattati con rituximab, un anticorpo monoclonale diretto contro il
recettore CD20 [Genetech, 2005]. La valutazione del rischio di sviluppare PML in seguito a trattamento con rituximab è attualmente oggetto di indagine.
2. RITUXIMAB: EFFICACIA E TOLLERABILITÁ
Rituximab è un anticorpo monoclonale che induce lisi e apoptosi delle cellule B sane e tumorali e sensibilizza quelle tumorali nei confronti dell’effetto citotossico della chemioterapia. In studi clinici di fase III su pazienti con linfoma non-Hodgkin (LNH) indolente o aggressivo a cellule B, l’associazione di rituximab con la chemioterapia convenzionale è risultata più efficace, come terapia di prima o seconda linea rispetto alla chemioterapia da sola, nel promuovere il miglioramento della salute del paziente e il prolungamento della sopravvivenza. Analogamente, in pazienti con leucemia linfocitica cronica (LLC), l’associazione di rituximab con la chemioterapia sembra più efficace della chemioterapia da sola come trattamento di prima o seconda linea. Inoltre, la terapia di mantenimento con rituximab determina un miglioramento significativo della salute e un prolungamento della sopravvivenza nei pazienti affetti da linfoma non-Hodgkin indolente a cellule B o da LLC. L’associazione di rituximab con ciclofosfamide, doxorubicina, vincristina e prednisone (CHOP) mostra la stessa efficacia di un trattamento di prima o seconda linea nel trattamento di LNH a cellule B grandi rispetto allo schema CHOP. Da solo rituximab, sia in monoterapia che in associazione con la chemioterapia, è generalmente ben tollerato dai pazienti affetti da LNH e da LLC. Esso rappresenta quindi un’importante alternativa come terapia di prima o seconda linea ed è compreso nelle linee guida per il trattamento di queste patologie [Risto et al., 2006].
2.1 Proprietà farmacologiche
Rituximab è un anticorpo monoclonale chimerico diretto contro gli antigeni di superficie CD20 espressi dalle cellule B sane, da più del 90% delle cellule B nel LNH e da circa il 14% delle cellule B nella LLC. Il farmaco induce lisi e apoptosi di tutte le cellule B CD20+
e sensibilizza le cellule B tumorali nei confronti dell’effetto citotossico della chemio-terapia. Alle dosi terapeutiche, rituximab causa una rapida e grave leucopenia da cellule B nella maggior parte dei pazienti, che sopravvive per oltre 6 mesi e guarisce entro 9-12 mesi dalla fine del trattamento. Questi risultati suggeriscono che la terapia di mantenimento con rituximab sia efficace per la soppressione delle cellule B tumorali [Risto et al., 2006]. In seguito alla somministrazione per via endovenosa di rituximab in pazienti con LNH a cellule B, la concentrazione ematica risulta direttamente proporzionale alla dose e inversamente correlata al livello assoluto di cellule B periferiche circolanti e alle misure di diffusione del tumore rispetto al basale. La farmacocinetica di rituximab è caratterizzata dall’accumulo di farmaco in seguito a somministrazioni ripetute e può avere profili diversi in base alla variabilità individuale. Le concentrazioni ematiche di rituximab rimangono a livelli terapeutici fino a sei mesi e, durante il periodo di trattamento con rituximab, il farmaco si trova a concentrazioni misurabili anche nel sistema nervoso centrale (SNC). La farmacocinetica di rituximab mostra profili simili anche in pazienti con LNH a cellule B diffuse o follicolari che non sono sottoposti a schemi di trattamento CHOP. Concentrazioni molto basse di rituximab sono presenti in pazienti con la forma linfomatosa di LLC a cellule B, rispetto a quelle dei pazienti con LNH a cellule B. [Risto et al., 2006]
2.2 Efficacia terapeutica
2.2.1 Linfoma non-Hodgkin indolente : negli studi di fase III su pazienti con LNH follicolare in stadio avanzato, non sottoposti ad alcuna chemioterapia, la somministrazione di rituximab 375 mg/m2 per via endovenosa per 6-8 cicli di chemioterapia è risultata molto più efficace nell’indurre la remissione completa a breve termine (2.5 anni) e della sopravvivenza libera da eventi (EFS) rispetto alla chemioterapia da sola. In associazione a ciclofosfamide, vincristina e prednisone (CVP), rituximab prolunga il tempo in cui si osserva una risposta alla terapia (più del doppio), il tempo libero da fallimento terapeutico
di circa quattro volte, la sopravvivenza libera da malattia (DFS) e rallenta la progressione della malattia più del doppio. Ad una valutazione di follow-up di tre anni questi effetti non hanno tuttavia determinato una sopravvivenza globale (Overall Survival, OS) significativamente superiore a quella osservata per la sola terapia CVP (89% vs 81%), sebbene entrambi i risultati siano clinicamente rilevanti. Tuttavia è importante sottolineare che i pazienti trattati con rituximab in associazione allo schema CVP presentano un tempo libero da sintomi della malattia o da tossicità significativamente superiore e sostanzialmente una migliore sopravvivenza aggiustata per parametri di qualità rispetto ai pazienti che assumono la sola CVP. Nei pazienti affetti da LNH follicolare avanzato, la terapia di prima linea con rituximab in associazione con mitoxantrone, clorambucile e prednisone (MCP) determina un aumento del tempo di sopravvivenza libera da progressione (PFS) e della OS rispetto allo schema MCP da solo [Hiddeman et al., 2005; Herold et al., 2005; Solal-Caligny et al., 2005; Salles et al., 2004; Baltazar et al., 2005]. In due studi clinici di fase III è stato osservato che l’aggiunta di una singola dose di rituximab ad ognuno dei sei cicli di terapia CHOP o a quattro cicli di fludarabina, ciclofosfamide e mitoxantrone (FCM), come terapia di seconda linea in pazienti con LNH follicolare refrattario o con recidive, migliora in modo significativo i tempi di remissione completa e della risposta oggettiva, e aumenta la PFS rispetto alla chemioterapia da sola. Un aumento significativo dell’OS di 4 anni è stato evidenziato anche da studi clinici sulla chemioterapia FCM [Forstpointner et al., 2004; Van Oers et al., 2005; Dreyling et al., 2005].
L’aumento significativo della sopravvivenza legato all’introduzione di rutuximab in associazione a chemioterapia standard di prima e seconda linea in pazienti con LNH follicolare è stato confermato anche in una meta-analisi di dati ottenuti in alcuni studi clinici randomizzati di fase III. In monoterapia, quattro somministrazioni settimanali di rituximab 375 mg/m2 per via endovenosa hanno determinato un aumento clinicamente rilevante dei tassi di remissione completa (complete response, CR) e di remissione
oggettiva (objective response, OR) in pazienti affetti da LNH a cellule linfoidi B della mucosa. Uno degli studi ha mostrato una sopravvivenza libera da fallimento terapeutico raddoppiata (p=0.001) in pazienti trattati con rituximab in monoterapia naive per la chemioterapia rispetto a pazienti precedentemente esposti a chemioterapia [Marcus et al., 2005; Shulz et al., 2005]. Allo stesso modo, la terapia di mantenimento con rituximab 375 mg/m2 (quattro somministrazioni per via endovenosa una volta alla settimana ogni sei mesi o una singola somministrazione ogni due o tre mesi, per più di due anni o fino alla comparsa di recidiva), ha mostrato un aumento significativo di CR e/o OR, del periodo di remissione, dell’EFS e del PFS dopo la terapia di induzione in pazienti affetti da LNH follicolare o LLC (forma linfomatosa), rispetto al ri-trattamento con rituximab (alla progressione, studi di fase II) o al non trattamento (studi di fase III). La terapia di mantenimento con rituximab determina un aumento del PFS e dell’OS di tre o quattro anni sia nei pazienti precedentemente trattati che in quelli non trattati con LNH follicolare, mentre i dati ottenuti sottoponendo i pazienti ad un nuovo trattamento non sono significativamente diversi (72% vs 68%) [Ghielmini et al., 2004; Hainsworth et al., 2005; Hochster et al., 2005].
2.2.2 LNH aggressivo : in diversi studi clinici di fase III l’associazione di rituximab 375 mg/m2 per via endovenosa con la terapia CHOP o una terapia simile, per 6-8 cicli, è risultata più efficace rispetto alla chemioterapia da sola come trattamento di prima scelta nei pazienti affetti da LNH a cellule B diffuse allo stadio avanzato o affetti da linfoma delle cellule mantellari (LCM). Indipendentemente dall’età dei pazienti con LNH a cellule B diffuse, l’introduzione del trattamento con rituximab aumenta in modo significativo la CR e la sopravvivenza libera da fallimento terapeutico di due o tre anni e l’OS da due a cinque anni (quest’ultimo effetto si riscontra solo nei pazienti a basso rischio). Un aumento di cinque anni del PFS è stato osservato anche in studi clinici condotti su pazienti anziani
sottoposti a trattamento con rituximab associato allo schema chemioterapico CHOP (età ≥ 60 anni) indipendentemente dalla prognosi della patologia. Nei pazienti più giovani (età < 60 anni) con malattia a basso rischio, rituximab, somministrato in associazione allo schema CHOP o ad una terapia simile, riduce il rischio relativo di fallimento terapeutico (del 64%) rispetto alla chemioterapia da sola [Feugier et al., 2005; Habermann et al., 2005; Pfreundschuh et al., 2005].
In due studi clinici di fase III randomizzati stato osservato un aumento significativo della sopravvivenza libera da fallimento terapeutico di un anno o dell’OS di quattro anni in pazienti affetti da LCM trattati con rituximab 375 mg/m2 per via endovenosa in associazione al trattamento CHOP di prima linea (sei cicli) o al trattamento FCM di seconda linea (quattro cicli), rispetto ai pazienti sottoposti alla chemioterapia da sola. Una meta-analisi dei dati ottenuti nei due studi conferma i benefici ottenuti in termini di OS in seguito all’aggiunta di rituximab alla chemioterapia in pazienti affetti da LCM. In entrambi gli studi i pazienti non erano stati precedentemente trattati e non presentano recidive o refrattarietà della LCM [Kaplan et al., 2005].
In uno studio clinico di fase III randomizzato è stato osservato che rituximab non migliora la risposta al trattamento o la sopravvivenza in pazienti affetti da LNH a cellule B aggressivo associato ad HIV, ma aumenta il rischio di morte per tutte le cause legate all’infezione, al contrario di quanto osservato negli studi precedenti di fase I e fase II che avevano evidenziato efficacia terapeutica in questa popolazione. La terapia di mantenimento con rituximab è risultata efficace nei pazienti con LNH a cellule B diffuse, solo se non compresa in un regime di induzione, e non è efficace in pazienti con LCM.
2.2.3 Leucemia linfocitica cronica : in uno studio clinico di fase II è stato osservato che in pazienti con LLC a cellule B la monoterapia standard con rituximab 375 mg/m2 (quattro somministrazioni per via endovenosa una volta alla settimana), è più efficace della
monoterapia con fludarabina. Tuttavia, da analisi retrospettive comparative di diversi studi clinici di fase II e III è emerso che rituximab 375 o 500 mg/m2 associato a sei cicli di fludarabina (associato o no a ciclofosfamide) migliora significativamente la risposta (CR e/o OR) e la sopravvivenza (da due a quattro anni di PFS e/o OS) in questi pazienti, rispetto a quanto osservato con la chemioterapia da sola [Byrd et al., 2003; Keating et al., 2005; O’brien et al., 2001; Wierda et al., 2005; Byrd et al., 2005., Rai et al., 2000].
2.3 Tollerabilità
Dagli studi clinici è stato rilevato che rituximab in monoterapia o in associazione alla chemioterapia è ben tollerato dai pazienti affetti da LNH a cellule B aggressivo o indolente e da quelli affetti da LLC a cellule B. Gli eventi avversi più comuni emersi negli studi clinici sono reazioni associate al sito di iniezione, eventi avversi ematologici e infezioni. Le reazioni correlate all’infusione, riscontrabili nella maggior parte dei pazienti, soprattutto entro due ore dalla prima somministrazione, consistono generalmente in una sintomatologia simil-influenzale, lieve o moderata, che si risolve con infusioni più lente di farmaco o con la sospensione del trattamento e divengono meno frequenti con le successive somministrazioni. Tra le reazioni gravi (di grado 3-4) è emersa la sindrome da liberazione di citochine, in circa il 10% dei pazienti, che spesso richiede specifici interventi terapeutici (analgesici, antistaminici, ossigeno, terapia endovenosa, broncodilatatori, vasocostrittori e/o corticosteroidi).
Rispetto alla chemioterapia classica, rituximab determina una minore incidenza di infezioni e di neutropenia grave. Inoltre, l’introduzione di rituximab non potenzia la tossicità della chemioterapia convenzionale in pazienti affetti da LNH a cellule B o da LLC a cellule B e il profilo di tollerabilità è simile nei pazienti affetti da LNH aggressivo e in quelli affetti da LNH indolente [Genentech, 2006; Roche, 2006]. Altri eventi avversi
rilevanti anche se rari sono rappresentati da reazioni letali associate all’infusione, dalla sindrome da lisi tumorale dalle reazioni mucocutanee gravi [Risto et al., 2006].
Nel 2006, una nota diffusa dalla FDA segnalava l’insorgenza di casi fatali di PML in pazienti che erano stati trattati con rituximab per la terapia del lupus eritematoso sistemico. A questa nota hanno fatto seguito delle “dear doctor letter” e l’aggiornamento della scheda tecnica di rituximab [FDA alert, 2006]. Da allora sempre maggiore attenzione è stata dedicata dai ricercatori e dai clinici alla possibile associazione tra PML e rituximab.
3. LEUCOENCEFALOPATIA MULTIFOCALE PROGRESSIVA
3.1 Epidemiologia
Prima della pandemia di AIDS l’incidenza di PML era di 0.15 casi per milione di abitanti/anno. Con il diffondersi dell’AIDS, l’incidenza si è quadruplicata [Holman et al., 1991]. Tra i farmaci più comunemente associati allo sviluppo di PML sono compresi i corticosteroidi (40% dei casi), seguiti da chemioterapici antitumorali (16%); nella metà dei pazienti non è stata identificata alcuna causa farmacologica [Eng et al., 2006]. Dopo l’AIDS, patologie linfoidi e mieloproliferative, tumori solidi e patologie immunodeficitarie congenite sono la causa principale di PML (4,5% dei casi) [Eng et al., 2006]. Raramente, malattie granulomatose croniche come tubercolosi o sarcoidosi causano PML.
Dato che approssimativamente l’85% dei casi di PML presenta un’infezione basale da HIV, la maggior parte dei dati disponibili derivano da studi di pazienti HIV-positivi con PML [Skromne et al.,2006-De Luca et al., 2001]. L’efficacia nell’ottenere la riduzione della carica virale di HIV e, conseguentemente, nel contrastare lo stato di immuno-soppressione è il fattore più importante in grado di determinare l’aumento della sopravvivenza nei pazienti con PML trattati con HAART rispetto a quanto rilevato da controlli storici prima della pandemia di AIDS [Berenguer et al., 2003, Silva et al., 2005-Elliot et al., 1997]. Prima dell’introduzione della HAART, l’incidenza di PML nei pazienti affetti da AIDS negli Stati Uniti e in Europa risultava costante [San-Andres et al., 2003, Sacktor et al., 2001]. L’introduzione della HAART ha favorito ad un aumento significativo del tempo medio di sopravvivenza, pari a 4.5 anni in pazienti affetti da AIDS con carica cellulare CD4 superiore a 100/µL, e a 3.4 anni in quelli con carica cellulare inferiore a 100/µL [Berenguer et al., 2003]. Ci cono evidenze di una riduzione della mortalità e di un prolungamento del periodo libero da PML in pazienti HIV-negativi con patologie
linfoproliferative, quali il morbo di Hodgkin e il linfoma non-Hodgkin [Garcia-Suarez et al., 2005]. Inoltre alcune terapie antineoplastiche quali i trattamenti con analoghi purinici e le chemioterapie a dosi elevate in associazione a trapianto di cellule staminali, sembrano ridurre l’incidenza di PML e promuovere un rallentamento dell’insorgenza della patologia (Tabella 1) [Garcia-Suarez et al., 2005]. L’anticorpo monoclonale natalizumab sembra essere associato al rischio di sviluppo di PML con un tasso dello 0.1% (circa 1 paziente su 1000 trattati) in un periodo di 18 mesi [Yousry et al., 2006].
Il JCV è diffuso in tutto il mondo. Circa l’80% della popolazione mondiale adulta presenta
anticorpi contro di esso ed è stata quindi soggetta ad una infezione asintomatica [Weber
Tabella 1: Farmaci e trattamenti associati a PML
Trattamento Farmaci Bersaglio molecolare
Glucocorticoidi orali Tutti Diversi meccanismi
Agenti alchilanti Ciclofosfamide
Carmustine Dacarbazina
DNA
Analoghi purinici Fludarabina
Cladribina Azatioprina
DNA
Antimetaboliti Metotrexato inibitore del metabolismo
dell’acido folico
Anticorpi monoclonali Rituximab CD20
Infliximab TNF-α Etanercept TNF-α Natalizumab VLA-4 Basiliximab IL-2Ra Daclixumab IL-2R Muromonab-cd3 CD3
Immunosoppressori Cidosporina cidofilina
Tacrolimus calcineurina
Sirolimus mTOR
and Major, 1997]. Studi di sierologia hanno rilevato una distinzione tra JCV e BKV (un poliomavirus della stessa famiglia di JCV), dato che non sussistono reazioni sierologiche crociate tra la proteina VP1 del capside di JCV e la proteina analoga di BKV [Hamilton et al., 2000]. La clonazione di entrambe le VP1 di JCV e BKV permette oggi un semplice, economico e rapido screening per mezzo di saggi immunoenzimatici (ELISA) [Hamilton et al., 2000, Weber et al., 1994]. Sono stati identificati almeno 14 genotipi di JCV. La genotipizzazione di JCV è stata utilizzata per studiare l’evoluzione umana, poiché si ritiene che il processo di divergenza evolutiva tra ospite (uomo) e virus sia progredito con la stessa velocità [Agostini et al., 1997]. Studi più recenti non sostengono queste ipotesi e suggeriscono una progressione della divergenza di JCV di circa due ordini di grandezza più veloce rispetto a quella del genoma umano [Shackelton et al., 2006].
3.2 Biologia
JCV è un virus a DNA circolare a doppio filamento e fa parte della famiglia dei poliomavirus. La particella virale ha un diametro di circa 45 nm e forma di icosaedro. Il capside virale contiene tre proteine denominate particelle virali (VP), VP1, VP2 e VP3. VP1 rappresenta il 75% delle proteine del capside. VP1 forma particelle virus-simili (VLPs) indipendentemente da VP2 e VP3 [Goldmann et al., 1999, Goldmann et al., 2000]. A scopo diagnostico VP1 può essere utilizzata come un antigene per il rilevamento di risposte umorali JCV-specifiche e/o risposte cellulo-mediate [Goldmann et al., 1999, Goldmann et al., 2000]. La prima sequenza di JCV isolata è stata il ceppo Mad con 5130 paia di basi [Frisque et al., 1984]. Il genoma circolare viene letto da una DNA polimerasi in entrambe le direzioni, cioè in senso “orario” e “antiorario” (Figura 1)[Weber and Major, 1997, Okada et al., 2001]. I geni ad induzione rapida (early genes) T e t sono trascritti da un singolo filamento di DNA a partire dal punto di origine della replicazione (ORI). L’antigene T (T-Ag) è una proteina multifunzionale che trasforma le cellule attraverso
l’interazione con vari geni che regolano la crescita cellulare [Khalili, 2001, White et al., 2005] ed è stato a lungo conosciuto come induttore di tumore cerebrale [Bollag et al., 1989, Dorries, 1997].
Figura 1: Mappa circolare del genoma di JCV. L’origine è affiancata dagli early genes a sinistra e dai geni
ad induzione lenta (delayed genes) a destra. Gli early genes “t”, “T” e le ultime tre varianti di splicing sono denominate T’e designate secondo il numero degli amminoacidi T’165, T’136 e T’135.
indica il codone d’inizio di t e T, indica il codone di stop di T.
indica il codone d’inizio degli ultimi geni; indica il codone di stop dell’ultimo gene VP1.
AP: agnoproteina, VP1: proteina virale 1, VP2: proteina virale 2, Vp3: proteina virale 3
I delayed genes che codificano per VP1, VP2, VP3 e agnoproteina sono letti nel filamento specifico [Frisque et al., 1984]. L’agnoproteina è un polipeptide con di 71 amminoacidi che agisce come inibitore della crescita cellulare e arresta il ciclo cellulare nella fase G2/M [Darbinyan et al., 2002]. Inoltre, questa proteina sopprime la replicazione del DNA indotta dalla proteina T e promuove l’assemblaggio del capside virale [Safak et al., 2001]. Le cellule infettate producono ulteriori proteine denominate T135, T136 e T165 che derivano
dalla sequenza del gene T attraverso uno splicing alternativo del mRNA virale precursore. Queste proteine alterano lo stato di fosforilazione delle proteine p107 e p130 inattivando i geni regolatori del ciclo cellulare e promuovendo la trasformazione cellulare indotta da JCV [Bollag et al., 2006]. Esse partecipano inoltre alla replicazione del DNA di JCV e regolano le funzioni della proteina T [Fisque, 2001]. La proteina T, a sua volta, interagisce con la proteina oncosoppressiva p53 e con la proteina pRb regolatrice del ciclo cellulare [Henson et al., 1995, Bollag et al., 2000]. In base a certi polimorfismi, la proteina p53 inibisce la replicazione del virus JC o non interferisce con la sua replicazione [Staib et al., 1996]. Le interazioni con queste proteine regolatrici possono determinare la lisi cellulare, l’induzione di cellule tumorali e la crescita incontrollata di queste.
La replicazione e le trascrizione del DNA sono regolate da interazioni di complessi cellulo-specifici di varie proteine e fattori che legano il DNA [Raj and Khalili, 1995; Safak and Khalili, 2001]. Il più importante gruppo di proteine è rappresentato dai fattori di trascrizione NF-kappa B e NF-1 [Devireddy et al., 2000-Sumner et al., 1996]. La possibilità di replicazione di JCV sembra dipendere dal profilo di espressione cellulare della famiglia dei fattori di trascrizione NF-1 ed in particolare dall’espressione di NF-1X (nota anche come NF-1D) [Monaco et al., 2001]. È stato recentemente dimostrato che c-Jun, un membro della famiglia delle proteine AP-1 leganti il DNA, regola l’attività di JCV legandosi a NF-1 [Ravichandran et al., 2006].
Nei modelli animali JCV ha dimostrato di poter indurre vari tumori cerebrali quali il glioblastoma multiforme (GBM) nella scimmia gufo, nel criceto e nel ratto e tumori neuro-ectodermici medulloblastoma/primitivi (PNET) in topi transgenici per gli early genes di JCV [Khalili, 2001, Ohsumi et al., 1986-Krynska et al., 1999]. Per mezzo della PCR e di altri metodi di amplificazione del DNA, il DNA di JCV è stato evidenziato in colonie cellulari, cellule stromali e linfociti delle tonsille [Ricciardiello et al., 2001, Monaco et al., 1998]. Queste ed altre osservazioni hanno permesso di ipotizzare un ruolo di JCV nello
sviluppo di varie tipologie di tumori, quali il carcinoma del colon, il glioblastoma multiforme, l’oligodendroglioma, il ganglioma, il medulloblastoma, il mesotelioma e i linfomi non-Hodgkin [Croul et al., 2003]. Tuttavia, attualmente non sono disponibili evidenze conclusive a sostegno di un ruolo eziopatogenetico di JCV nello sviluppo di questi o altri tumori [Newcomb et al., 2004, Gillison et al., 2007]. Per quanto riguarda i tumori astrogliali dell’uomo, sebbene in pazienti positivi agli anticorpi IgG anti-VP1 di JCV l’odds ratio per lo sviluppo di patologie neoplastiche sia leggermente più alto di 1, le evidenze definitive attuali non sono sufficienti a stabilire un’associazione tra questi tumori e JCV [Rollison et al., 2003]. Tuttavia, dati siero-epidemiologici sembrano suggerire un aumento del rischio di sviluppo di linfoma non-Hodgkin in presenza di aumenti dei titoli anticorpali per JCV aumentati [Rollison et al., 2006]. Resta in ogni caso da dimostrare una relazione tra questo dato e un ruolo eziologico diretto di JCV nello sviluppo del linfoma non-Hodgkin. Inoltre, un polimorfismo nel codone 72 dell’esone 4 della proteina p53 sembra essere associato allo sviluppo di PML in pazienti HIV-positivi o con AIDS conclamato [Power et al., 2000].
JCV si lega ad almeno due recettori sulla superficie cellulare. Uno presenta residui di acido sialico legati in posizione terminale alfa-2,6 [Liu et al., 1998]. Questi residui sono stati trovati anche in altre sialoglicoproteine, quali glicoproteina acida alfa-1, fetuina (una glicoproteina prodotta a livello epatico e deputata al controllo delle concentrazioni extracellulari del calcio) e recettore della transferrina [Komagone et al., 2002]. Nel tessuto cerebrale umano sano, i recettori dotati di questi residui sono presenti negli oligodendrociti e negli astrociti ma non nei neuroni corticali e la loro espressione è più elevata nei linfociti B, rispetto ai linfociti T [Eash et al., 2004]. I livelli più alti di espressione nelle cellule B rispetto ai linfociti T situati nella milza e nelle tonsille umane sane, rafforzano ulteriormente l’ipotesi di una captazione virale attraverso la via respiratoria con persistenza nei tessuti linfoidi e diffusione al cervello per mezzo delle cellule B [Eash et al., 2004,
Sabath and Major, 2002]. Diversamente da SV-40, i virioni JC penetrano nelle cellule gliali attraverso l’endocitosi mediata da recettori clatrina-dipendenti [Pho et al., 2000]. Il sottotipo recettoriale 2A della serotonina (5-HT2A), caratterizzato da elevata espressione in aree cerebrali prive di barriera ematoencefalica, ovvero, nel plesso corioideo e nell’area postrema, è stato indicato come un possibile recettore per la penetrazione cerebrale di JCV [Elphick et al., 2004]. Il recettore 5-HT2A è espresso in cellule astrogliali normali, e si ritiene che esso sia deputato alla regolazione di funzioni gliali specifiche, quali la proliferazione degli astrociti e il controllo dello sviluppo delle vie serotoninergiche centrali, o alla modulazione della microcircolazione ematica attraverso la liberazione di molecole vasoattive da parte delle cellule astrogliali [Post et al., 1999]. È importante sottolineare che la proteina caveolina 1, una proteina strutturale particolarmente espressa nella caveole in complessi con il recettore 5-HT2A, è coinvolta nell’endocitosi di JCV [Bhatnagar et al., 2004]. Sembra quindi sussistere una stretta correlazione tra l’espressione di recettori per JCV contenenti residui di acido sialico nelle cellule e la loro suscettibilità alle infezioni da JCV [Eash et al., 2004].
3.3 Patogenesi
La patogenesi della PML può essere suddivisa in tre fasi. La prima fase è quella di un’infezione primaria, non apparente. Le possibili vie di infezione sono la via respiratoria e/o quella oro-fecale [Zheng et al., 2004]. La seconda fase dell’infezione è quella di latenza nell’ospite umano. Il tessuto di latenza più probabile per JCV è rappresentato dal rene, che rappresenta la via di escrezione di JCV dall’organismo [Hogan et al., 1980-Chang et al., 2002]. La struttura genomica della variante di JCV ritrovata più comunemente nel rene è stata considerata come archetipo [Kitamura et al., 1990]. Altri probabili tessuti di latenza virale sono il midollo osseo e la milza [Houff et al., 1988]. La terza e ultima fase, quella produttiva, è probabilmente innescata da alterazioni immunitarie e molecolari della regione
regolatrice de DNA virale e determina una disseminazione ematogena di JCV nel cervello [Weber and Major, 1997].
3.4 Immunità umorale
L’infezione da JCV induce una risposta immunitaria umorale con produzione di IgG, dirette prevalentemente contro la proteina VP1, costituente del capside [Weber et al., 1997, Rollison et al., 2003, Knowles and Sasnauskas, 2003]. Sembra che venga indotta contro JCV anche una risposta mediata da IgM. Il ruolo di questa risposta anticorpale è ancora sconosciuto, ma essa potrebbe rappresentare un indicatore di stimolazione antigenica attiva da parte di JCV in soggetti sani o essere il risultato di una reazione crociata aspecifica [Knowles et al., 1992]. In tutti i pazienti con PML descritti in letteratura è stato possibile rilevare anticorpi IgG specifici anti-JCV nel siero [Weber et al., 1997, Knowles et al., 1995, Sindic et al., 1997]. Gli anticorpi non rappresentano una protezione contro lo sviluppo di PML. Il test ELISA, basato sulla VP1 ricombinante di JCV come antigene, è in grado di evidenziare una sieropositività dell’84.5% [Weber et al., 1997]. Sebbene i titoli di IgG non siano significativamente diversi tra i soggetti di controllo (pazienti sani o con sclerosi multipla) e quelli con PML, la presenza intratecale di anticorpi IgG specifici contro VP1 di JCV può essere utilizzata per confermare la diagnosi di PML [Weber, 2001, Weber et al., 1997]. Come in altre infezioni virali del SNC, la produzione intratecale di anticorpi anti-JCV si sviluppa col tempo ed eccede il limite superiore (1.5) dell’indice dell’anticorpo, due o tre settimane dopo l’inizio della patologia [Sindic et al., 1997]. La diagnosi di PML attraverso l’evidenza intratecale di anticorpi IgG può essere ottenuta con una sensibilità del 76% e una specificità del 96,8% [Weber et al., 1997].
Nel 1961 Richardson ipotizzò che la causa della PML potesse essere attribuita ad una compromissione del sistema immunitario [Richardson, 1961]. L’ipotesi si basava sull’osservazione di casi che mostravano anergia per reazioni di ipersensibilità di tipo ritardato nei confronti di antigeni come la tossina del tetano e il 2,4-dinitrofluorobenzene [Ellison, 1969, Knight et al., 1972]. Nel 1980, Willoughby e colleghi [Willoughby et al., 1980] osservarono una riduzione della capacità proliferativa dei linfociti in presenza di mitogeni, quali fitoemoagglutinina o concanavalina A, in 7 pazienti con PML. Inoltre, un’evidenza clinica ulteriore a sostegno del ruolo dell’immunoreattività cellulare deriva dalla comparsa preferenziale di PML in pazienti con patologie note per essere caratterizzate da una risposta immunologica cellulare alterata (ad esempio pazienti con linfoma o immunodepressi per trapianto d’organo). La comparsa di PML in pazienti con ipo- o agammaglobulinemia è comunque rara. In pazienti affetti da HIV la probabilità di sviluppare PML è inversamente correlata alla conta dei linfociti CD4+ [Berger et al., 1998]. Frye e colleghi [Frye et al., 1997] hanno osservato una ridotta proliferazione dei linfociti in pazienti con PML utilizzando virioni purificati di JCV. Metodi di clonazione molecolare e di espressione ricombinante di VP1 hanno permesso di ottenere quantità sufficienti di antigene stabile per analizzare la risposta immunitaria cellulare e umorale [Goldamann et al., 2000]. In pazienti affetti da PML, con o senza infezione da HIV, si osserva una ridotta proliferazione di linfociti e una ridotta produzione di interferone-γ (TH1) in seguito a stimolazione con VP1. La produzione di IL-10 (TH2) in pazienti con PML risulta elevata in pazienti HIV-positivi. Queste osservazioni suggeriscono una soppressione delle funzioni dei linfociti T-helper di tipo 1 [Weber et al., 2001]. Durante la reazione immunitaria contro l’infezione virale, i linfociti CD4+ riconoscono le proteine extracellulari del virus che sono state convertite in peptidi mediante digestione esogena e presentate dal complesso maggiore di istocompatibilità II (MHC-II) sulla superficie delle cellule presentanti l’antigene. I linfociti T citotossici CD8+ (CTLs) riconoscono i peptidi
virali sintetizzati nell’ambiente intracellulare, i quali vengono degradati a livello endogeno e sono presentati insieme al complesso di istocompatibilità di classe I sulla membrana di cellule infettate dal virus. I pazienti con risposta immunitaria specifica dei linfociti citotossici CD8+, che riconoscono MHC di classe I in associazione con l’antigene HLA 0201 di MHC di classe I, contro un epitopo JCV-VP1 specifico (p100), mostrano un tempo di sopravvivenza più prolungato [Koralnik, 2002, Lima et al., 2007]. In uno studio prospettico, Koralnik e colleghi [Koralnik et al., 2002] hanno rafforzato l’ipotesi di un ruolo protettivo dei CTLs JCV-specifici. Infatti 13 di 15 pazienti affetti da PML con CTLs JCV-specifici hanno sviluppato una PML “cronica” o quiescente, mentre 9 degli 11 pazienti senza CTLs JCV-specifici hanno avuto un decorso rapido della patologia e una sopravvivenza ridotta. L’identificazione precoce di CTLs JCV-specifici nella PML sembra avere un valore predittivo positivo per il controllo della PML [Du Pasquier et al., 2004]. La frequenza di linfociti T citotossici epitopo-specifici per JCV A*0201 non è significativa-mente differente tra individui sani, pazienti HIV-positivi e pazienti HIV-positivi con PML, e non può essere quindi utilizzata per diagnosticare la PML o monitorarne l’evoluzione [Koralnik, 2002].
3.6 Segni clinici e sintomi
Segni clinici e sintomi di PML non sono specifici. In circa il 25% dei casi, la PML si manifesta come fase iniziale dell’AIDS [Berger et al., 1998]. A causa del numero ridotto dei pazienti descritti e del carattere retrospettivo di tutti gli studi pubblicati finora, è stato variamente descritto un numero elevato di segni e sintomi quali manifestazioni iniziali della PML (Tabelle 2 e 3). [Berger et al., 1998, Brooks and Walker, 1984, Berguer et al., 2003, Berger and Concha, 1995, Weber et al., 1994]. Il sintomo più comune è la perdita di forza in un arto nel 52% dei casi. Approssimativamente nel 45% dei casi la PML viene diagnosticata in seguito all’insorgenza di deficit cognitivi. Circa il 30% dei pazienti
presenta altri disturbi del linguaggio e/o deficit della visione. Circa il 20% dei pazienti presenta alterazione della coordinazione nel movimento degli arti. Approssimativamente il 12% di tutti i pazienti presenta anche attacchi epilettici o cefalea, mentre il 10% dei pazienti ha alterazioni sensoriali (vedi Tabella 2). I segni clinici sono stati raccolti retrospettivamente in uno studio multicentrico spagnolo e sono stati confrontati con i risultati ottenuti precedentemente da Berenguer e colleghi (Tabella 3) [Berger et al., 1998, Berenguer et al., 2003]. Anche in questo caso la paresi o persino la paralisi e l’atassia emergono come i segni più comuni per la diagnosi di PML.
Tabella 2: Frequenza (%) dei segni clinici in pazienti affetti da AIDS con PML
Sintomi Frequenza (%)
Berger et al., 1998 Berenguer et al., 2003
Paresi o paralisi 54 67
Anomalie della deambulazione 28 64
Disartria 24 –
Afasia 19 –
Danno dei nervi cranici – 31
Anomalie della visione 17 20
3.7 Diagnosi
Tabella 3: Frequenza dei sintomi in pazienti affetti da PML rispetto ai pazienti affetti da sclerosi multipla
Brooks e Walker, 1984 Berger et al., 1997 von Einsiedel et al, 1993 Fong et al., 1995 Gillespie et al., 1991 Weber et al., 1994 Berenguer et al., 2003 PML MS Matthews, 1998 Sintomi Mediana Sensoriali 6 8 7 7 18 4 19 11 50 Crisi epilettiche 6 - 20 14 11 22 13 14 4 Cefalea 7 16 7 - 23 11 - 16 17 Alterazioni della coordinazione dei movimenti degli arti 13 28 40 11 26 11 44 22 47 Deficit della visione 33 24 20 36 30 36 20 36 26 Deficit del linguaggio 17 16 53 7 31 36 47 31 0.8 Mono-emiparesi 33 48 60 46 67 54 70 52 80 Deficit cognitivi 36 24 40 54 66 61 - 45 16
Nelle prime sette colonne sono riportati i sintomi così come osservati in case series retrospettive di PML. L’ultima colonna mostra la frequenza dei sintomi iniziali nella sclerosi multipla (MS). Si osserva una maggiore incidenza di crisi epilettiche e di alterazioni del linguaggio per la PML rispetto alla MS.
Fino a qualche tempo fa, la PML non era mai stata considerata come diagnosi differenziale o patologia concomitante rispetto alla sclerosi multipla (MS), ma lo sviluppo inatteso di PML in due pazienti trattati per MS ha reso necessario stabilire criteri e mezzi per identificare la PML in pazienti con MS trattati con natalizumab, un anticorpo monoclonale diretto contro VLA-4, un’integrina a4/b1 [Langer-Gould et al., 2005, Kleinschmidt-De Masters and Tyler, 2005, Yousry et al., 2006]. Attacchi epilettici, disturbi del linguaggio o afasia rappresentano sintomi nuovi e rari in pazienti con MS, con frequenza pari al 2.3%, 4% e 0.8% rispettivamente, e sono suggestivi di PML nel 20-30% dei casi (Tabella 2). Il miglior metodo non invasivo di diagnosi di PML in pazienti immunodepressi è la MRI [Whiteman et al., 1993, Thurnher et al., 1997]. In circa il 90% dei pazienti non sottoposti a una ricostituzione del sistema immunitario, le lesioni non assorbono il gadolinio e non sono occupanti spazio (Figura 2).
Le aree prevalentemente colpite comprendono il lobo frontale paraventricolare e subcorticale e la sostanza bianca del lobo parieto-occipitale, seguite in ordine decrescente da tronco cerebrale, cervelletto, talamo, gangli della base, corpo calloso e, raramente, midollo spinale nella zona cervicale o toracica [Whiteman et al., 1993, Chang et al., 1997]. Segni di una lesione occupante spazio sono stati trovati in circa il 10% dei casi e sono suggestivi di prognosi grave [Post et al., 1999].
Le tecniche diagnostiche Diffusion-weighted imaging (DWI) e Diffusion tensor imaging (DTI) evidenziano aree anomale di diffusione correlate con la progressione della patologia. Con l’uso della spettroscopia protonica a risonanza magnetica (H-MRS), le lesioni mostrano una riduzione del contenuto di N-acetil-aspartato (NAA) e creatina, mentre composti contenenti colina (ES. lipidi di membrana) e lattato sono presenti in concentrazione elevata [Chang et al., 1997, Iranzo et al., 1999].
Figura 2: Immagini ottenute per mezzo di TAC e MRI eseguite in un paziente affetto da AIDS e la
cui autopsia ha confermato la diagnosi di PML e toxoplasmosi cerebrale. Nell’immagine in alto a sinistra, la TAC mostra piccole lesioni ed evidenzia un’area più piccola rispetto a quanto mostrato dalla MRI. Nella colonna centrale si può osservare l’immagine MRI in modalità T1 che evidenzia una lesione di grandi dimensioni ed ipointensa nella regione destra del lobo occipitale (a livello del giro angolare e del solco intraparietale). A destra invece, l’immagine in modalità T2 rivela la presenza di un interessamento multifocale tipico della PML con ulteriori lesioni della sostanza bianca nella parte sinistra dei lobi frontale, temporale e occipitale. Si nota che le lesioni non colpiscono la materia grigia. Nella fila in basso, la TAC mostra un aumento del contrasto nel peduncolo cerebrale destro, analogamente a quanto ottenuto con il gadolinio nella colonna centrale. Queste lesioni caratterizzate da aumento del contrasto corrispondono alla toxoplasmosi cerebrale evidenziata in fase di autopsia. Nell’immagine in modalità T2 di destra, una ulteriore lesione iperintensa causata dalla PML può essere evidenziata nella sostanza bianca nella regione posteriore destra del giro.
La sensibilità della H-MRS è elevata, ma la sua specificità bassa, per cui il valore predittivo di un risultato positivo è insufficiente per differenziare la PML da lesioni di altra natura, quali le aree demielinizzate nella MS, nei gliomi, nei linfomi, o negli ascessi cerebrali [Simone et al., 1998]. La descrizione dettagliata delle varie caratteristiche di imaging della PML e la loro differenzazione da quelle ascrivibili a nuove lesioni della MS riportate da Yousry e colleghi [Yousry et al.,2006] possono essere riassunte come segue: a) le nuove lesioni dovute alla PML sono principalmente diffuse e subcorticali, mentre nella MS le lesioni sono focali e localizzate nella sostanza bianca;
b) i margini delle lesioni nella PML sono poco definiti e con forma irregolare, mentre nella MS i margini sono netti, talvolta digitiformi (Dawson finger) (Figura 3);
c) nella PML le lesioni crescono asimmetricamente e in modo diffuso, sono confinate alla sostanza bianca e non colpiscono la corteccia, mentre nella MS le lesioni sono focali, si ingrandiscono nell’arco di giorni o settimane e decrescono in tempi più prolungati; d) nell’immagine in modalità T2, le lesioni acute della MS mostrano un’area centrale molto marcata (iperintensa) circondata da un anello meno evidente (isointenso) con una leggera
Figura 3: MRI in paziente con MS nella quale si
evidenziano le tipiche lesioni digitiformi (Dawson finger)
iperintensità al di fuori dell’anello mentre la lesione cronica è iperintensa senza strutture anulari; nella PML le lesioni acute tendono invece ad apparire diffusamente iperintense e possono mostrare solo un’intensità da lieve a moderata nelle aree di recente formazione; e) nelle immagini in modalità T1 contrastate con gadolinio, le lesioni acute della MS risaltano in maniera omogenea e presentano margini ben definiti, mentre le lesioni subacute mostrano una maggiore evidenza della struttura anulare. Fino a qualche anno fa, si sarebbe potuto ragionevolmente argomentare che le lesioni della PML non risultano generalmente evidenziate. Tuttavia in pazienti trattati con HAART per diagnosi recente di AIDS è talvolta possibile osservare un potenziamento periferico o anche denso (Figura 4) [Yousry et al., 2006].
L’introduzione in terapia di natalizumab e lo sviluppo inatteso della PML in 3 pazienti (2 con MS o 1 con morbo di Crohn) trattati con questo anticorpo monoclonale ha stimolato l’analisi retrospettiva di una popolazione ampia di pazienti esposti, che ha suggerito un rischio di PML pari a circa 1 caso su 1000 pazienti trattati per 18 mesi [Yousry et al., 2006]. In due dei tre pazienti con PML associata a natalizumab descritti inizialmente per i quali erano disponibili i campioni di fluido cerebrospinale (CSF), è stato evidenziato il DNA del JCV. Questa osservazione non è stata riscontrata in
Figura 4: PML infiammatoria. Lesioni demielinizzate estese nel lobo fronto-parietale destro rivelate dall’imaging FLAIR (A), ma non captanti gadolinio come si osserva nell’immagine in modalità T1 (B). Lesione allargata dopo 5 settimane di trattamento con HAART rivelate dall’imaging FLAIR (C), con lesioni captanti gadolinio puntiformi (freccia) (D).
34 pazienti della stessa coorte, valutati retrospettivamente, che erano stati trattati con natalizumab e avevano manifestato sintomi suggestivi di PML [Yousry et al., 2006]. I meccanismi tramite i quali natalizumab potrebbe determinare l’insorgenza di PML sono molteplici [Stuve et al., 1998, Niino et al., 2006]. Natalizumab causa una deplezione durevole dei linfociti, sia CD4 che CD8-positivi nel fluido cerebrospinale. Inoltre, la capacità delle cellule bianche del sangue di migrare appare ridotta, mentre il profilo di espressione delle diverse cellule del sistema immunitario è regolato in modo diverso, con profili unici per ciascun paziente, e la soglia di attivazione delle cellule immunitarie è attenuata. La viremia, o più precisamente, la rilevazione del DNA di JCV nel plasma, si verifica in circa l’1.3% degli individui sani e può aumentare fino al 10-15% negli individui HIV-positivi, mentre raggiunge il 33-50% dei pazienti HIV-positivi con PML [Dubois et al., 1998, Weber, 2001]. In base ad esperienze molto limitate, relative a due pazienti con PML trattati con natalizumab, la viremia di JCV può precedere o coincidere con l’insorgenza dei sintomi e dei segni della PML [Sandborn et al., 2005; Langer-Gould and Steinman, 2006]. I dati ottenuti in studi su pazienti affetti da HIV suggeriscono al massimo una possibilità del 50% di diagnosticare PML attraverso misurazioni in serie della carica virale di JCV nel plasma [Dubois et al., 1998, Weber, 2001]. Questa bassa probabilità di evidenziare nel plasma la carica virale del virus JC ha comportato il fatto che, per più di una decade, l’analisi del CSF con la PCR abbia rappresentato il metodo di scelta per la diagnosi non- o minimamente invasiva di PML [Dubois et al., 1998, Weber, 2001, Moret et al., 2006-Cinque et al., 1993]. L’analisi del CSF evidenzia una pleiocitosi solo nel 14% dei casi, mentre la sintesi intratecale di IgG è stata riscontrata nel 29% dei pazienti [Weber et al., 1997]. Nei pazienti non affetti da AIDS, la PCR per JCV presenta una sensibilità dell’80% e una specificità del 95% nella diagnosi di PML [Weber, 1999]. Tuttavia evidenze più recenti suggeriscono una sensibilità inferiore, in un intervallo compreso tra 58% e 81% nei pazienti sottoposti ad HAART, determinando una riduzione del valore
predittivo negativo dell’89% [Bossolasco et al., 2005, Marzocchetti et al., 2005]. Livelli simili di sensibilità (78%) e di specificità (96.8%) possono essere ottenute dalla ricerca della presenza di anticorpi IgG sintetizzati a livello intratecale e rivolti contro la proteina VP1 di JCV [Weber et al., 1997, Guillaume et al., 2000]. Inoltre nel corso del tempo la percentuale dei pazienti positivi alle IgG prodotte anti-VP1 è andata aumentando, rendendo questi saggi immunoenzimatici una metodologia più affidabile per confermare la diagnosi di PML, anche nei casi in cui con la PCR non sia possibile evidenziare livelli rilevanti di JCV dopo l’eradicazione del virus dal SNC [Giudici et al., 2000].
3.8 Terapia
Il primo farmaco al quale inizialmente era stato attribuito un potenziale terapeutico nella PML è stato citarabina o ARA C. Studi clinici successivi non hanno tuttavia confermato la sua efficacia [Hall et al., 1998, Bauer et al., 1973]. Anche l’uso di interferone alfa-2a non ha mostrato di essere efficace in pazienti con PML in studi clinici pilota [Berger et al., 1992, Weck et al, 1988]. Queste evidenze contrastano con quelle ottenute in alcuni case reports e in un’analisi retrospettiva, ma nel complesso i risultati disponibili orientano verso l’assenza di effetti dell’interferone alfa-2a nel decorso della PML [Huang et al., 1998, Greschwind et al., 2001-Re et al., 1999]. Evidenze aneddotiche suggeriscono una risposta favorevole della PML al trattamento con interleuchina-2 (IL-2). Tuttavia non sono disponibili dati clinici sufficienti a sostegno di queste conclusioni [Jaeckle et al., 1997, Kunschner and Scott, 2005].
Risultati preliminari ottenuti in vitro avevano suggerito un effetto antivirale di cidofovir [Andrei et al., 1997]. Prove di efficacia scarse, o analisi retrospettive ottenute da case reports suggeriscono una risposta almeno parziale della PML a cidofovir ed una sopravvivenza prolungata associata al trattamento con questo farmaco antivirale [De Luca et al., 2001, Antinori et al., 2003, Blick et al., 1998-De Luca et al., 1999]. Al contrario,
un’analisi retrospettiva più ampia, condotta su 35 casi di PML, ha mostrato una sopravvivenza media inferiore associata al trattamento con HAART e cidofovir rispetto ai casi trattati con solo HAART [Wyen et al., 2004]. Uno studio prospettico di fase 1 in 17 pazienti HIV-positivi con PML non ha mostrato alcun effetto di cidofovir nel prolungare la sopravvivenza [Marra et al., 2002]. In un secondo studio prospettico su 24 pazienti è stato osservato che la sopravvivenza non era associata alle diverse modalità di trattamento, cioè, HAART contro HAART + cidofovir, ma con livelli plasmatici di RNA di HIV-1 ≤ 500 copie/ml, e alla conta delle cellule T CD4+ [Marra et al., 2002]. Dati simili sono stati ottenuti in uno studio osservazionale monocentrico condotto su 46 pazienti, che ha confrontato 22 pazienti trattati solo con HAART e 24 pazienti con HAART e cidofovir [Gasnault et al., 2001]. Nel complesso, i dati attualmente disponibili suggeriscono un’associazione tra sopravvivenza prolungata e parametri immunologici (conta delle cellule CD4+) e virologici (livelli plasmatici dell’RNA di HIV-1, carica virale di JC) piuttosto che con le modalità di trattamento. Pochi case report di efficacia del trattamento con cidofovir in pazienti affetti da PML con tumori ematologici [Viallard et al., 2005] o l’assenza di qualsiasi efficacia [Houston et al., 2001, Terrier et al., 2007] sostengono fermamente un bias osservazionale (sovra segnalazione di risultati positivi e sotto-segnalazione di quelli negativi) quale causa del presunto effetto benefico di cidofovir nella PML. Dati ottenuti con camptotecina, un inibitore della topoisomerasi I, hanno dimostrato che questo farmaco è scarsamente efficace nell’inibire la replicazione di JCV in modelli di coltura cellulare [Kerr et al., 1993]. Un case report suggerisce un prolungamento della sopravvivenza in un paziente con lupus eritematoso sistemico controllato con steroidi e trattato con camptotecina [Vollmer-Haase et al., 1997]. Una serie di nove pazienti trattati con topotecano sembrava mostrare un modesto beneficio, benché associato ad una grave tossicità ematologica [Royal et al., 2003]. Anche per cidofovir e camptotecina, in un bilancio delle evidenze disponibili, non si rilevano prove di efficacia sufficienti.
La comprensione del meccanismo di trasmigrazione del JCV virus attraverso la BBB è essenziale per lo sviluppo di strategie terapeutiche che limitano l’entrata del JCV nel cervello, prevenendo l’entrata di JCV nel sistema nervoso centrale. Un recente studio ha dimostrato che il poliomavirus chimerico JC utilizza il recettore 5HT2A per infettare le cellule SVG-A, un sub-clone di cellule umane gliali fetali (Elphick et al 2004). Altschuler and Kast (2005) hanno suggerito che farmaci antipsicotici quali risperidone, ziprasidone e olanzapina con un’attività antagonista del recettore 5HT2A significativamente più potente possono essere utili nel trattamento o nella prevenzione della PML. Dato che metà della popolazione sana adulta elimina grandi quantità di JCV nelle urine (Agostini et al, 2001; Agostini et al, 1997; Agostini et al, 1996; Shah et al, 1997) questo fornisce un’opportunità unica per valutare la sicurezza e la tollerabilità di farmaci quali risperidone e mirtazapina per l’attività anti-JCV, utilizzando la clearance urinaria di JCV come marker surrogato di efficacia nella PML (Focosi et al, 2007b). Alcuni case reports hanno suggerito un beneficio sulla sopravvivenza in pazienti con PML, HIV-negativi, trattati con risperidone (Focosi et al, 2007) o mitrazepam (Owczarczyk et al, 2007; Verma et al, 2007; Vulliemoz et al, 2006), dimostrando l’eliminazione di JCV dal sangue (Focosi et al, 2007). E’ possibile che i bloccanti del recettore 5HT2A siano efficaci nel controllo della replicazione JCV in vivo. Comunque, non è chiaro se l’effetto benefico osservato in questi pazienti sia il risultato di una variazione del trattamento immunosoppressore che ha portato a ricostituzione immune o sia dato dal contributo dei bloccanti del recettore 5HT2A quali risperidone o mirtazapina. Uno studio successivo ha dimostrato che cellule umane endoteliali cerebrali microvascolari possono essere infettate con JCV indipendentemente dal recettore 5HT2A (Chapagain et al, 2007). Per valutare la capacità di un antagonista del recettore 5HT2A, risperidone, per inibire l’infezione JCV, Moti e coll (2008), hanno trattato primariamente cellule gliali fetali umane in-vitro con risperidone per 24 h e inoculate con JCV. Non è stata osservata una significativa differenza nelle copie del genoma JCV o nei trascritti di mRNA e
l’espressione della proteina nel trattamento nivee nelle cellule primary human fetal glials (PHFG) trattate con risperidone. Questi dati indicano che risperidone non inibisce l’attacco, l’internalizzazione e la replicazione di JCV nelle cellule PHFG e i bloccanti del recettore 5HT2A possono risultare inefficaci nel trattamento della PML.
In conclusione, la terapia ideale della PML in pazienti con AIDS è una terapia antiretrovirale, che diminuisce significativamente la carica virale di HIV-1 RNA nel plasma e possibilmente nel liquido cerebrospinale (CSF), oltre ad un miglioramento della funzione immunitaria cellulare [Berenguer et al., 2003, De Luca et al., 2001, Antinori et al., 2003, De Luca et al., 2000, Giudici et al., 2000, Yiannoutsos et al., 1999].
3.9 Prognosi
Una conta dei CD4 di 100/mm3 o più elevata, una carica virale bassa di JC nel CSF pari a 100 copie/µl o inferiore, e la presenza di cellule T citotossiche specifiche per JCV in pazienti HLA-A2+ e HIV-1 positivi sono associati ad un prolungamento della soprav-vivenza [Koralnik et al., 1999, Koralnik et al., 2002, Yiannoutsos et al., 1999, Taoufik et al., 1998, Garcia De Viedma et al., 2002]. Più del 10% dei pazienti con PML e nuovamente trattato con HAART per l’HIV sviluppa una sindrome infiammatoria da ricostituzione immunitaria (IRIS) [Venkataramana et al., 2006, Safdar et al., 2002, Martinez et al., 2006]. Questa sindrome è caratterizzata da un rapido deterioramento clinico insieme ad una massiva reazione infiammatoria nel parenchima cerebrale [Berenguer et al., 2003, Miralles et al., 2001]. Valutazioni che hanno impiegato immunocolorazioni del tessuto cerebrale hanno mostrato un numero di infiltrazioni di cellule T CD3+ significativamente aumentato e di CD8+ nelle lesioni captanti il gadolinio rispetto alle non captanti [Huang et al., 2007].
4. RITUXIMAB E LEUCOENCEFALOPATIA MULTIFOCALE PROGRESSIVA
Il trattamento con rituximab è stato associato ad infezioni virali come complicanza del trattamento. Nel febbraio 2006, dopo 9 anni dall’approvazione della Food and Drug Administration (FDA) per l’uso clinico, nel foglietto illustrativo di rituximab sono state introdotte nuove informazioni riguardanti pazienti affetti da linfoma non-Hodgkin (LNH) che hanno sviluppato gravi infezioni virali in seguito al trattamento con questo farmaco. Le infezioni comprendono epatite B, infezioni da citomegalovirus, infezioni da virus herpes simplex, varicella zoster, virus West Nile e virus JC [Aksoy et al., 2007]. Nel 2006 e nel 2007, la FDA, la European Medicines Agency, la World Health Organization e l’azienda produttrice hanno divulgato avvisi di allerta sulla sicurezza del farmaco riportando il caso di due pazienti affetti da lupus eritematoso sistemico (LES) che hanno sviluppato PML in seguito a trattamento con rituximab e con un altro farmaco immunosoppressore [FDA, 2009; EMEA, 2009; WHO, 2009; Genentech, 2006]. Nel settembre 2008, la FDA e l’azienda produttrice di rituximab hanno divulgato alcune “Dear Health Care Professional Letter” descrivendo il caso di un terzo paziente affetto da artrite reumatoide che è deceduto a causa di PML 18 mesi dopo essere stato sottoposto a trattamento con rituximab, corticosteroidi e metotrexato [Genentech, 2008; Genentech and Biogen, 2009].
Uno studio condotto da Carson e coll. (2009) ha pubblicato una revisione dei casi di PML associati a rituximab documentabili attraverso la letteratura e la consultazione di banche dati. Sono stati descritti 57 casi di pazienti HIV-negativi che hanno sviluppato PML in seguito al trattamento con rituximab nell’ambito del progetto “Research on Adverse Drug Events and Reports” (RADAR). Il progetto RADAR è costituito da un team multidisciplinare sostenuto dall’NIH, focalizzato sull’identificazione, la valutazione e la divulgazione di informazioni che descrivono eventi avversi rari e potenzialmente fatali. I casi di PML sono stati individuati in pazienti trattati con rituximab dai clinici di 12 centri
oncologici o ospedali universitari di Stati Uniti, Italia e Australia (22 casi), attraverso una revisione dei casi segnalati all’FDA (11 casi), utilizzando il database dell’azienda produttrice (30 casi) e tramite pubblicazioni (18 casi). Il periodo di ricerca era compreso tra la prima data di approvazione di rituximab da parte dell’FDA (1997) al 31 dicembre 2008. Eventuali casi duplicati sono stati identificati in base all’età, al sesso e alla patologia. I criteri di inclusione comprendevano: terapia con rituximab precedente alla diagnosi di PML o ai suoi sintomi; conferma della diagnosi di PML sulla base dell’esame istologico del tessuto cerebrale o tramite risonanza magnetica che mostrasse lesioni compatibili con un processo di demielinizzazione; rilevazione di DNA del JCV nel liquido cerebrospinale (CSF) con tecnica PCR; nessuna evidenza di infezione da HIV.
L’età mediana dei 57 pazienti descritti era di 61 anni (range 30-89 anni). La diagnosi primaria comprendeva: disturbo linfoproliferativo delle cellule B (52 pazienti), lupus erite-matoso sistemico (2 pazienti), artrite reumatoide (1 paziente), pancitopenia autoimmune (1 paziente) e porpora trombocitopenica immune (1 paziente). Due pazienti con patologia linfoproliferativa delle cellule B avevano sviluppato anemia emolitica autoimmune. Sette pazienti affetti da malattia linfoproliferativa erano stati sottoposti a trapianto ematopoietico di cellule staminali (tre allogenico e quattro autotrapianto). Tra questi sette pazienti, uno aveva ricevuto la terapia con un analogo purinico e tutti erano stati trattati sia con farmaci alchilanti che con corticosteroidi. Un paziente aveva sviluppato una malattia linfopro-liferativa dopo il trapianto renale.
I 49 pazienti con PML, non sottoposti a trapianto in precedenza, erano stati trattati con analoghi purinici (46%), agenti alchilanti (81%) e corticosteroidi (75%). Un paziente affetto da LNH con anemia emolitica autoimmune aveva assunto in precedenza soltanto corticosteroidi e rituximab, un paziente con pancitopenia idiopatica autoimmune era stato trattato con corticosteroidi, azatioprina, e rituximab e un paziente affetto da porpora trombocitopenica immune aveva ricevuto corticosteroidi, danazolo, immunoglobuline per
via endovenosa, azatioprina e romiplostim. Le terapie effettuate in precedenza per i due pazienti con lupus eritematoso sistemico comprendevano corticosteroidi e farmaci antineo-plastici mentre per il paziente con artrite reumatoide la terapia comprendeva cortico-steroidi, metotressato, un chemioterapico contenente platino e un inibitore del TNF-a. La diagnosi di PML era preceduta da una mediana di sei dosi di rituximab (range 1-28 dosi). Il periodo di tempo mediano dalla prima somministrazione di rituximab alla diagnosi di PML era 16 mesi (range 1-90 mesi) mentre dall’ultima somministrazione di rituximab alla diagnosi di PML era di 5.5 mesi (range 0.3-66 mesi).
I sintomi iniziali della PML comprendevano confusione/disorientamento (54% dei pazienti), emiparesi/debolezza motoria (33%), perdita di coordinazione motoria (25%), disturbi del linguaggio (21%) o della visione (18%). La diagnosi è stata confermata tramite risonanza magnetica e rilevamento del JCV nel CSF (54%) e dalla biopsia o autopsia nei rimanenti casi. Studi quantitativi su cellule T, disponibili per 14 pazienti, hanno identificato linfopenia CD4 (conta linfocitica CD4+ minore di 500 cellule/µL) (9 pazienti) o rapporto CD4/CD8 diminuito (9 pazienti). Il periodo mediano fra l’ultima dose di rituximab e la diagnosi di PML era minore nei pazienti che avevano una conta linfocitica CD4+ <500 cellule/µL rispetto a quelli con valori superiori (3 vs 17 mesi). Un paziente trattato con rituximab che non aveva ricevuto in precedenza un trapianto ematopoietico di cellule staminali, analoghi purinici o farmaci alchilanti aveva una conta cellulare CD4+ e un rapporto CD4/CD8 normale. Campioni di midollo osseo fissati in paraffina ottenuti da tre pazienti precedentemente trattati con rituximab presentavano JCV rilevabile tramite PCR. L’incidenza dei casi fatali è stata del 90%; 100% fra i casi di PML diagnosticati entro tre mesi dall’ultima dose di rituximab contro l’84% dei casi di PML diagnosticati più di tre mesi dopo l’ultima dose di rituximab. I trattamenti per la PML comprendevano citarabina, terapie antivirali o immunologiche. Dei cinque pazienti non deceduti, due non ricevevano terapia, uno assumeva citarabina, un altro mirtazapina e l’ultimo era trattato
con cidofovir, infusioni di linfociti, citarabina e risperidone. Questi pazienti presentavano postumi quali deficit neuronale con afasia motoria, emiparesi e disturbi visivi.
Questo studio rappresenta il primo ‘case series’ di PML sviluppata in pazienti HIV-negativi trattati con rituximab. Non è stato finora possibile stimare con precisione l’incidenza di questa reazione avversa in soggetti affetti da linfoma per la sottosegnalazione dei casi di PML in pazienti trattati con rituximab e per la mancanza di informazioni sul numero effettivo dei pazienti con linfoma trattati con questo farmaco. E’ verosimile che la somministrazione di rituximab aumenti il rischio di insorgenza di PML, sebbene il rischio assoluto sia probabilmente basso.
5. OBIETTIVI
Questa tesi si pone i seguenti obiettivi:
1) Descrivere i casi di PML osservati presso l’Unità Operativa (UO) di Ematologia dell’Azienda Ospedaliera Universitaria Pisana (AOUP) in pazienti affetti da linfoma non-Hodgkin esposti a trattamento con rituximab.
2) Valutare l’incidenza di PML in pazienti affetti da linfoma non-Hodgkin esposti a rituximab attraverso l’analisi del database dell’UO di Ematologia dell’AOUP.
3) Confrontare l’incidenza di PML nei pazienti affetti da linfoma non-Hodgkin esposti a rituximab in cura presso l’UO di Ematologia dell’AOUP con l’incidenza di PML descritta in letteratura per altre popolazioni di pazienti.
3) Valutare il rischio di PML in pazienti affetti da linfoma non-Hodgkin esposti a rituximab rispetto ai non esposti attraverso l’analisi del database dell’UO di Ematologia dell’AOUP.
6. METODI
Nella prima fase della ricerca sono stati valutati retrospettivamente i registri informatizzati dell’UO di Ematologia dell’AOUP, contenenti dati demografici e clinici di tutti i pazienti ricoverati dal 1994 (n = 12.291). Di ciascun paziente sono state recuperate le seguenti informazioni: età, sesso, motivo di accesso alle cure, diagnosi di patologie ematologiche, data della diagnosi delle patologie reumatologiche, trattamenti farmacologici, eventi avversi, necessità di consulenze specialistiche neurologiche. Sono stati analizzati i dati disponibili fino ad ottobre 2008 alla ricerca di possibili casi di PML. Di tutti i casi sospetti sono state recuperate le cartelle cliniche complete per verificare la diagnosi. Le informazioni contenute nelle cartelle hanno permesso la descrizione dei casi confermati. Nella seconda fase della ricerca sono stati selezionati tutti i pazienti affetti da linfoma non-Hodgkin esposti a trattamento con rituximab in monoterapia o nel contesto di protocolli di trattamento multipli. Sono stati esclusi dall’analisi pazienti HIV-positivi. All’interno di questa popolazione dono stati individuati i casi di PML con diagnosi documentata da risonanza magnetica (RM) e positività al virus JC alla biopsia cerebrale stereotattiche e/o positività JCV nel fluido cerebrospinale, i cui sintomi fossero insorti almeno un mese dopo la prima dose di rituximab somministrata. Per questi pazienti è stato calcolato l’incidence
rate di PML in base al periodo di esposizione a rituximab calcolato all’ultimo follow up
disponibile. Inoltre, abbiamo esaminato la letteratura inglese utilizzando PubMed e EMBASE senza limiti di tempo, per gli studi che hanno valutato l’incidenza di PML in popolazioni differenti per confrontare i nostri dati con le osservazioni precedenti. Sono stati selezionati solo gli studi che hanno riportato l’incidence rate o dati nei quali l’incidence rate poteva essere stimata.
Nella terza fase della tesi è stato valutato il rischio di PML in pazienti affetti da NHL, HIV-negativi esposti a regime chemioterapico standard con o senza rituximab, per determinare se l’uso di rituximab abbia aumentato l’incidenza di PML. I pazienti sono stati