Università degli Studi di Ferrara
DOTTORATO DI RICERCA IN
SCIENZE FARMACEUTICHE
CICLO XXV
COORDINATORE Prof. MANFREDINI STEFANO
Identification and synthesis of
3,4-isoxazolediamides as new class of
Hsp90 inhibitors
Settore Scientifico Disciplinare CHIM/06
Tutore Dottorando
Dott. Marchetti Paolo Dott. Mangiola Stefania
i
Contents
1
INTRODUCTION
1
1.1 Heat shock proteins in health and disease 2
1.2 Functions of molecular chaperone 3
1.3 Heat shock proteins classification 4
1.4 Hsp90 7
1.4.1 Molecular anatomy of Hsp90 8
1.4.2 ATPase cycle 10
1.5 Oncogenic Hsp90 client proteins and signaling pathways 15
1.5.1 Growth Factor Receptor 12
1.5.2 Steroid receptors 13
1.5.3 Bcr-Abl 13
1.5.4 Flt3 13
1.5.5 Protein Kinase 14
1.6 A high-affinity conformation of tumor Hsp90 confers drug
selectivity 15
1.7 Inhibitors of Hsp90 C-terminal ATP-binding domain 17
1.7.1 Novobiocin and analogues 17
1.7.2 Other C-terminal inhibitors 18
1.8 Inhibitors of Hsp90 N-terminal ATP binding domain 18
1.8.1 Geldanamycin and its derivatives 19
1.8.2 Radicicol and its derivatives 21
1.8.3 Purine-based derivatives 22
1.8.4 o-Aminobenzoic acid derivatives and resorcinol
derivatives 24
ii
2
AIM OF THESIS
32
3
CHEMISTRY
36
3.1 Synthesis of 3,4-isoxazolediamides scaffold 37
3.1.1 Synthesis of chloro-resorcinol derivatives 37
3.1.2 Modification at C-3 38
3.1.3 Synthesis of isopropyl-resorcinol derivatives 38
3.1.4 Modification at C-4 39
3.1.5 Synthesis of 4-aminoisoxazole 42
3.2 Attempts to synthesize 4,5,6,7-tetrahydro-isoxazole-[4,5-c]pyridine
scaffold 43
3.3 Synthesis 4,5,6,7-tetrahydro-isoxazlo-[4,5-c]pyridine scaffold 45
3.3.1 Synthesis of resorcinol acid 46
4
RESULTS AND DISCUSSION
48
4.1 In vitro studies of 3,4-isoxazolediamides scaffold 49
4.1.1 Analysis of the degradation of Hsp90 client proteins in A431
cells 59
4.2 In vivo studies of 3,4-isoxazolediamides scaffold 61
4.3 In vitro studies of 4,5,6,7-tetrahydro-isoxazolo-[4,5-c]pyridine
scaffold 64
5
CONCLUSION
67
6
EXPERIMENTAL SECTION
69
6.1 Biological section 70
6.2 Molecular modeling 72
6.3 Materials and methods 73
6.4 Experimental procedures 74
iii
List of Figures
1.1 Structural domain organization of human Hsp90 protein 8
1.2 ATP-dependent molecular clamp 10
1.3 Hsp90 cycle 15
1.4 Hsp90 client proteins 12
1.5 Model for tumor selectivity of Hsp90 inhibitors and Hsp90-dependent malignant
progression 15
1.6 Chemical structure of novobiocin and coumercyn A1 17
1.7 Chemical structure of A4 and DHN2 18
1.8 Chemical structure of cisplatin and EGCG 18
1.9 Chemical structure of geldanamycin 19
1.10 Chemical structure of geldanamycin derivatives 20
1.11 Chemical structure of radicicol 21
1.12 Chemical structure of radicicol derivatives 21
1.13 Chemical structure of PU3 and PU24FCI 22
1.14 Chemical structure of PU-H71 and CUDC-305 23
1.15 Chemical structure of CNF-2024/BIIB021 and derivatives 24
1.16 Chemical structure of SNX-2112 and SNX-5422 24
1.17 Chemical structure of benzoisoxazole derivatives 25
1.18 Chemical structure of resorcinol derivatives 25
1.19 Chemical structure of other N-terminal domain inhibitors 26
1.20 Chemical structure of pyrazole derivatives 26
1.21 Chemical structure of VER49009 27
1.22 X-ray structure of VER49009 and VER50589 28
1.23 Chemical structure of VER50589 29
1.24 Chemical structure of NVP-AUY922 30
2.1 General structure of new Hsp90 inhibitors used in these study 33
iv
4.1 36 co-crystallized in Hsp90 active site and docking pose of compound 91 in Hsp90
active site 50
4.2 123b co-crystallized in Hsp90 active site and docking pose of compound 58 in Hsp90
active site 53
4.3 Analysis of Hsp90 client protein levels and Hsp70 in A431 tumor cells 60
4.4 Antitumor activity of the compounds 91 and 115 62
4.5 Analysis of Hsp90 client protein levels (EGFR, Akt, CDK-4, and Hsp70) in A431
tumor xenografts 63
v
List of Schemes
3.1 Synthesis of chloro-resorcinol intermediate 44a 37
3.2 Synthesis of chloro-resorcinol intermediate 44b,c 38
3.3 Synthesis of isopropyl-resorcinol intermediate 44d 39
3.4 Synthesis of 3,4-isoxazolediamides inhibitors 40
3.5 Synthesis of morpholine and piperazine derivatives 41
3.6 Synthesis of amines-isoxazole derivates 123a-d 42
3.7 Attempts to synthesize 4,5,6,7-tetrahydro-isoxazole-[4,5-c]pyridine scaffold 43
3.8 Synthesis of 4,5,6,7-tetrahydro-isoxazole-[4,5-c]pyridine 45
3.9 Synthesis of chloro-resorcinol acid 46
vi
List of Tables
1.1 The most families of molecular chaperones 4
1.2 Members of the family of 90-kDa molecular chaperones 7
1.3 Relationship between Hsp90 client proteins and hallmarks of cancer 14 1.4 Values of IG50 of VER-49009 and VER-50589 in different cells line 29
1.5 Hsp90-binding drugs: many classes of natural, semisynthetic and synthetic inhibitors of
Hsp90 31
2.1 Data of G-score of structures proposed 34
4.1 Data for isoxazole-3,4 diamides 54
4.2 Data for isoxazole-4-alkyl/cycloalkyl amides 55
4.3 Data for isoxazole-4-aryl/heteroaryl amides 57
4.4 Data for 4-amino isoxazole 59
4.5 Data of cytotoxicity on A431 epidermoid carcinoma cells of the compounds tested in
vivo 61
4.6 Antitumor activity of compounds 91 and 115 62
4.7 Data of binding on Hsp90 and citotoxicity on NCI-H460 non small cell lung carcinoma
cells 65
vii
List of Abbreviations
Aha1: activator of Hsp90 ATPase Akt: Protein Kinase B
Boc: tert-butoxycarbonyl Cdk: Cyclin-dependent kinases DCM: dichloromethane
Cdc37: cell division cycle DMF: dimethylformamide DMSO: dimethylsulfoxide
EDC: N‟-(3-dimethylaminopropyl)-N-ethyl-carbodiimide EGF-R: epidermal growth factor receptor
EtOH: ethanol
Flt3: FMS-like tyrosine kinase 3 FP: fluorescence polarization GRP: glucose regulated protein94
HER-2: Human Epidermal Growth Factor Receptor 2 HOBt: 1-Hydroxybenzotriazole
Hop: Hsp70-organizing protein
IGF-R: insulin-like growth factor-1 receptor IP: immunophilins
kDa: kilo Dalton
PDGF-R: platelet-derived growth factor receptor PhMe: toluene
PI3K: phosphatidyl inositil-3 kinase TFA: trifluoracetic acid
TPR: tetratricopeptide repeat
TRAP: tumor necrosis factor receptor-associated protein p-TSA: p-toluene sulfonic acid
1
Chapter 1
Introduction
Contents
1.1 Heat shock proteins in health and disease 1.2 Functions of molecular chaperone
1.3 Heat shock proteins classification 1.4 Hsp90
1.4.1 Molecular anatomy of Hsp90 1.4.2 ATPase cycle
1.5 Oncogenic Hsp90 client proteins and signaling pathways
1.5.1 Growth Factor Receptors 1.5.2 Steroid receptors
1.5.3 Bcr-Abl 1.5.4 Flt3
1.5.5 Protein Kinase
1.6 A high-affinity conformation of tumor Hsp90 confers drug selectivity 1.7 Inhibitors of Hsp90 C-terminal ATP-binding domain
1.7.1 Novobiocin and analogues 1.7.2 Other C-terminal inhibitors
1.8 Inhibitors of Hsp90 N-terminal ATP binding domain
1.8.1 Geldanamycin and its derivatives 1.8.2 Radicicol and its derivatives 1.8.3 Purine-based derivatives
1.8.4 o-Aminobenzoic acid derivatives and resorcinol derivatives 1.8.5 Pyrazoles and isoxazoles derivatives
2
1.1 Heat shock proteins in health and disease
Heat Shock Proteins (Hsps) are molecular chaperones that have emerged over the last few years as one of the hottest and most interesting topics in biology. It has also become increasingly recognized that they play an important role in oncogenesis and cell death.1,2
Among chaperones, heat shock proteins are the most important.The heat shock response was discovered in 1962 by Ritossa, who observed a pattern of Drosophila salivary gland chromosome puffs that were induced in response to transient exposures to elevated temperatures.Since then, efforts from a large number of investigators have shown that the heat shock response is ubiquitous and highly conserved in all organisms (from bacteria to plants and animals) as an essential defense mechanism for protection of cells from a wide range of harmful conditions. Stress can be any sudden change in the cellular environment, to which the cell is not prepared to respond, such as heat shock, hypoxia, alcohols, inhibitors of energy metabolism, heavy metals, oxidative stress, fever, inflammation, DNA damage or UV radiation.3 The rationale behind this phenomenon is that after stress there is an increased need for the chaperone function of Hsp, which triggers their induction. This need is caused by the increased amount of damaged proteins, by the inhibition of their elimination via the proteasome as well as by the damage of the chaperones themselves. Hsp induction might help to renature chaperones and, therefore, Hsp induction might lead to a „cascading amplification‟ of available chaperone activity.3-5
Cancer is a disease characterized by genetic instability. Although identification of novel therapeutic agents via molecular targeting offers the promise of great specificity coupled with reduced systemic toxicity, specific inhibition of individual proteins or signaling pathways faces the potential peril of being subverted by the inherent genetic plasticity of cancer cells. Cancer cells are very adept at adapting to noxious environments.
If one assumes that cancer cells are always under moderate to severe stress of one type or another, an approach to this apparent dilemma might be to target the basic machinery that allows cancer cells to adapt so successfully to stress. Cells respond to stress by increasing synthesis of a number of molecular chaperones (also known as heat shock proteins, or Hsps, because they were first observed in cells exposed to elevated temperature).6
A recent trend in cancer therapy has been to develop agents that “target” a single molecular alteration. As most cancers are a result of multiple transformation-specific regulatory alterations, targeting one abnormality may be insufficient in reversing the transformed
3
phenotype. Chaperones are proteins that allow cancer cells to tolerate the components of dysregulated pathways that otherwise would be lethal; thus, their inactivation may result in targeting multiple molecular alterations.7,8
1.2 Functions of molecular chaperones
Molecular chaperones support protein synthesis in vivo. Their most important roles are: facilitate the folding of nascent proteins into a stable and functional conformation, maintain conformation, stability and function of client proteins within the cell,9 protect other proteins against aggregation,
maintain the cellular homeostasis,
help to transport proteins from the cytoplasm into different intracellular compartments,
maintain proper protein folding, and facilitate the assembly of protein polymers,10 chaperones do not determine the tertiary structure of the folding proteins, but help
them find their structure more efficiently.11
Instead, under conditions of stress, where protein folding/assembly events may be compromised, the increased expression and accumulation of the stress proteins facilitates the ability of cells to both repair and synthesize new proteins to replace those that were damaged after the particular metabolic insult. Further, chaperones are upregulated in response to cellular stresses and serve as essential regulators of proteins by preventing misfolding. In the case of acute misfolding and aggregation, proteins may be targeted to the proteasome for degradation.12,13
4
1.3 Heat shock proteins classification
Heat shock proteins are classified into six major families based on their molecular mass: Hsp100 (100-110 kDa), Hsp90 (83-90 kDa), Hsp70 (66-78 kDa), Hsp60, Hsp 40 and the small Hsp (15-30 kDa) (Table 1.1). Family members of Hsps are expressed either constitutively or regulated inductively, and are present in different subcellular compartments. High molecular weight Hsps are ATP-dependent chaperones, whereas small HSPs act in an ATP-independent fashion. The most studied stress inducible Hsps are Hsp90, Hsp70 and Hsp27.14
Table 1.1 The most families of molecular chaperones. Eukaryotic molecular
chaperones Functions of chaperone
Hsp27, the small heat-shock proteins Prevent aggregation of proteins and the
release of proteins by aggregation
Hsp40 Help the folding proteins
Hsp60 Prevent aggregation of proteins and
help the folding proteins
Hsp70,grp78 Prevent aggregation of proteins and
help the folding proteins
Hsp 90, grp94 Prevent aggregation of proteins
Hsp100 Release proteins by aggregations
sHsp (small Heat shock proteins)
Small heat shock proteins have a molecular mass between 15 and 30 kDa. They exist as high molecular weight complexes in vertebrates, Drosophila, yeasts, fungi, and plants. The small mammalian Hsps are oligomeric structures of about 32 subunits, corresponding to a molecular mass of 800 kDa. They are present in the cytosol of most cells and tissues even in the absence of stress factors such as elevated temperatures.
All these proteins contain a rather conservative so called α-crystallin domain, containing 80-100 residues located as a rule in the C-terminal part of these proteins.15 Additional features attributed to small Hsps range from RNA storage in heat shock granules to inhibition of apoptosis, actin polymerization and contribution to the optical properties of the eye lens in the case of α-crystallin. At the moment, it is unclear how these seemingly different functions can be explained by a common mechanism. However, as most of the observed phenomena involve non-native protein, the repeatedly reported chaperone properties of Hsp might be a key feature for further understanding of their function.16
5
Hsp40
Also known as chaperone DnaJ in E. Coli or Hsp40 (heat shock protein 40 kD) in eukaryotic cells, Hsp40 is a molecular chaperone protein and is involved in stabilization processes and correct folding of nascent proteins. It is expressed in a wide variety of organisms from bacteria to humans.
This family of proteins contains a 70 amino acid consensus sequence known as the J domain. The J domain interacts with Hsp70 and plays a role in regulating the ATPase activity of Hsp70.17
Hsp60
Hsp60 (heat shock protein 60 kDa) is a mitochondrial chaperonin that is typically held responsible for the transportation and refolding of proteins from the cytoplasm into the mitochondrial matrix. In addition to its role as a heat shock protein, Hsp60 functions as a chaperonin to assist in folding linear amino acid chains into their respective three-dimensional structure. Hsp60 are divided into two general groups: the chaperonines of the first group are expressed in Eubacteria, in mitochondrial, in chloroplasts and in most Archae, instead the chaperonines of the second group are found in cytosol of Eukaryotes and Archaebacteria. Under normal physiological conditions, Hsp60 is a 60 kDa oligomer composed of monomers that form a complex arranged as two stacked heptameric rings. This double ring structure forms a large central cavity in which the unfolded protein binds via hydrophobic interactions. Each subunit of Hsp60 has three domains: the apical domain, the equatorial domain, and the intermediate domain. Through the extensive study of groEL, Hsp60‟s bacterial homolog, Hsp60 has been deemed essential in the synthesis and transportation of essential mitochondrial proteins from the cell's cytoplasm into the mitochondrial matrix.
Hsp70
Hsp70 is a conserved molecular chaperone that assists a wide range of folding processes, including the folding and assembly of newly synthesized proteins, refolding of misfolded and aggregated proteins, membrane translocation of organellar and secretory proteins, and control of the activity of regulatory proteins. The role of Hsp70s in the
6
folding of non-native proteins can be divided into three related activities: prevention of aggregation, promotion of folding to the native state, and solubilization and refolding of aggregated proteins.18
All such functions are mediated by interaction of extended, hydrophobic regions of substrate proteins with the Hsp70 C-terminal substrate-binding domain (SBD). Hsp70 proteins all consist of the same working parts: a highly conserved NH2-terminal ATPase domain of 44 kDa and a COOH-terminal region of 25 kDa, divided into a conserved substrate binding domain of 15 kDa and a less-conserved immediate COOH-terminal domain of 10 kDa.19 The dynamic closing and reopening of the base onto the lid is regulated by ATP hydrolysis in the nucleotide binding domain. In the mammalian system, the molecular chaperones Hsp70 and Hsp90 are involved in the folding and maturation of key regulatory proteins, like steroid hormone receptors, transcription factors, and kinases, some of which are involved in cancer progression. Hsp70 and Hsp90 form a multichaperone complex, in which both proteins are connected by a third one called Hop.20
Hsp90
Heat shock protein 90 (Hsp90) is one of the most abundant chaperone proteins, and has been recently shown to interact with a variety of proteins involved in cell proliferation. Of interest to cancer researchers, Hsp90 is constitutively expressed at 2 to 10 fold higher levels in tumor cells compared to their normal counterparts, suggesting that it may play a critical regulatory role in tumor cell growth and/or survival.21
The following section will focus on detailed description of the structure, function and clients oncogenic proteins of Hsp90.
Hsp100
The Hsp100 family of proteins has a wide distribution in both prokaryotes and eukaryotes. Members of the Hsp100 family were described first as components of the 2-subunit bacterial Clp protease system. The large-2-subunit ClpA represents an adenosine triphosphate (ATP)–dependent unfoldase, whereas the small-subunit ClpP is the protease. ClpA alone has no proteolytic activity, but it is able to prevent target proteins from aggregation. Interestingly, many ClpA-related proteins were characterized in bacteria and eukaryotes as stress-induced proteins, and hence, they are summarized as members of the Hsp100 family. A proteolytic subunit (ClpP) is only found in bacteria associated mainly or
7
exclusively with ClpA protein, whereas in eukaryotes, only the large subunit with chaperone function is observed. A peculiarity of the Hsp100 chaperones is their capability to promote dissociation of aggregated proteins in an ATP-dependent manner.22 Hsp100 (Clp family in E. coli) proteins are a family with a great diversity of functions, such as increased tolerance to high temperatures, promotion of proteolysis of specific cellular substrates and regulation of transcription. HSP100/Clp proteins are also synthesized in a variety of specific patterns and, in eukaryotes, are localized to different subcellular compartments. Recent data suggest that a common ability to disassemble higher order protein structures and aggregates unifies the molecular functions of this diverse family.23 Hsp100 have been studied in vivo and in vitro for their ability to target and unfold tagged and misfolded proteins.
1.4 Hsp90
The 90-kDa heat shock protein, Hsp90, is a highly conserved molecular chaperone protein found in bacteria and all eukaryotes, exhibiting a 40% sequence identity between human and Escherichia coli protein.24-28 Under non-stress conditions, Hsp90 is present as 1-2% of the total cytosolic protein content, whereas under stress it is increased up to 4-6%.29 This renders it one of the most abundant proteins of a typical eukaryotic proteome and has been shown to be essential for cell survival. Hsp90 is an ATP-dependent chaperone essential for the maturation and activity of a varied group of proteins involved in signal transduction, cell cycle regulation and apoptosis.30
All the Hsp90 family members share a common general structural plan that likely reflects functional similarities in their mode of action. Currently, four forms are known in humans: the two major cytoplasmic isoforms Hsp90α (inducible form) and Hsp90β (constitutive form), glucose regulated protein94 (Grp94) in the endoplasmic reticulum and tumor necrosis factor receptor-associated protein1 (TRAP1/Hsp75) in the mitochondrial matrix. The four isomer forms are listed in Table 1.2.
Table 1.2 Members of the family of 90-kDa molecular chaperones.
Name Location in cells
Hsp90α, HSP90β Cytoplasm Hsp75/TRAP-1 Mitochondrial
8
The cytoplasmic forms exist predominantly as dimer within the cell and Hsp90 functions as a dimer, both homo and heterodimers, to maintain the appropriate folding and conformation of many other proteins.31
1.4.1 Molecular anatomy of Hsp90
Crystallization of full-length Hsp90 was first reported in the early 1990s.32,33 Functional Hsp90 is a homodimeric protein composed of two identical and symmetrical subunits.34 Each monomer is divided into three highly conserved domains common to all members of the Hsp90 family, separated by a highly charged region of varying length.35 These include the N-terminal domain (24-28 kDa), the C-terminal domain (11-15 kDa) and the middle domain (38-44 kDa) (Figure 1.1).36
Figure 1.1 Structural domain organisation of human Hsp90 protein. The Hsp90 monomer is comprised of three domains: the N-terminal domain responsible for ATP-binding and inhibitor of Hsp90 as geldanamycin, radicicol (red), a core domain (green), and a C-terminal domain that facilitates homodimerization (blue). In eukaryotes, a "charged linker" region, that connects the N-terminus with the middle domain (not shown in figure).
The most studied is the 25kDa N-terminal domain, a proteolytically resistant core domain of approximately 220 amino acid residues containing both binding and ATP-hydrolytic activities. The three-dimensional crystal structure of human and yeast N-terminal fragments revealed the presence of an eight-stranded anti-parallel β-sheet and nine α-helices, which configure an α/β sandwich module and delimit the nucleotide-binding
9
pocket. This fold differs from the typical ATP-binding sites and shares high 3D homology with members of the ATPase/kinase GHHL (Gyrase B, Hsp90, Histidine Kinases, MutL) superfamily, in addition to other members of the Hsp90 family. These proteins are unrelated in function, but all of them require ATPase activity and contain an unusual adenine-nucleotide-binding pocket know as “Bergerat fold” for binding ATP.37,38
A second ATP binding site is near the C-terminal domain, a 12kDa structural lobe that provides strong dimerization interface, necessary for both constitutive homodimerization and target substrate binding, which is absolutely essential for the implementation of HSP90 chaperone activities. The C-terminal of Hsp90 is the site of dimerization. This region contains a pentapeptide domain (MEEVD) implicated in binding to co-chaperones of Hsp90 such as Hop and Sti1 which containing tetratricopeptide repeats (TPR). Thus, the C-terminal domain is also involved in the formation of active Hsp90 multiprotein complexes.39-43
Most of the Hsp90 family members contain a highly charged linker region that joins the N-terminal with the flexible middle domain of 35kDa. This middle domain plays a crucial role in modulating ATP hydrolysis, through interaction with the N-terminal pocket. The isolated ATP-domain shows low intrinsic ATP-ase activity, which is enhanced by the cooperation of C-terminal sequence.44
10
1.4.2 ATPase cycle
The structural mechanism for the chaperone activity of Hsp90 has been likened to a „molecular clamp‟. In the absence of bound nucleotide, the N-termini of the Hsp90 homodimer maintain an open-state, facilitating the „capture‟ of client proteins. Association with ATP induces modest changes in the conformation of Hsp90 that permit a transitory interaction between the opposing N-terminal domains. This produces the closed form of Hsp90 where clamping of the substrate protein occurs (Figure 1.2). It is through this ATPase-driven cycle that Hsp90, with the assistance of several co-chaperones, induces the activation of its „clientelle‟.
Figure 1.2 ATP-dependent molecular clamp: ATP-driven molecular clamp cycle of Hsp90. C-termin domain (C), N-termin domain (N), middle domain (M).
In eukaryotic plasma, Hsp90 is considered to mediate the folding, stabilization, activation and assembly of its client proteins, including steroid receptors, protein kinases and transcription factors. Hsp90 is not capable of autonomously functioning as a protein chaperone. Instead, it serves at the core of various multiprotein complexes that incorporate other chaperones, such as Hsp70, and an assortment of co-chaperones. Three dynamic steps have been observed in this assembly process (Figure 1.3). In a typical chaperone cycle, a client protein initially binds to a Hsp70/Hsp40 which is transferred onto the ADP-bound Hsp90 via the TPR (tetratricopeptide repeat) to form an early complex. Then Hsp90 with the co-chaperone Hop binds to this early complex to form an intermediate complex,
11
while the protein Hip binds to hsp70. When ADP is replaced by ATP, Hsp90 under goes a conformational change which releases Hsp70/Hsp40 and Hop, thus allowing the ATP- dependent association of other co-chaperones, including p50, p23, CDC37, AHA1 which increases the ATPase activity of HSP90and the immunophilins (IP) to form a mature
complex.45 When the ATP-binding site of Hsp90 is occupied by competitive inhibitors, the formation of the mature complex is disrupted and the client proteins can be degraded through the ubiquitination-proteasome pathway.46,47
Figure 1.3 Hsp90 cycle: GM = geldanamycin analogue; 40 = HSP40; 70 = HSP70; IP = immunophillin; HIP = HSP70-interacting protein; HOP=HSP70/HSP90 organizing protein. Cdc37 and Aha1 are not shown.
1.5. Oncogenic Hsp90 client proteins and signaling
pathways
By the early 1990s, several groups reported the observation that Hsps in general, and Hsp90 in particular, were over-expressed in a wide variety of cancer.48 Cancer is a disease associated with genetic instability which allows cancer cells to acquire distinguishing characteristics, including self-sufficiency in growth signaling, resistance to apoptosis, insensitivity to growth inhibitory signaling, sustained angiogenesis, tissue invasion and metastasis, and limit less proliferative potential. Many signaling proteins
12
(more than 100 are known) related to these hall marks of cancer are client proteins of Hsp90. Most of these proteins play important roles in the control of cell cycle, growth and apoptosis and their dysregulated function might lead to transformation. Examples include such EGFR, HER-2, Akt, Raf-1, Cdk4, mutant p53, the estrogen and androgen receptor, mutant Raf, Bcr-Abl and Flt3 (Figure 1.4 and Table 1.3). The following section will focus on certain critical oncogenic proteins and pathways that are affected by Hsp90.
Figure 1.4 Hsp90 client proteins. Hsp90 client proteins regulate multiple signal transduction pathways that are deregulated in cancers. Hsp90 client proteins (shown in bold) include key components of the mitogenic signaling pathway that drives cell-cycle progression, as well as survival signal transduction pathways that inhibit apoptosis. Hsp90 client proteins include growth factor receptors (HER-2, IGF-1R, EGF-R and PDGF-R), signaling kinases (Akt and Raf-1), cell-cycle regulators (cdk4) and nuclear steroid receptors (AR and ER).
1.5.1. Growth Factor Receptors
EGFR and HER-2 are receptor tyrosine kinases of HER family that play critical roles in cell proliferation. Overexpression or mutation of either protein has been recorded in a variety of malignancies, including glioblastoma, breast and ovarian cancer. In the past, great efforts have been made in developing HER-2 and EGFR inhibitors (Herceptin, Tarceva, Iressa, Erbitux), but beneficial responses were limited. Studies in resistant cell lines revealed that major downstream signaling pathways, including Akt and Raf-1 pathways, were still active due to the expression of compensatory receptor tyrosine kinases or other mutation. These adaptations bypassed the suppression of the target and maintained
13
tumor growth, indicating that tumor cells can circumvent inhibition of one target and activate an alternative pathway to proliferate and survive. Hsp90 inhibitors inhibit tumor cell growth in HER-2 cell and may provide more robust and long-lasting antitumor effects by inhibiting multiple signaling pathways.49
1.5.2. Steroid Receptors
Hsp90 inhibitors also inactivate steroid receptors, such as the androgen receptor and estrogen receptor, which, when bound to their cognate ligands, translocate to the nucleus and act as transcription factors.
1.5.3. Bcr-Abl
The fusion protein Bcr-Abl, known as Philadelphia Chromosome, is expressed in approximately 95% of cases of Chronic Myeloid Leukemia (CML) and is the target of the first small molecule tyrosine kinase inhibitor, Imatinib (Gleevec). Patients become resistant to the drug due to mutations in the kinase domain. Both wild type and mutant Bcr-Abl are Hsp90 clients and its inhibition causes degradation of Bcr-Abl and suppresses tumor growth in a variety of hematopoietic tumor lines. 49
1.5.4. Flt3
FMS-like tyrosine kinase 3 (Flt3) is a receptor tyrosine kinase which plays a crucial role in several hematopoietic malignancies as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL) and myeloid lymphoblastic leukemia (MLL). It can mutate in two different points, both mutations cause constitutive Flt3 activation and are associated with poor prognosis. Even in this case both wild type and mutant proteins are sensitive to Hsp90 inhibitors which induce cell cycle arrest and apoptosis.49
14
1.5.5. Protein Kinase
Hsp90 inhibitors inactivate multiple kinases, such as Raf-1, Akt and cdk4. Inactivation of the Ras-Raf-1-Mek-ERK and phosphatidyl inositol-3 kinase-Akt pathways by Hsp90 inhibitors causes the downregulation of cyclin D1 and the functional inactivation of Cdk4, both of which are important for the G1-S cell cycle transition (Figure 4).50
PI3K/Akt Pathway
PI3K/Akt pathway has a central role in cell proliferation and survival and it has attracted a lot of attention in drug development for anticancer therapy. Akt is abnormally activated in human tumors including breast, prostate, lung, pancreatic, ovarian and colorectal carcinomas. Several studies have shown that Akt relies on Hsp90 for its stability and activity: in animal models, Hsp90 inhibitors block the growth of tumors with active Akt when administered at doses capable of suppressing Akt phosphorylation.49
Ras/Raf/MEK
The Ras/Raf/MEK pathways occupies center stage in cell proliferation and survival; mutations of Ras and Raf are the most common in human tumors (35% in melanomas and 70% in papillary thyroid tumors). Raf isoforms and its downstream effector, MEK, are both HSP90 clients, and Hsp90 inhibitors deplete both proteins from tumor cells.49
Table 1.3 Relationship between Hsp90 client proteins and hallmarks of cancer.
Hallmark of cancer Hsp90 client protein
Evasion of apoptosis Akt, Rip, p53, Survivin, Apaf-1, Bcl-2,
IGF-IR
Sustained angiogenesis VEGFR, HIF1, Akt, Fit-3,FAK, Src
Limitless replicative potential n-TERT, telomerase
Tissue invasion and metastasis c-MET, MMP2
Self-sufficiency in growth signals EGFR/Her-2, Raf, Bcr-Abl, ErbB-2, Src,
Akt, MEK
Insensitivity to anti-growth signals Plk-1,Cdk4,Cdk6, Myt-1,cyclin D
Therefore, Hsp90 inhibitors simultaneously destabilize many oncoproteins in multiple signaling pathways, suggesting that the inhibition of Hsp90 could be particularly beneficial in attacking late-stage cancer cells, which can easily circumvent the blockade of a single target or pathway. Because of the wide array of Hsp90 client proteins, Hsp90 inhibitors can
15
be used to target diverse cancers in which an Hsp90 client protein is necessary for cancer proliferation, survival or progression.
1.6. A high-affinity conformation of tumor Hsp90 confers
drug selectivity
Figure 1.5 Model for tumor selectivity of Hsp90 inhibitors and Hsp90-dependent malignant progression.
Even though Hsp90 is an abundant protein in the cell, Hsp90 inhibitor drug selectively destroys tumor cells over normal cells. Hsp90 in normal cells exists in an uncomplexed form („latent state‟) that has low affinity for Hsp90 inhibitor drugs, which accumulate poorly in normal tissues, and normal cells exhibit poor drug sensitivity. By contrast, the Hsp90 in cancer cells is involved in the active chaperoning of overexpressed oncoproteins and exists in a complexed form („activated state‟) with co-chaperone proteins (include Hsp70, Hop, p23, cdc37, immunophilins and Aha1). Complexed Hsp90 in cancer cells exhibits high-affinity binding to Hsp90 inhibitor drugs, which accumulate in tumor tissues, and tumor cells exhibit good drug sensitivity. This model predicts that the accumulation of mutant proteins in advanced cancer would further increase Hsp90 usage and make tumor cells more Hsp90 dependent. Furthermore, this model suggests that the high-affinity change of Hsp90 can be driven by the overexpression of oncoproteins, as well as by stressful conditions in normal cells.4,51-54
The Figure 1.5 schematically depicts Hsp90 and its possible interaction partners and does not represent timely complexes.
17
Box 1: Potential mechanisms of selectivity of Hsp90 inhibitors for cancer versus normal cells:
Hsp90 inhibitors cause simultaneous combinatorial depletion of oncogenic client proteins, including kinases, hormone receptors and transcription factors.
Cancer cells might be especially dependent on Hsp90 to ensure the correct folding and function of the large quantities of mutated and overexpressed oncoproteins.
Malignant cells are likely to acquire greater dependence on these cancer-causing client proteins compared with normal cells through the process of multistep oncogenesis, selection and „oncogene addiction‟.
Cancer cells might also be especially dependent on Hsp90 and other molecular chaperones to survive the hostile tumor microenvironment owing to hypoxia, nutrient deprivation, acidosis, and so on.
Effects of Hsp90 inhibitors on multiple oncogenic clients and pathways will cause antagonism of all six „hallmark traits‟ of cancer. By analogy with its role in morphological evolution, Hsp90 might act as a buffer against mutations that accumulate in cancer cells, and its inhibition might uncover synthetic lethal effects.
Some Hsp90 inhibitors, such as 17AAG, might accumulate to a great extent in cancer versus normal cells and tissues.
New evidence shows that Hsp90 is present in cancer cells in heightened multichaperone complexes with increased ATPase activity and 100-fold greater binding affinity for 17AAG compared with the largely uncomplexed and less-active form of Hsp90 that is found in normal cells.25
18
1.7 Inhibitors of Hsp90 C-terminal ATP-binding domain
1.7.1 Novobiocin and analogues
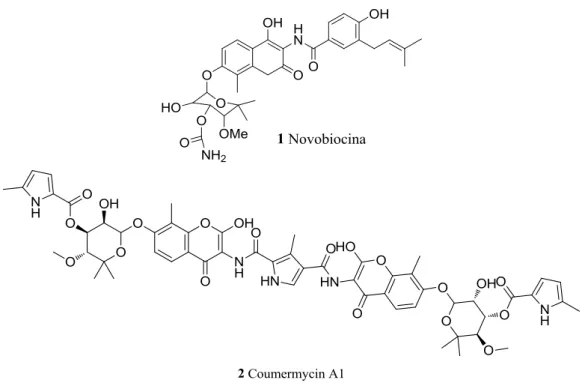
Novobiocin (1, NB) and coumermycin A1 (2) (Figure 1.6) are members of the coumermycin family of antibiotics isolated from Streptomyces strains and are well-established inhibitors of DNA gyrase and topoisomerase II.
Figure 1.6 Chemical structure of novobiocin and coumercyn A1.
They also interact with Hsp90 causing in vitro and in vivo depletion of key regulatory Hsp90-dependent kinases including v-Src and Raf-1 at 700 μM and 70 μM, respectively. These compounds inhibit HSP90 by disrupting or preventing formation of dimer required for Hsp90 function.
In order to improve the affinity for HSP90 and the potency against cancer cells, two research groups synthesized more analogues based on the coumarin scaffold.
NB derivatives A4 (3) and DHN2 (4) (Figure 1.7), both lacking the 4-hydroxyl on the coumarin moiety, showed improved IC50 values of 10 μM and 0.5 μM against SKBr3 cells,
19
Figure 1.7 Chemical structure of A4 and DHN2.
1.7.2 Other C-terminal inhibitors
Figure 1.8 Chemical structure of cisplatin and EGCG.
Cisplatin (5), a platinum-based anticancer drug known to crosslink DNA, and epigallocatechin-3-gallate (EGCG, 6), the major component of green tea, are able to bind the C-terminal domain. Cisplatin interacts in a region proximal to the C-terminal nucleotide binding site; EGCG inhibits transcription mediated by aryl hydrocarbon receptor (AhR) through binding to Hsp90.
1.8 Inhibitors of Hsp90 N-terminal ATP binding domain
Two general classes of natural product inhibitors of Hsp90 have been discovered which bind to the N-terminal ATP pocket, and are based on geldanamycin (GM) and radicicol (RD). Interestingly, each was originally isolated as an antibiotic from fermentation broths. They affect Hsp90 chaperone function in a similar manner and possess comparable biological activity.20
1.8.1 Geldanamycin and its derivatives
Figure 1.9 Chemical structure of geldanamycin.
Geldanamycin (GM, 7) is a benzoquinone ansamycin that was first isolated as an antibiotic in 1970 by scientists at Upjohn by fermentation of Streptomyces
hygroscopicus.57
The crystal structure showed that GM binds tightly to the ATP pocket of the N-terminal domain and inhibits Hsp90-mediated protein conformation/refolding, resulting in a depletion of oncogenic kinases through the proteosomal degradation of immature protein. This process subsequently down-regulates expression of many oncogenes in cancer cells.58,59 Its structure includes a benzoquinone ring fused to a macrocyclic ansa ring. The benzoquinone ring is found near the entrance of the binding pocket and the ansa ring is directed towards the bottom of the pocket. When bound to Hsp90, GM adopts a C-shaped conformation similar to that of ADP. GM shows a potency in the micromolar concentration range in vivo (Kd = 1.2 μM) that increases 50 to 100-fold in vitro.60,61
GM is a potent anticancer antibiotic but its potential clinical utility is hampered by its severe toxicity. First, it exhibits severe hepatotoxicity, which has been associated with the benzoquinone ring and imposes strict dosing limitations. Secondly, it is metabolically and chemically unstable. Also, it has very low solubility in aqueous media resulting in formulations requiring DMSO.
For this reason, a substantial effort has been made to modify its structure, generating a number of analogues in attempts to improve safety, stability, potency and water solubility.62
21
Figure 1.10 Chemical structure of geldanamycin derivatives.
The site of focus is the 17 position of the benzoquinone group to generate 17-allylamino- 17-demethoxy geldanamycin (17-AAG, 8). 17-AAG shows a better toxicity profile and was the first Hsp90 inhibitor to enter clinical trials and is now in phase II. This molecule has several limitations, such as poor solubility and limited bioavailability.54 To overcome some of these issues two more water soluble GA derivatives, 17-DMAG (17-dimethylamino-ethyl-GA, 9) and the hydroquinone of 17-AAG-prodrug (called IP-504, 10) were synthesized, which are reported to be in phase-I/Ib and phase-I/II clinical trials, respectively. In fact, studies revealed that 10 can be isolated as the hydrochloride salt, an intravenously administered small molecule.63 Protonation of the aniline nitrogen in 17-AAG hydroquinone decreases electron density in the aromatic ring, thus reducing the oxidative potential of the hydroquinone. Infinity is developing one drug candidates in its Hsp90 chaperone inhibitor program: IPI-493 (11), which is admistered orally.64
22
1.8.2 Radicicol and its derivatives
Figure 1.11 Chemical structure of radicicol.
Radicicol (12, in Figure 1.11) belongs to the macrocyclic lactone antibiotics originally extracted from the filtrate of fungus Monosporiumbonorden and it binds human Hsp90 with nanomolar affinity in vitro (Kd = 19 nM).65 Although radicicol is structurally dissimilar from benzoquinone ansamycins, it can also competitively bind to the N-terminal domain of Hsp90 to disrupt Hsp90 complex formation.66 Radicicol showed more potent antitumor activity than geldenamycins in vitro, but weaker activity in vivo, which could be explained by the fact that the presence of epoxy and α, β, , δ-unsaturated carbonyl groups reduced the stability of radicicol. In the other side, structure–activity relationship analysis indicated that radicicol possesses phenolic ring structures that may be necessary for its function.67
With the aim to improve the pharmacological properties of RD, several scaffold modifications have been analyzed.
23
Some analogues include cycloproparadicicol 13 and some radicicol oxime derivatives of general structure 14 (Figure 1.12) were designed to reduce the reactivity of the epoxide group. These results indicate that the oxime moiety plays a significant role in sustaining the stability and enhancing the biological activity of radicicoloxime analogue. All analogues show the propensity to adopt the bioactive conformation: among them Pochonin A (15) and D (16), two natural products, have an IC50 value of 80 nM and 90 nM,
respectively. In contrast to geldanamycin and its derivatives, radicicol has no hepatotoxicity, which implies that hepatotoxicity of benzoquinone ansamycins is not a common characteristic of all Hsp90inhibitors. Thus, radicicol and its analogues are promising drug candidates for further development as non-benzoquinone ansamycin Hsp90 inhibitors. Until now no compound of this class has yet entered clinical trials.68,69
1.8.3 Purines-based derivatives
Figure 1.13 Chemical structure of PU3 and PU24FCI.
The purine scaffold is another important class of Hsp90 N-terminal domain inhibitors.70 This class (PU-class) was empirically designed by Chiosis and coworkers following the idea to mimic the unique shape adopted by ATP when bound to the N-terminal nucleotide pocket of Hsp90: the adenine ring was envisioned to mimic the adenine ring of ATP, while the benzene moiety was decorated to capture the same network of hydrogen bonds in which the quinone ring of geldanamycin is involved.
The first synthesized derivative of this class was PU3 (17) with an affinity of 10-20 μM and antiproliferative activity against several cancer lines. The weakly active PU3 was an important starting point for the development of clinically useful Hsp90 inhibitors and the purine scaffold was adopted for further optimisation by several investigators. Further
24
efforts focused at improving the potency of this agent, have led to the synthesis of several compounds with improved activity in both biochemical and cellular assays.
Initial optimisation led to the identification of PU24FCl (18), a selective inhibitor of tumor Hsp90 that exhibits antitumor activities in both in vitro and in vivo models of cancer. The biological effects of PU24FCl are demonstrated in the 2-6 µM concentration range.71 The synthesis of subsequent structure-based optimization was resulted in the discovery of the potent, water-soluble inhibitor PU-H71 (19 in Figure 1.14).
Figure 1.14 Chemical structure of PU-H71 and CUDC-305.
Until now PU-H71 remains the most active derivative in the PU series and it is scheduled to enter phase I clinical trial.
Recently Bao et al. described compound CUDC-305 (20) similar to arylsulfanyl adenine; this molecule has IC50 of 100 nM and blocks proliferation of a panel of cancer cell lines;
moreover, it has good pharmacological properties as oral bioavailability, blood-brain barrier penetration, and tumor retention.72
A series of purine with improved physicochemical properties was discovered by Kasibhatla et al. by rearrangement of substituents around the purine ring. They shift the aryl binding moiety from the C-8 to the N-9 position and the NH2 group from the 6 to the 2
position so as to re-establish the overall six-bond distance between the 6-NH2 group and
25
Figure 1.15 Chemical structure of CNF-2024/BIIB021 and derivatives.
Compounds of this structural series (in particular 21 and 22 in Figure 1.15) show significant binding selectivity, preferring interaction with the activated form of the Hsp90 complex, which is reflected in increased tumor cell retention and more effective tumor cell killing.75,76 CNF2024/BIIB021 (21) was advanced into phase I-II clinical trials.77
1.8.4 o-Aminobenzoic acid derivatives and resorcinol derivatives
Figure 1.16 Chemical structure of SNX-2112 and SNX-5422.
New classes of Hsp90 inhibitors unrelated to any previously known scaffold were found through screening of focused compound libraries against sets of ATP-binding proteins. Huang and coworkers identified a novel class of indol- 4-one and indazol-4-one derived 2- aminobenzamides that potently inhibit Hsp90. The crystal structure of the complexes between compound of this series and Hsp90 shows that these molecules mimic the carbamate/Asp93/water molecules interactions established by GA, as the benzamide group overlays the carbamate of GA and the rest of the molecule closely maps other parts of the ansamycin ring. The best compound of this series, SNX-2112 (23), show a high affinity (Kd= 16 nM) and its prodrug SNX-5422 (recently acquired by Pfizer
PF-26
04929113) (24) is orally bioavailable, efficacious in a broad range of cancer cell lines and entered phase I clinical trials.78,79
Figure 1.17 Chemical structure of benzoisoxazole derivatives.
The benzisoxazole compound 25 identified (Figure 1.17), using high-throughput screening and optimisation, by Wyeth. It has high binding affinity (IC50 = 0.03 μM) and
submicromolar IC50 values against a panel of cancer lines.
Figure 1.18 Chemical structure of resorcinols derivatives.
Other resorcinols, AT13387 (26) by Astex and ganetespib (27) by Synta Pharmaceuticals Corp. (chemical structure not yet disclosed), entered recently into clinical trials.81
Other interesting molecules are AICAR (28), which destabilizes multiple client proteins in vivo and shows antiproliferative activity in multiple tumor cell lines, 2- naphthol compounds 29, 30 and tetrahydrobenzopyrimidine molecule 31 with submicromolar inhibitory activity.78
27
Figure 1.19 Chemical structure of other N-terminal domain inhibitors.
1.8.5 Pyrazoles and isoxazoles derivatives
Figure 1.20 Chemical structure of Pyrazole derivatives.
This class of synthetic Hsp90 inhibitors was identified by high-throughput screening of a library of 50,000 compounds.81 Wokman and co-workers identified CCT018159 (32) with an IC50 of 7.1 μM in yeast ATPase assay and antiproliferative
activity against HCT116 human colon cancer cells.82 The structure-based elaboration of this hit resulted in a series of active analougues. Further, replacement of the chlorine of the resorcinol ring in CCT018159 by an ethyl group in CCT072440 (33) results in an ATPase activity 2- to 3-fold more potent than 32 (Figure 1.20).83
After extensive optimization, Vernalis found VER49009 (34) with in vitro potency comparable to 17-AAG (IC50 = 0.14 μM).
28
Figure 1.21 Chemical structure of VER49009.
Vernalis strategy to optimize the in vivo activity in diaripirazole series was focused on the structure-activity relationships (SAR) in three distinct areas of the molecule and was guided by structural information of ligands (such as VER-49009 in Figure1.21) bound to the ATP binding site of Hsp90. Crystallographic data shows that this family of compounds binds to the ATP pocket in the N-terminal domain similar to RD.84 The resorcinol groups are clearly central to the binding mode of the compound, particularly the 2‟-hydroxyl, which makes a hydrogen bond to the residue Asp93, a residue, which has been previously identified to be critical for ATP binding. To evaluate the importance of the resorcinol OH moieties in the binding of this class of inhibitor, several O-methylated analogues of this class were synthesized. As demonstrated O-methylation caused a large drop in binding affinity compound, reduced potency and cellular activity and thus demonstrated no advantage over the free resorcinol.
Additional interactions between the adenine ring of ATP and protein residues of the binding pocket are bridged by several water molecules. Several of those water interacts with Asp93 and the pyrazole N2 of the inhibitor. This water is part of a network with Asp93 and two other waters that are seen in all reported crystal structures of Hsp90.85 Beside the importance of the key interaction of the resorcinol with Asp93 no less important is the hydrogen bond from the 5-amide substituent to Gly97.86 Crystallographic data shows that the amide group at C5 of the pyrazole ring forms a salt bridge to Lys58 as well as hydrogen bonds to two nearby water molecules (Figure 1.22).
29
Figura 1.22 X-ray structure of VER-49009 (Figure A) and VER50589 (Figura B) bound to the ATP binding site of human Hsp90α. H-bonds are shown as dotted blu line; green: protein residues involved in polar interactions; cyan: protein residues involved in non-polar interactions; blue ball: molecole of water.
The high complementarities and tight packing between Hsp90 and the inhibitor in this area leave room only for small modifications of the inhibitor‟s core, but a wide range of substituents can be placed off of the pyrazole ring and the 5‟-position of the resorcinol.87
Vernalis focuses his work on three main areas:
1) modification of the central pyrazole heterocycle,
2) the optimization of the 5‟substituent on the resorcinol ring,
3) incorporation of a solubilizing group on the 4-arylpyrazole substituents.
1) Modification of the central pyrazole heterocycle
As shown in Figure 22 A and B, examination of the binding for the pyrazole ring in ligands such as 34 demonstrated that the nitrogen adjacent to the resorcinol (N1) is involved in hydrogen bonding with the key Asp93/water network at the base of the pocket as an H-bond acceptor, but pyrazoles can exist in tautomeric forms, and when N1 is protonated, this represents a form which is unlikely to bind well to Hsp90. Further, the interaction between N2 and the amide backbone carbonyl of Gly97 is not in the ideal geometry. For this reason optimisations led to a series of isoxazole-resorcinol analogues 35 (Figure 1.23) eliminating the possibility of different tautomeric forms of the pyrazole ring
30
and pyrazole was substituted with an alternative hydrogen-bond acceptor such as oxygen of an isoxazole. Isoxazoles and pyrazoles differ in that the former does not bear hydrogen on a hetero atom and can therefore not be substituted. As a consequence, the isoxazole is also smaller in size and should still be accommodated in the binding site. In addition, there is no possibility of the existence of isomeric forms that can exist with the pyrazole.
Figure 1.23 Chemical structure of VER50589.
The cytotoxic activity of VER-50589 was higher than VER-49009 on all cell lines tested, in Table1.4 we can observe the values of IG50:
Tabella 1.4 Values of IG50 of VER-49009 and VER-50589 in different
cells line.
Cell type Cell line IG50 (nmol/L)
VER-49009 VER-50589 Melanoma SKMEL. 2 SKMEL.5 1.093± 111.2 163.3 ± 14.5 62.0 ± 4.3 125 ± 22.6 Colon cancer HCT116 BEneg BE2 HT29 357.0± 0.003 372.5 ± 29.5 422.5 ± 46.2 4.600.0±611.0 115± 0.005 36.7 ± 2.7 35.0± 3.5 32.7±0.3 Ovaric cancer CH1 376.7±26.0 32.7±0.3 Brain cancer MB-231 BT20 570.0±0.04 550 ±0.09 58.8±6.4 59.0 ±12.7
Endothelial cell HUVEC 444.0±91.9 19.0±2.4
Unlike 17-AAG, but as with CCT018159, cellular potency of these analogues was independent of P-glycoprotein expression.88 P-glycoprotein (P-gp) is a plasma membrane protein which acts as a localized drug transport mechanism, actively exporting drugs out of the cell. The effects of P-gp on the distribution, metabolism and excretion of drugs in the body is great. P-gp activity, for example, decreases the intracellular concentration of cancer drugs, enabling resistance to develop to them.
31
2) The optimization of the 5’substituent on the resorcinol ring
The substituents in this area have the potential to induce a conformational change in the protein by forming a helix between residues Ile104 and Ala111. In fact, there is considerable plasticity in this region of the structure, which is at the entrance to the ATP binding site. Some of the residues seen as important for binding to ligands are in this loop.89 This structural change creates a lipophilic pocket in the ATP binding site of the Hsp90 protein. As previously demonstrated the removal of ethyl or chloro from this position resulted in a 20-fold decrease in binding affinity for ligands closely related to 34. This loss in potency is rationalized by structural information showing that the ethyl and chloro protrudes into a hydrophobic pocket created by residues Phe138, Leu107, Val150, Met98, Val186, and Leu103. The size of the hydrophobic pocket is large enough to accommodate a bulkier substitution at the C5 position of the dihydroxyphenyl ring.90 Replacement of the chlorine of the resorcinol ring in 34 or 35 by an isopropyl group results in an additional hydrophobic interaction with Leu107 in the flexible lipophilic pocket. Analogues with an 5‟ isopropyl group demonstrated excellent potency in cell growth inhibition assays.87
3) Incorporation of a solubilizing group on the 4-arylpyrazole substituents.
Subsequently introduction of a solubilizing group led to a series of potent Hsp90 inhibitors with good pharmacokinetic properties. Further hydrophobic interactions are also seen with Thr109 and Gly135 when the methoxy group of VER-49009 or VER-50589 is replaced with the morpholino moiety of VER-52296, now acquired by Novartis NVP-AUY922 (Figure 1.24), which showed GI50 = 9 nM in antiproliferation assays against a
panel of human cancer cell lines and has now entered phase II clinical trials.91
32
Table 1.5 shown a short summary of the characteristics of Hsp90 inhibitors described
above.
Table 1.5 Hsp90-binding drugs: many classes of natural, semisynthetic and
synthetic inhibitors of Hsp 90 have been discovered and some of them have been exploited in clinical trials for different types of tumours.
Binding site Chemical
class Inhibitors Properties
N-terminal ATP-binding pocket Benzoquinone ansamycin GA 17AAG 17DMGA IPI-504 IPI-493 Natural Semisynthetic Semisynthetic Semisynthetic Semisynthetic N-terminal
ATP-binding pocket Macrolide
Radicicol Cycloproparadicol Pochonin A Pochonin D Natural Semisynthetic Semisynthetic Semisynthetic N-terminal ATP-binding pocket Purine Scaffold PU3 PU24FC1 PU-H71 CUDC-305 CNF-2024/BIIB021 Synthetic Synthetic Synthetic Synthetic Synthetic N-terminal ATP-binding pocket Pyrazole Isoxazole CCT018159 CCT072440 VER49009 VER50589 NVP-AUY922 Synthetic Synthetic Synthetic Synthetic Synthetic C-terminal Coumarin Novobiocin Coumermycin A1 A4 DHN2 Natural Natural Synthetic Synthetic
33
Chapter 2
Aim of the thesis
As previously described, the 4,5-diaryl-isoxazole class represents the most investigated class of Hsp90 inhibitors. Compounds belonging to this class show biological activity in the micromolar range and high selectivity for tumor cells versus normal cells. However, to date, no Hsp90 inhibitors in clinical trials meet all of the requirements of safety and stability. Some of the drugs under clinical investigation have showed toxicity toward liver, eyes, stomach-intestine, and heart.92,93
In this work, we identify NVP-AUY922, belonging to the class of 4,5-diaryl-isoxazoles and currently in Phase II clinical trials (Figure 1.25), as lead compound. When a molecule is identified as a "lead", the aim of the medicinal chemist is the optimization of the compound by modifying the structure to:
1. improve pharmacological properties (such as affinity and efficacy);
2. modify the physicochemical properties (such as solubility, permeability and chemical stability);
3. reduce, if necessary, any toxicity or side effects;
4. optimize pharmacokinetic characteristics (such as bioavailability, metabolic stability and duration of action).
Therefore, the aim of this work is the design and synthesis of more potent and selective Hsp90 inhibitors. It is important to remember that the project research has been developed in collaboration with pharmaceutical industry, Sigma Tau. Therefore the importance to make patentable the products synthetized.
Our attention was focused mainly on the C-4 position of the isoxazole scaffold, where we found, contrary to the literature, that a phenyl moiety at C-4 position is not a prerequisite for the activity on Hsp90.94
In fact, during our preliminary studies we found that the derivative with a bromine in C-4 of the isoxazole maintain the binding activity (ability to bind Hsp90 (FP): IC50 = 0,15 µM
34
we focused our work in a detailed structural investigation on a new class of 3,4-isoxazolediamides (Figure 2.1) where we found that compounds with a nitrogen atom directly attached to the heterocycle ring possess in vitro Hsp90 inhibitory properties comparable to the 4,5-diaryl-isoxazole derivatives.
Figure 2.1 General structure of new Hsp90 inhibitors used in these study.
In particular:
the feasibility of the project was initially investigated by preparing a small explorative series of C-4 amides bearing a chloro-resorcinol residue at the C-5 position. Meanwhile the amides at C-3 and C-4 of the isoxazole ring were varied as shown in Figure 2.1. The 5-chloro substitution was chosen at the beginning for its ease of synthesis. The synthetic route for these compounds allowed to obtain the final products faster and in a greater amount; moreover, 5-unsubstituted resorcinols are known to be less active with respect to the substituted ones;95
then, we moved to explore the potential of the new scaffold investigating a lot of additional substitution at the C-4 amide portion. Given that substitution of the chlorine atom at the resorcinol moiety with an isopropyl group was previously documented to improve cytotoxic activity of the 4,5-diarylisoxazole, such a type of substitution has been also utilized by us for the new compounds;
furthermore, a C-4 amino series was synthetized to better substantiate our structure activity investigation. In fact, amide derivatives are more potent than the corresponding amine derivatives.
In continuation of our efforts in search of potential antitumor agents,94 in order to obtain new Hsp90 inhibitors, different molecules have been specifically designed to interact with the whole binding pocket of Hsp90 (general scaffolds are shown in Table
1.7). In particular, docking experiments in collaboration with the pharmaceutical industry
35
results of these experiments are expressed in "G-Score", which is a measure of the free energy of the binding site-ligand complex. From the analysis of these sets of molecules compound 127 reported the highest value of G-score (Table 2.1).
Table 2.1 Data of G-score of structures proposed.
Compounds R GScore 123 -9.31 124 -8.4 125 -11.2 126 -10.74 127 -12.26 128 -11.2 129 -10.91 130 -10.41 131 -11.36
Based on the predicted activity, we planned to synthesize a series of derivatives of compound 127 as novel Hsp90 inhibitors.
36
Figure 2.2 New 4,5,6,7-tetrahydro-isoxazolo-[4,5-c]pyridine scaffold.
As described in this chapter, compounds that contain an isoxazole nucleus (Figure 1.18,
1.24 and 1.25), have shown potent and selective inhibition of Hsp90.87,94,96,97 The presence of this heterocyclic moiety seem to play an important role in the interaction of this class of compounds with the ATP-binding site of Hsp90.98 So we became interested in the synthesis of new hits containing the 4,5,6,7-tetrahydro-isoxazolo-[4,5-c]pyridine scaffold. In particular we focused on the synthesis of compounds bearing in N-5 resorcinol moiety and different ester/amide groups in C-3 due to their potential role in the interaction with the Hsp90 protein (Figure 2.2).
37
Chapter 3
Chemistry
Contents
3.1 Synthesis of 3,4-isoxazolediamides scaffold
3.1.1 Synthesis of chloro-resorcinol derivatives 3.1.2 Modification at C-3
3.1.3 Synthesis of isopropyl-resorcinol derivatives 3.1.4 Modification at C-4
3.1.5 Synthesis of 4-aminoisoxazole
3.2 Attempts to synthesise 4,5,6,7-tetrahydro-isoxazole-[4,5-c]pyridine scaffold 3.3 Synthesis 4,5,6,7-tetrahydro-isoxazlo-[4,5-c]pyridine scaffold
38
3.1 Synthesis of 3,4-isoxazolediamides scaffold
3.1.1 Synthesis of chloro-resorcinol derivatives
The route used for the synthesis of 44a is shown in Scheme 3.1 Acetophenone 40 was obtained by regioselective Friedel-Crafts acylation of the commercially available 4-chlororesorcinol 39. Both phenolic functions were protected with benzyl groups, generating 41 that was reacted with diethyl oxalate to afford keto-enol ester 41. The isoxazole heterocycle 43 was obtained by reaction of 42 with hydroxylamina hydrochloride in ethanol. Finally, reaction of the ethyl ester moiety of 43 with ethylamine provided the desired ethylamide 44a.
Scheme 3.1 Synthesis of chloro-resorcinol intermediate 44aa
aReagents and conditions: (a) AcOH, BF
3 xOEt2; (b) BnBr, K2CO3, MeCN; (c) (CO2Et)2, EtO-Na+, EtOH ;
39
3.1.2 Modification at C-3
Compounds 44b,c were prepared starting from the appropriate ester derivative with the following sequence of reactions (a) hydrolysis of ester moiety, (b) activation of resulting carboxylic acid, and (c) reaction with the proper commercially amines (Scheme
3.2).
Scheme 3.2 Synthesis of chloro-resorcinol intermediate 44b,ca
aReagents and conditions: (a) LiOH, H
2O, MeOH, 50-60°C; AcOH, BF3xOEt2; (b) i: SOCl2, 80°C; ii: NHR,
TEA, 0°C.
3.1.3 Synthesis of isopropyl-resorcinol derivatives
For the synthesis of the isopropyl-substituted intermediate 44d was used the same route outlined in Scheme 3.3
Starting from the commercially available 2,4-dihydroxyacetophenone, this was protected with benzyl groups to give compound 48. A Wittig reaction, followed by hydrogenation generate compound 50. The reduction of the styrene functionality in 50 caused concomitant debenzylation. So 51 was obtained by Friedel-Crafts acylation of the isopropyl derivatives 50. The reaction occurs in a regioselective way only in presence of the free phenolic function. Then, the resorcinolic derivative was protected by the same route as that used for the intermediate 48. The intermediate 53 was obtained by reaction 52 with diethyl oxalate that was reacted with hydroxylamina hydrochloride in ethanol to give
54. Finally, reaction of the ethyl ester moiety of 54 with ethylamine provided the desired
40
Scheme 3.3 Synthesis of isopropyl-resorcinol intermediate 44da
aReagents and conditions: (a) BnBr, K
2CO3, MeCN; (b) CH3P+(Ph)3Br-, NaH, toluene; (c) Pd/C, H2; (d)
AcOH, BF3xOEt2; (e) BnBr, K2CO3, MeCN; (f) (CO2Et)2, EtO-Na+, EtOH; (g) NH2OHxHCl, EtOH; (h)
EtNH2/MEOH, EtOH.
3.1.4 Modification at C-4
The intermediates 44a-d were used for the preparation of amides 57-116 (see
Tables 4.1, 4.2, 4.3) as shown in Scheme 3.4. The 4-nitroisoxazoles 55a-d were
synthesized by chemoselective nitration of the isoxazoles 44a-d using concentrated nitric acid and acetic anhydride. Reduction of the nitro compounds 55a-d to give 56a-d was done using zinc powder and ammonium chloride in a mixture of water and tetrahydrofuran as solvents. Reaction of the amines 56a-d with the proper commercially available acyl chloride gave the corresponding dibenzylated amides that, after deprotection of phenolic groups with boron trichloride, furnished the desired amides 57-116.