UNIVERSITÀ DEGLI STUDI DI GENOVA
SCUOLA IN SCIENZE MEDICHE E FARMACEUTICHE CORSO DI
LAUREA MAGISTRALE IN MEDICINA E CHIRURGIA
“CRITERI PER IL TRATTAMENTO CHIRURGICO
ORTOPEDICO DEI SARCOMI OSSEI NELLA PELVI
NEL PAZIENTE IN ETÀ EVOLUTIVA”
Relatore: Chiar.mo Prof. Lamberto FELLI
Correlatore: Prof. Silvio BOERO
Candidato: Roberto FACCHI
Mtr. 4140456
ANNO ACCADEMICO 2019/2020
Dedico questo elaborato ai miei amati nonni Mario, Elisabetta e Livia; e al nonno Natale che sicuramente starà sorridendo fiero ed orgoglioso di questo mio percorso.
INDICE
INTRODUZIONE ... 1
CAPITOLO 1 – TUMORI OSSEI MALIGNI PRIMITIVI DELL’ETÀ EVOLUTIVA ... 3
1.1 – CRITERI DI STADIAZIONE ... 4
1.1.1 – IL SISTEMA DI STADIAZIONE CHIRURGICO MSTS ... 5
1.2 - OSTEOSARCOMA ... 9
1.2.1 – EPIDEMIOLOGIA ... 10
1.2.2 – EZIOLOGIA E GENETICA ... 11
1.2.3 – SEGNI CLINICI E SINTOMI ... 12
1.2.4 – DIAGNOSI STRUMENTALE E DI LABORATORIO ... 13
1.2.5 – ANATOMIA PATOLOGICA ... 15
1.2.6 – FATTORI PROGNOSTICI ... 15
1.3 – SARCOMA DI EWING ... 17
1.3.1 - EPIDEMIOLOGIA ... 17
1.3.2 – SEGNI CLINICI E SINTOMI ... 18
1.3.3 – DIAGNOSI STRUMENTALE E DI LABORATORIO ... 18
1.3.4 – ANATOMIA PATOLOGICA ... 20
1.3.5 – FATTORI PROGNOSTICI ... 20
CAPITOLO 2 – ITER DIAGNOSTICO MULTIDISCIPLINARE E TECNICHE BIOPTICHE NEI SARCOMI DELLA PELVI ... 22
2.1 - VALUTAZIONE CLINICA E RADIOLOGICA INIZIALE ... 22
2.2 - INDICAZIONI ALLA BIOPSIA E TECNICHE DI SVOLGIMENTO ... 24
CAPITOLO 3 – INDICAZIONI PER IL TRATTAMENTO DEI SARCOMI DELL’OSSO ... 27
3.1 – OSTEOSARCOMA ... 28
3.1.1– LA TERAPIA MEDICA ... 29
3.1.2– LA TERAPIA DELLE METASTASI ... 32
3.2 – SARCOMA DI EWING ... 34
3.2.1- LA TERAPIA MEDICA ... 34
CAPITOLO 4 – LA TERAPIA CHIRURGIA ... 37
4.1 - PIANIFICAZIONE PREOPERATORIA E RIVALUTAZIONE TRAMITE IMMAGINI RADIOGRAFICHE ... 37
4.2 – LE PROCEDURE CHIRURGICHE ... 39
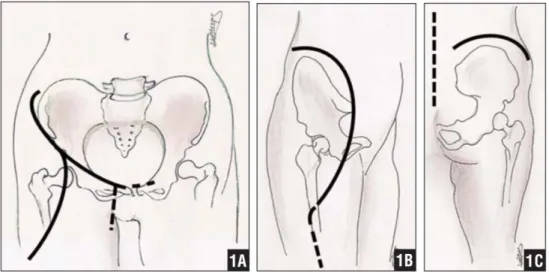
4.2.1 – EMIPELVECTOMIA ESTERNA ... 39
4.2.2 – EMIPELVECOTMIA INTERNA: TIPOLOGIE DI RESEZIONE DELLA PELVI ... 42
4.3 - RESEZIONE VERSUS AMPUTAZIONE ... 54
4.4 - COMPLICANZE OPERATORIE E POSTOPERATORIE ... 55
4.4.1 – COMPLICANZE VASCOLARI E VISCERALI ... 56
4.4.2 – COMPLICANZE INFETTIVE ... 58
4.4.3 – COMPLICANZE NERVOSE ... 59
4.4.4 – COMPLICANZE MECCANICHE ... 60
CAPITOLO 5 – LO STUDIO CLINICO ... 62
5.1 – INTRODUZIONE E SCOPO DELLO STUDIO ... 62
5.2 – MATERIALI E METODI ... 64 5.3 – RISULTATI ... 68 5.4 – DISCUSSIONE ... 72 5.5 – CONCLUSIONI ... 74 BIBLIOGRAFIA ... 76 RINGRAZIAMENTI ... 86
1
INTRODUZIONE
Il mondo dell’ortopedia pediatrica affronta quotidianamente sfide ricorrenti, cercando di restare al passo con la scienza moderna, che richiede maggior qualità e miglior risultati. Il significato della parola “ortopedico” deriva dalle parole greche Orthos (diritto) e Paidòs (bambino). Il legame vincolante tra questa disciplina e la salvaguardia della vita dei piccoli pazienti, è quindi imprescindibile. La citazione di Ippocrate “la vita è così breve,
il mestiere così tanto da imparare” racchiude l’essenza della vita dell’ortopedico
pediatrico. La genetica e l’istologia, legate alla crescita biologica del bambino, ci spingono verso il futuro aprendo nuove porte per la ricerca e per una chirurgia sempre più tecnologica, rendendo questa specialità particolarmente dinamica [1].
Dall'inizio degli anni '70 sono stati compiuti progressi sostanziali nel trattamento dei tumori muscoloscheletrici, con miglioramenti nelle capacità chirurgiche, radiologiche, chemio e radioterapiche [2]. I sarcomi ossei primari della pelvi rappresentano dal 5% al 10% di tutti i tumori ossei maligni, i più comuni dei quali sono condrosarcomi, nell’adulto, e i sarcomi di Ewing e osteosarcomi, nell’età evolutiva. La prognosi e la sopravvivenza dei pazienti con sarcomi ossei in questa posizione sono molto meno favorevoli rispetto ai pazienti con tumori agli arti [3].
La resezione dei tumori ossei del bacino è una procedura estremamente complessa per l’ortopedico oncologo. Dalla sua prima descrizione da parte di Enneking e Dunham, l’emipelvectomia interna ha ampiamente sostituito l’amputazione del quarto posteriore (emipelvectomia esterna) come procedura per i sarcomi ossei pelvici. Le difficoltà nella resezione, nella ricostruzione e nella riabilitazione sono strettamente correlate all’estensione del tumore. Varie tecniche ricostruttive, come la ricostruzione biologica (mediante alloinnesti o autotrapianti da tibia, perone o cresta iliaca), la trasposizione dell'anca, la pseudo-artrodesi ischiofemorale, iliosacrale o ileofemorale; la procedura Girdlestone modificata e la ricostruzione endoprotesica, sono state proposte per stabilizzare l’anca e garantire l’outcome funzionale migliore [4].
2
Tuttavia, in tutti questi metodi ricostruttivi, sono stati riportati tassi significativi di complicanze precoci e tardive. Le complicanze precoci, come infezioni e lussazioni, potrebbero ritardare la chemioterapia adiuvante, con possibili effetti negativi sulla prognosi del paziente. Le complicanze tardive richiedono spesso una revisione chirurgica complessa con il rischio di compromettere ulteriormente la capacità funzionale residua [5].
Questo elaborato si pone l’obiettivo di esaminare indicazioni, tecniche, limitazioni, pro e contro e complicazioni delle diverse opzioni di resezione e ricostruzione dell’anello pelvico, nel contesto di un settore in costante crescita come è quello della chirurgia conservativa dell’arto. La rarità di questo quadro patologico ha reso difficile reperire una serie di pazienti soddisfacente per lo studio. Ciononostante, è stato possibile individuare dalla letteratura un pool di 34 casi, ai quali sono stati aggiunti 2 pazienti afferenti al reparto di Ortopedia e Traumatologia dell’Ospedale Pediatrico Giannina Gaslini di Genova, andando a costituire la popolazione sperimentale. Il risultato di questa analisi retrospettiva è frutto delle nozioni acquisite durante il percorso di studio, dall’approfondimento personale della materia e dalla insostituibile esperienza clinica dei medici del reparto di Ortopedia e Traumatologia del Gaslini.
3
CAPITOLO 1 – TUMORI OSSEI MALIGNI
PRIMITIVI DELL’ETÀ EVOLUTIVA
I tumori primitivi dell’osso sono rari e costituiscono meno dello 0,2% delle neoplasie maligne registrate nella banca dati EUROCARE (European Cancer Registry based study on survival and care of cancer patients). A causa della loro complessità per la presentazione clinica, radiologica ed istologica, i tumori del sistema scheletrico rappresentano un’entità patologica di difficile approccio. Le nuove opzioni terapeutiche cercano di integrare maggiormente radioterapia, chemioterapia e chirurgia, dando vita a schemi di trattamento personalizzati con lo scopo di assicurare a ciascun paziente il miglior outcome possibile [6].
I differenti sottotipi di tumore osseo mostrano valori diversi di incidenza, non superando mai il tasso di 0,3 casi ogni 100'000 abitanti. Sono circa 10 volte meno comuni dei tumori dei tessuti molli e la prevalenza di neoplasie secondarie dell’osso (metastasi scheletriche) è circa 25 volte superiore rispetto ai sarcomi primitivi [7].
Secondo i dati italiani, i sarcomi ossei rappresentano 3-4% delle neoplasie infantili e dell’età adolescenziale, con un’incidenza annua pari a 0,8 nuovi casi ogni 100'000 abitanti. L’incidenza specifica per età ha una distribuzione bimodale con due picchi, uno nell’intervallo 10-19 anni e l’altro nei pazienti con età superiore ai 65 anni. Dei tre sottotipi più frequenti, l’osteosarcoma ha un picco tra i 10 e 25 anni mentre il sarcoma di Ewing presenta una curva con andamento bimodale, 3-6 anni e 15-25 anni. Il condrosarcoma, invece, colpisce prevalentemente i pazienti con età superiore ai 65 anni [8].
I sottotipi istologici più frequente diagnosticati sono il condrosarcoma (30% negli uomini e 29% nelle donne), l’osteosarcoma (17% negli uomini e 16% nelle donne), il sarcoma di Ewing (14% sia negli uomini che nelle donne) ed il condroma (8% negli uomini e 5% nelle donne) [9]. Tra gli istotipi rari dei sarcomi ossei (<5%) citiamo: il cordoma, il fibrosarcoma, il sarcoma pleomorfo indifferenziato (precedentemente noto come
4
istiocitoma fibroso maligno dell’osso) e il fibroma dell’osso. L’adamantinoma e i tumori vascolari maligni primitivi dell’osso sono tumori ancora più rari (<1%) [8, 10].
1.1 – CRITERI DI STADIAZIONE
Dalla sua organizzazione nel 1959, l'American Joint Committee on Cancer (AJCC) si è assunta la responsabilità di sviluppare sistemi di stadiazione clinicamente utili per molti tipi di cancro. L'intento è di designare "lo stadio di un cancro, in vari momenti nel tempo, e il correlato decorso naturale di questo particolare tipo di cancro", allo scopo di “fornire un modo in cui queste informazioni possano essere prontamente comunicate ad altri, per decidere adeguatamente il trattamento e per indirizzare la prognosi”. A tal fine, il sistema TNM, dove T rappresenta l'estensione locale della malattia del tumore primario, N l'estensione nodale e M l’estensione metastatica, insieme con l'estensione anatomica, l'analisi istopatologica e il grado del tumore sono stati considerati come fattori determinanti. Il sistema AJCC per i tumori ossei è quindi basato su quattro importanti fattori rappresentati dalle lettere T, N, M e G (grado istologico) [11]. A ciascuna lettera è poi assegnato un numero che ha il compito di descrivere il grado di progressione della patologia tumorale. Una volta associato il corrispondente codice TxNxMxGx, al tumore può essere assegnato lo stadio corrispondente, con lo stadio IA che corrisponde alla neoplasia più localizzata, fino allo stadio IVC (Tabella 1.1) che corrisponde a quella più aggressiva e avanzata [12].
Un altro sistema di stadiazione chirurgica per i sarcomi muscolo-scheletrici, definito come sistema MSTS (Musculoskeletal Tumor Society) (Tabella 1.2), è più logicamente completo e si basa sulla valutazione del grado chirurgico (G), dell'estensione locale (T) e della presenza o assenza di metastasi regionali o distanti (M). I sarcomi per i quali questo sistema è stato progettato sono quelli derivanti dal tessuto connettivo mesenchimale del sistema muscolo-scheletrico [11].
5 Tabella 1.1. Classificazione AJCC
1.1.1 – IL SISTEMA DI STADIAZIONE CHIRURGICO MSTS
Un sistema di stadiazione chirurgico per il sarcoma dovrebbe:1. Incorporare i fattori prognostici più significativi in un sistema che descriva i gradi progressivi di rischio a cui è soggetto un paziente;
2. Delineare fasi progressive della malattia che hanno implicazioni specifiche per la gestione chirurgica;
3. Fornire linee guida per l'uso di terapie aggiuntive;
Il sistema MSTS fu organizzato da W. Enneking e rappresenta, ancora oggi, il modello di riferimento globale per la stadiazione dei sarcomi ossei.
6
Grado chirurgico (G)
Dal punto di vista della pianificazione chirurgica, le neoplasie di qualsiasi istogenesi sono suddivise in due gradi: basso (G1) e alto (G2). La maggior parte delle lesioni di basso grado può essere gestita con procedure relativamente conservative, mentre quelle di alto grado richiedono procedure più aggressive per raggiungere l'obiettivo primario, ossia una resezione completa della neoplasia e il suo controllo locale. In generale, le lesioni di basso grado corrispondono agli stadi I o II di Broder e hanno un basso rischio di metastasi (<25%). Istologicamente sono ben differenziati, hanno poche mitosi e moderata atipia citologica. Quando si verificano nell'osso, c'è una tendenza alla circoscrizione da parte del nuovo osso reattivo. Le lesioni di alto grado (III e IV di Broder) hanno un'incidenza significativamente maggiore di metastasi. Sono caratterizzati da una scarsa differenziazione, alto tasso mitotico, necrosi e invasione microvascolare.
Il grado chirurgico può differire leggermente dal grado puramente istologico in considerazione delle caratteristiche cliniche e radiografiche. Ciascuna lesione viene, infine, valutata in base alle proprie caratteristiche clinico-patologiche [11].
L’estensione locale (T)
Proprio come il grado chirurgico, l’estensione locale è una misura dell'aggressività biologica complessiva di una lesione e indica quale tipo di margine chirurgico è appropriato. Indica, inoltre, come sia più probabile ottenere gli obiettivi della procedura chirurgica [15, 16] o anche se il margine desiderato possa essere raggiunto. Il fattore
Stage
Tumor
Metastases
Grade
IA
T1
M0
G1
IB
T2
M0
G1
IIA
T1
M0
G2
IIB
T2
M0
G2
III
T1 or T2
M1
G1 or G2
Tabella 1.2. Classificazione MSTS7
principale nel determinare come viene realizzato un margine chirurgico è se la lesione si trova all'interno di un compartimento anatomico ben delineato o se si sta infiltrando in modo diffuso in piani e spazi scarsamente delimitati. Pertanto, i due stadi sono suddivisi a seconda che la lesione sia intracompartimentale (A) o extra-compartimentale (B). Sia la dimensione della lesione che la sua distanza fisica dalle strutture vitali sono correlate alla compartimentalizzazione, ma non sono determinanti nella pianificazione chirurgica [15]. Sebbene le lesioni più grandi abbiano maggiori probabilità di diventare extracompartimentali, vi sono sia grandi lesioni intracompartimentali sia piccole lesioni extracompartimentali. Allo stesso modo, una lesione può essere separata solo di pochi millimetri da un nervo o un vaso maggiore e tuttavia essere contenuta da un setto fasciale che fornisce un adeguato piano di dissezione, senza sacrificare le strutture adiacenti. Poiché i micronoduli satellitari si trovano abitualmente nelle zone pseudocapsulari e reattive, per tutti i sarcomi, queste zone devono essere considerate parte integrante della lesione. Il fatto che la lesione e la sua reazione siano contenute o meno all'interno di un compartimento anatomico ben definito indica più accuratamente la fattibilità di una procedura locale, rispetto alle dimensioni o alla vicinanza a strutture vitali. I compartimenti anatomici sono definiti: intraosseo, sottocutaneo intra-articolare, paraosseo e intrafasciale. I siti anatomici extracompartimentali sono elencati nella colonna di destra della Tabella 1.3. Una lesione è extracompartimentale se insorge in questi tessuti o se si estende secondariamente in essi, da un sito intracompartimentale originale.
8
La manipolazione chirurgica di una lesione, senza la sua completa rimozione, pone i piani tissutali a rischio di recidiva. Pertanto, il rischio è quello di convertire la maggior parte delle lesioni intracompartimentali in lesioni extracompartimentali [11].
Estensione regionale o distante (M)
La presenza o l'assenza di metastasi è il terzo fattore principale, correlato sia alla prognosi che alla pianificazione chirurgica. Nei sarcomi, sia le metastasi ematogene al polmone e sia le metastasi regionali, meno comuni, ai linfonodi hanno lo stesso significato prognostico minaccioso. Indicano, infatti, il fallimento del controllo locale e ridotte possibilità di sopravvivenza.
L'articolazione del sistema di stadiazione chirurgica è basata su una procedura che valuta i margini ottenuti al termine dell’intervento, come indici di recidiva locale prevedibili. Vengono riconosciuti quattro tipi di margini basati sulla relazione del margine chirurgico con la neoplasia e la sua zona reattiva pseudocapsulare:
1. Intralesionale: viene ottenuto mediante una procedura in cui la dissezione passa all'interno della lesione. Il tumore macroscopico o microscopico è lasciato ai margini della ferita e vi è una contaminazione su tutti i piani dei tessuti esposti. Più comunemente, le procedure intralesionali locali vengono eseguite come biopsia incisionale diagnostica, o curettage di una lesione presumibilmente benigna o, ancora, la rimozione subtotale di una lesione.
2. Marginale: si ottiene con una procedura in cui la lesione viene rimossa in un unico pezzo. Il piano di dissezione passa attraverso la pseudocapsula o il tessuto reattivo attorno alla lesione e, quando eseguito per lesioni maligne, lascia una malattia microscopica al margine della ferita in un'alta percentuale. L'escissione marginale è solitamente descritta come biopsia escissionale o "shell out" di una presunta lesione benigna.
3. Ampio: viene ottenuto mediante una procedura in cui la lesione, la sua pseudocapsula e/o la zona reattiva e una cuffia circostante di tessuto normale vengono presi in un unico blocco. Il piano di dissezione è interamente nel tessuto normale ma all'interno del compartimento coinvolto. Non viene fatto alcuno sforzo per rimuovere l'intera lunghezza del muscolo coinvolto. La procedura
9
corrisponde probabilmente a quella che viene definita "escissione locale ampia", "escissione in blocco" o "escissione radicale in blocco". Un ampio margine è attutato per le lesioni allo stadio IA, poiché le lesioni allo stadio IB, di solito, coinvolgono in combinazione ossa, tessuti molli e strutture neurovascolari ed è più probabile l’attuazione dell'amputazione.
4. Radicale: si ottiene con una procedura in cui la lesione, la pseudocapsula, la zona reattiva e tutti i muscoli, o l'osso coinvolti, vengono rimossi come un blocco. Un margine radicale è definitivo per le lesioni allo stadio II. Una resezione locale radicale può spesso essere eseguita per una lesione in stadio IIA. Se una lesione coinvolge più di un compartimento, o si estende o sorge nei piani o negli spazi extracompartimentali, il contenimento compartimentale viene perso e non è solitamente possibile ottenere un margine radicale con tecniche conservative. Pertanto, si esegue l'amputazione per ottenere un margine radicale nelle lesioni in stadio IIB. Spesso per ottenere tale margine è necessaria la disarticolazione o l’amputazione prossimale all'articolazione [11].
1.2 - OSTEOSARCOMA
L’osteosarcoma è un tumore maligno primitivo dell’osso, formato da cellule fusate mesenchimali secernenti matrice osteoide immatura. Questa sua peculiarità garantisce una diagnosi certa. Infatti, qualsiasi neoplasia maligna nella quale ci sia dimostrazione, anche minima, di produzione da parte di cellule mesenchimali di matrice osteoide viene riconosciuta come osteosarcoma. Nessun tumore primitivo dell’osso possiede un’eterogeneità radiologica, anatomica, istologica, citologica e biologica maggiore dell’osteosarcoma [17, 18].
Nonostante la maggior parte degli osteosarcomi (99%) origini come neoplasia solitaria, in rare occasioni, i pazienti possono presentare un coinvolgimento scheletrico multiplo con lesioni che mostrano lo stesso grado di sviluppo. Se questo accade in assenza di un altro sito di malattia metastatica o in assenza di malattie ossee predisponenti, l’osteosarcoma viene definito come multifocale. È, invece, detto sincrono se l’osteosarcoma multifocale è associato a lesioni scheletriche multiple, identificate entro 6 mesi dalla presentazione primaria. Infine, si definiscono osteosarcomi metacroni, le
10
forme multifocali che presentano un intervallo di tempo, tra la diagnosi della lesione primaria e quella delle successive lesioni, superiore ai 6 mesi [19].
Nei pazienti in età evolutiva (<21anni), gli osteosarcomi secondari sono generalmente inferiori al 3% e sono, invece, molto più frequenti nei pazienti in età adulta (56%). Diverse sono le lesioni preesistenti correlate all’insorgenza di osteosarcoma secondario: malattia di Paget ossea, osteogenesi imperfetta, radioterapia, infarti ossei, encondroma solitario, encondromatosi multipla ereditaria, osteocondroma solitario, osteomielite cronica, fibroma non ossificante, displasia fibrosa, cisti ossea aneurismatica, impianti metallici, osteopoichilosi ed osteopetrosi [20].
1.2.1 – EPIDEMIOLOGIA
In Italia, l’incidenza annua dell’osteosarcoma è di 0,17 casi ogni 100'000 abitanti, ossia il 22% dei tumori maligni primitivi dell’osso. Questo è il secondo tumore per frequenza subito dopo il condrosarcoma [8].
L’osteosarcoma è il tumore primitivo dell’osso più frequente dell’età evolutiva (48%). La massima incidenza di malattia si osserva nella seconda decade di vita con il 70 -75 % dei casi tra i 10-25 anni. Sebbene l’osteosarcoma possa interessare quasi tutte le età, i pazienti di età maggiore dei 60 anni raramente vengono colpiti. Nella popolazione femminile, il picco di incidenza si osserva in età precoce rispetto alla popolazione maschile. Il fenomeno sarebbe correlato strettamente alla curva di crescita [21, 22]. Gli osteosarcomi dimostrano, tuttavia, una predilezione per il sesso maschile, con un rapporto maschi : femmine di circa 1,3/1,7:1 [19, 21, 22].
Nell’osteosarcoma esistono diverse eccezioni alla normale distribuzione di incidenza della malattia, in particolare, esistono sottotipi di malattia che colpiscono prevalentemente l’età adulta. L’osteosarcoma periostale ne è un esempio. L’incidenza è distribuita su un largo range della popolazione (8-69 anni), ma la maggioranza dei casi viene diagnosticata nei soggetti di età compresa tra i 15 ed i 40 anni (con picco a 30 anni). Inoltre, colpisce prevalentemente le pazienti di sesso femminile con un rapporto femmine : maschi di 2:1. Anche gli osteosarcomi secondari si manifestano in fasce di età più elevate. Le forme secondarie alla malattia di Paget sono diagnosticate prevalentemente tra la sesta e la ottava decade. Gli osteosarcomi secondari a radioterapia presentano un picco di incidenza compreso tra i 35 ed i 45 anni, a seguito del trattamento con terapia
11
radiante al quale i pazienti sono stati sottoposti in età infantile, adolescenziale o nella prima età adulta. Ciò è giustificato dal periodo di latenza di 13-15 anni che intercorre tra l’esposizione alle radiazioni ionizzanti e lo sviluppo della neoplasia maligna primitiva dell’osso [19]. L’osteosarcoma è in grado di colpire qualsiasi componente scheletrica con, però, una prevalenza delle ossa lunghe degli arti (80%). Il femore è l’osso più comunemente colpito (40-45%), seguito da tibia (15-20%) ed omero (10-15%). Questa distribuzione anatomica non è casuale, infatti, l’osteosarcoma si sviluppa prevalentemente a livello delle fisi a maggior sviluppo (femore distale 35%, tibia prossimale 20% ed omero prossimale 10%) e circa nel 50% dei casi è coinvolto il ginocchio. La distribuzione degli osteosarcomi localizzati alle ossa lunghe è prevalentemente metafisaria (80-90%), anche se la neoplasia può originare dalle diafisi (10-15%). Sono piuttosto rari a livello di rachide, pelvi (4-10%), cranio e perone, meno a livello di avambraccio, mano, piede, coste, clavicola scapola e sterno [19-21].
1.2.2 – EZIOLOGIA E GENETICA
L’eziologia, come del resto in tutti i tumori maligni dell’osso, resta ancora incerta. In letteratura sono stati documentati alcuni rari casi sporadici di osteosarcoma familiare, senza però portare all’identificazione di mutazioni genetiche predisponenti [21].
Il panorama genomico dell’osteosarcoma è diverso da quello degli altri tumori infantili. È caratterizzato da un numero elevato di varianti strutturali (circa 200), come traslocazioni, mutazioni, amplificazioni o delezioni. Nella maggior parte dei casi di osteosarcoma sono presenti alterazioni genomiche in TP53 attraverso meccanismi che portano alla perdita della sua funzione con inattivazione biallelica. Mutazioni in questo gene si trovano in circa il 70% dei pazienti con sindrome di Li-Fraumeni e ciò spiegherebbe una possibile correlazione tra le due patologie [19].
Un altro gene, comunemente inattivato nell’osteosarcoma per delezione o mutazione, è l’RB1che determina, normalmente, l’insorgenza del retinoblastoma. Bambini affetti da retinoblastoma, qualora sopravvivessero al tumore originario, presentano quindi un elevato rischio di sviluppare l’osteosarcoma con un tasso di incidenza del 38% dei malati. Il rischio aumenta nei pazienti con diagnosi di malattia bilaterale e nei pazienti affetti da forme familiari (40%) [19, 21].
12
Tra i fattori predisponenti il rischio di insorgenza della neoplasia spiccano la malattia di Paget e l’esposizione a radiazioni ionizzanti. L’incidenza totale dei sarcomi associati a radiazioni, nei pazienti sottoposti a radioterapia a dosi comprese tra 2500-7200 cGy, è meno dell’1%. Tuttavia, nei bambini affetti da tumore, trattati con radioterapia ed agenti anchilanti, il rischio di sviluppare osteosarcoma è 133 volte superiore alla popolazione generale. La popolazione affetta da malattia di Paget, presenta normalmente un rischio pari all’1% (variabile tra l’1 e il 2,5 %), con un aumento fino al 10% in caso di malattia poliostotica (osteite deformante) [19, 21, 22].
Secondo numerosi studi, l’osteosarcoma sembrerebbe più frequente in bambini e adolescenti con anamnesi positiva per anormale altezza e peso alla nascita, anche se nessun autore è riuscito a dimostrarne una correlazione. Il fatto che i sarcomi dell’osso colpiscano maggiormente il sesso maschile, che vanta un periodo di sviluppo prolungato e altezze maggiori rispetto al sesso femminile, e che il picco di incidenza sia nell’intervallo di età 10-15 anni ha fatto presupporre che vi sia una stretta correlazione tra l’età evolutiva e la patogenesi della maggior parte dei sarcomi ossei. A supporto di questa tesi, inoltre, si osserva come i sarcomi ossei abbiano un’origine principalmente a livello delle fisi delle ossa lunghe a maggior potenziale di crescita (femore e tibia), ovvero le sedi anatomiche con il picco più elevato di incidenza [22].
1.2.3 – SEGNI CLINICI E SINTOMI
Il dolore è il sintomo più frequente e precoce dei tumori ossei maligni. La crescita tumorale è un processo lento ed indolore e permette alla massa neoplastica di progredire in modo asintomatico fino alla distruzione locale dell’osso e/o all’erosione della corticale. Si genera quindi una debolezza strutturale che può portare, come primo segno di presentazione della neoplasia (15%), a fratture patologiche o da stress. Originariamente, il dolore è intermittente e di grado moderato, ma nella totalità dei casi evolve nell’arco di settimane o mesi aumentando l’intensità, severità e frequenza. La durata media dei sintomi prima di raggiungere la diagnosi definitiva è di circa 3 mesi. Il dolore solitamente diventa profondo, invalidante, fisso e di natura meccanica con scarsa, o assente, risposta alla terapia con antinfiammatori ed analgesici vari [19, 21, 22].
Il segno più frequente nei pazienti con osteosarcoma è il riscontro di una massa locale nel contesto dei tessuti molli, manifestazione dell’espansione del tumore oltre l’area di
13
distruzione della corticale ossea. La massa spesso visibile all’ispezione e palpazione può anche rimanere celata a seconda della localizzazione del tumore. I tumori localizzati profondamente nella coscia, o le masse pelviche, sono difficili da esaminare e possono non venire identificati. In caso di diagnosi ritardata, la massa tumorale extraossea può raggiungere grandi dimensioni provocando edema locale, senso di gonfiore, stasi venosa e diminuzione dell’escursione articolare [15, 22].
1.2.4 – DIAGNOSI STRUMENTALE E DI LABORATORIO
L’iter diagnostico dovrebbe sempre iniziare con l’esecuzione di una radiografia tradizionale nelle due proiezioni ortogonali del segmento corporeo interessato dalla sintomatologia. Il tipico aspetto radiologico di un osteosarcoma è quello di una lesione intraossea caratterizzata da aree di aumentata radiodensità intramidollare e lisi ossea non marginata, aree di distribuzione della corticale e neoformazione ossea periostale. Nel caso di tumore extraosseo, sarà visibile una massa nel contesto dei tessuti molli caratterizzata da una produzione di matrice calcifica e da una reazione periostale. Nelle fasi inziali è presente un’osteolisi permeativa ed infiltrante a limiti non ben definiti che ben presto raggiunge la corticale e la distrugge con una chiara e visibile reazione del periostio che tenta di circoscrivere il tumore (“reazione a bulbo di cipolla”). Con il superamento della corticale il periostio viene distrutto e rimane solo una reazione alla periferia del tumore a livello metafisario (triangolo di Codman). A questo punto, il tessuto patologico si espande rapidamente nei tessuti molli vicini, rendendolo ben visibile alla tomografia assiale computerizzata (TC) o alla risonanza magnetica (RMN) [19].
Nel 90% dei casi, gli osteosarcomi hanno già invaso i tessuti molli alla diagnosi. L’estensione extraossea è meglio inquadrabile con tecniche di imaging come la TC o la RMN. È, tuttavia, possibile riscontrare segni di invasione anche grazie alla radiografia semplice. Infatti, il riscontro di densità soffici ed irregolari nel contesto dei tessuti molli è un reperto radiologico indice di produzione di tessuto osseo in sede extraossea [21, 22]. Quando la radiografia tradizionale mostra una massa con ossificazioni “a nuvola” diffuse nei tessuti molli, che circondano l’osso colpito, si tratta di caratteristiche radiologiche tipiche degli osteosarcomi juxtacorticali o periferici (parostali e periostali). Gli
osteosarcomi parostali sono generalmente delle neoplasie radiopache di grandi
14
collegati alla corticale mediante un’attaccatura ampia e sessile che occasionalmente può apparire interrotta da una sottile linea radiolucente o “piano di clivaggio” che è indice della presenza di periostio ritenuto nella neoplasia. Man mano che il tumore cresce, forma una massa ossificata prominente che ingloba la superficie corticale esterna e determina un assottigliamento della corticale. Ciò può determinare la perdita di dettaglio radiologico dell’osso sano e del canale midollare. L’ossificazione nell’osteosarcoma parostale è più accentuata in prossimità della corticale, divenendo meno matura verso la periferia. La matrice tumorale può apparire omogenea e presentare aree cistiche radiolucenti. L’osteosarcoma periostale è un tumore di dimensioni ridotte confinato nella superficie ossea che presenta un’attaccatura sessile aderente alla corticale lungo tutta l’estensione della neoplasia. È comune il riscontro di spiculazioni sottili e raggiate, perpendicolari al tumore e aggettanti al di fuori della superficie corticale (reazione a “sole radiante”). Sia i tumori parostali, che quelli periostali, non mostrano alcun coinvolgimento del canale midollare, caratteristica tipica degli osteosarcomi centrali [19].
Gli esami di laboratorio non sono diagnostici, ma è stato dimostrato che alti livelli di fosfatasi alcalina (ALP) e latticodeidrogenasi (LDH) sono degli indicatori di prognosi infausta [23, 24].
Una volta eseguita la radiografia, e riscontrata una neoplasia ossea, è necessario passare ad imaging di secondo livello. Nell’osteosarcoma è meglio la prescrizione di una risonanza magnetica (RMN). La RMN è indicata come tecnica “gold standard”, in quanto permette di valutare l’eventuale invasione nel midollo osseo, le dimensioni della massa nei tessuti molli ed i suoi rapporti con le strutture vitali vicine. Gli osteosarcomi sono ipointensi nella sequenza T1 ed iperintensi nella sequenza T2 e STIR. Solitamente sono lesioni che appaiono come omogenee, circondati da un edema peritumorale e con un forte enhancement dopo la somministrazione di contrasto. La TC è inferiore in termini di potere diagnostico ma fornisce informazioni utili aggiuntive riguardo all’integrità della corticale e l’eventuale presenza di frattura. Una TC total body è utile nell’individuare eventuali metastasi e fornire una stadiazione tumorale il più accurata possibile. Un’indagine mediante scintigrafia ossea o tomografia ad emissioni di positroni (PET) è raccomandabile al fine di identificare la malattia metastatica ossea o dei tessuti molli. Studi combinati PET/TC hanno dimostrato una certa accuratezza nel determinare
15
l’estensione dell’osteonecrosi istologica post-chemioterapia neoadiuvante, offrendosi come possibile alternativa alla biopsia [21, 22].
Nonostante le nuove tecniche non invasive abbiano portato notevoli vantaggi nello studio della patologia tumorale ossea, la diagnosi definitiva per osteosarcoma, ad oggi, deve essere attuata mediante la biopsia. Il prelievo bioptico può essere effettuato mediante agobiopsia sotto guida TC o ecografica, oppure “a cielo aperto” [22].
1.2.5 – ANATOMIA PATOLOGICA
Nonostante la grande varietà di presentazioni istologiche e stromali osservabili negli osteosarcomi, il criterio essenziale e distintivo della neoplasia è la presenza di una franca popolazione di cellule sarcomatose secernenti matrice osteoide. Tuttavia, una percentuale pari al 25% dei tumori presenta caratteristiche istologiche confuse, tali da rendere necessaria un'accurata analisi dei reperti clinico-radiologici per raggiungere una diagnosi corretta. All'esame istologico sarà possibile osservare cellule mesenchimali maligne fusate o poliedriche con nuclei pleomorfi, figure mitotiche sparse e livelli di anaplasia variabile. Riconosciamo numerosi sottotipi di osteosarcoma: osteoblastico, condroblastico, fibroblastico, a piccole cellule, teleangectasico, alto grado, superficiale, extrascheletrico ed altre forme come l’osteosarcoma parostale e periostale. Sulla base dell’aspetto istologico, i sottotipi di osteosarcoma possono essere raggruppati in tre categorie: alto grado, grado intermedio e basso grado [18]. Ciò permette di instaurare terapie differenti. Nelle forme a basso grado si interviene solo chirurgicamente e la prognosi di solito è buona. L’osteosarcoma periostale con aspetto istologico condroblastico è l’unico che viene classificato di grado intermedio. La terapia può o meno richiedere dei cicli di chemioterapia sistemica in associazione alla chirurgia. La maggior parte dei sottotipi di osteosarcoma sono di alto grado e devono essere considerati come micrometastatici già alla diagnosi e, quindi, trattati con una terapia che combini la chemioterapia sistemica ed il trattamento chirurgico di resezione del tumore [21, 22].
1.2.6 – FATTORI PROGNOSTICI
Le variabili prognostiche significative per l’osteosarcoma risultano essere numerose e non sempre condivise da tutti gli esperti. L'estensione del tumore è probabilmente il più significativo tra gli indicatori di prognosi. La presenza di evidenza all'epoca della
16
diagnosi di diffusione regionale o metastatica della malattia è un predittore affidabile di diminuita sopravvivenza del paziente. Per diffusione regionale si intende sia la disseminazione nel sistema linfatico sia la presenza di skip metastases intraossee. Allo stesso modo, le malattie ossee multifocali dovrebbero essere considerate alla stregua delle altre metastasi ematogene, almeno nei pazienti in età evolutiva [19, 21, 22].
Il grado istologico, sicuramente, deve essere considerato come uno dei fattori prognostici principali. I pazienti con tumori a basso grado presentano una sopravvivenza nettamente superiore ai pazienti con neoplasia ad alto grado, con tassi di sopravvivenza a 5 anni pari al 90%, nel primo caso, e al 68% nel secondo [25].
Il sito anatomico di origine del tumore rappresenta nell’osteosarcoma un fattore prognostico abbastanza significativo. I dati sulla mortalità per specifici siti anatomici non sono univoci, ma dalla letteratura si evince che nelle neoplasie localizzate all'estremità più distali (es. tibia) vi è un vantaggio sostanziale in termini di sopravvivenza, rispetto ai tumori più prossimali (es. femore, omero) e/o centrali (pelvi) [19]. Anche la dimensione del tumore è stata indicata in diversi studi come un fattore prognostico significativo. Sfortunatamente, i criteri utili a determinare se un tumore sia da considerarsi di grosse o piccole dimensioni differiscono tra i diversi autori rendendo impossibile una vera e propria analisi comparativa dei dati sulla sopravvivenza. Nella maggior parte dei casi, gli osteosarcomi classificati come di grandi dimensioni hanno mostrato una prognosi significativamente peggiore rispetto a quelli classificati come di piccole dimensioni [26]. Sia la frattura patologica sia il genere sono da tempo fonte di dibattito tra i diversi esperti. Nel primo caso, studi recenti hanno confermato l'ipotesi secondo la quale la presenza alla diagnosi di tale elemento porti a una riduzione del tasso di sopravvivenza a 5 anni pari al 29%. Nel secondo caso, alcuni dati hanno riportato un tasso di sopravvivenza nettamente migliori nelle pazienti di sesso femminile, ma analisi multivariate hanno fallito nel dimostrare la validità di genere ed età come fattori prognostici [19, 26, 27].
Infine, la necrosi tumorale post chemioterapia neoadiuvante possiede il più alto grado di significato prognostico subito dopo stadio e grado del tumore. Pazienti che hanno dimostrato una necrosi tumorale post chemioterapia pre-operatoria maggiore del 90% (good responders) presentano una sopravvivenza a 5 anni tra il 75 e 80%, nettamente superiore ai pazienti che invece mostrano una risposta inferiore a tale valore [28].
17
1.3 – SARCOMA DI EWING
Il sarcoma di Ewing è una neoplasia maligna, ad alto grado, composta da cellule tumorali piccole e rotonde, a istogenesi incerta, che ha origine in primis nell’osso e meno comunemente nei tessuti molli. Nonostante il sarcoma di Ewing sia una neoplasia rara, nell’età evolutiva rappresenta il tumore maligno primitivo dell’osso più frequente, subito dopo l’osteosarcoma. Oggi è classificato come un tumore periferico primitivo neuroectodermico (PNET), ovvero, un gruppo di neoplasie che si ritiene derivi da elementi cellulari della cresta neurale. Ha una netta predilezione per la seconda decade di vita: circa nel 90% dei casi, si manifesta in soggetti tra i 5 e 25 anni [29, 30].
1.3.1 - EPIDEMIOLOGIA
Secondo i dati italiani, il sarcoma di Ewing rappresenta il 15% di tutte le neoplasie maligne primitive dell’osso, con un’incidenza annua media di 0.12 casi ogni 100.000 abitanti. Questi dati si allineano a quelli negli USA, dove l'incidenza di sarcoma di Ewing, nei bambini di razza caucasica di età inferiore ai 15 anni, è di 0.17 casi ogni 100.000 abitanti all'anno. I bambini caucasici mostrano un'incidenza di sarcoma di Ewing 6 volte maggiore rispetto ai bambini neri ed asiatici. Questa particolare distribuzione razziale potrebbe suggerire l'esistenza di influenze genetiche maggiori nella popolazione caucasica, rispetto a quella afroamericana o africana o asiatica, dove i dati indicano dei tassi nettamente più bassi sull’incidenza di questa patologia [29]. Il sarcoma di Ewing è caratterizzato da una predilezione di genere per il sesso maschile, con un rapporto maschio-femmina di 1.7: 1[8], come la maggior parte dei tumori ossei primitivi [30]. Ogni segmento scheletrico può essere virtualmente colpito dal sarcoma di Ewing. Nella maggior parte dei casi si localizza a livello delle ossa lunghe (50-55%), con al primo posto il femore (25%), seguito da tibia e perone (15%). Le ossa piatte del cingolo pelvico sono colpite nel 25% dei casi, mentre le coste e il cingolo scapolare per il 5-10% dei casi. Le ossa corte del cranio, delle mani e dei piedi sono raramente interessate dalla malattia, presentando generalmente un outcome superiore in termini di sopravvivenza e funzionalità residua. Come detto, si ha una prevalenza a livello delle ossa lunghe soprattutto in sede diafisaria. Tuttavia, il coinvolgimento metafisario e metadiafisario è due volte più frequente (metadiafisario 44%, diafisario 33%, metafisario 15%) e spesso
18
di osserva una malattia estesa che coinvolge l’intero segmento osseo. La localizzazione mataepifisaria (6%) o epifisaria (2%) è piuttosto rara [19].
1.3.2 – SEGNI CLINICI E SINTOMI
Un numero elevato di pazienti affetti da sarcoma di Ewing si presenta con sintomi sistemici come febbricola intermittente, leucocitosi, VES elevata ed anemia, insieme a sintomi locali come ipertermia cutanea ed eritema, dilatazione venosa, nonché una massa o un rigonfiamento soffice e palpabile. Questi sintomi possono facilmente essere confusi con quelli di un’osteomielite [19].
Come spesso accade anche in altre neoplasie ossee, dolore e rigonfiamento del sito di malattia sono i principali sintomi di presentazione della maggior parte dei pazienti affetti da sarcoma di Ewing. Tuttavia, lo sviluppo graduale e la natura aspecifica dei sintomi spesso contribuiscono al ritardo tra presentazione clinica e diagnosi, come comunemente osservato nei pazienti affetti da sarcoma di Ewing [19, 31].
Il coinvolgimento del sistema nervoso centrale è raro nel sarcoma di Ewing e nella maggior parte dei casi è successivo a disseminazione ematogena del tumore o a invasione locale a partenza da un osso limitrofo [19, 29].
1.3.3 – DIAGNOSI STRUMENTALE E DI LABORATORIO
I dati laboratoristici riscontrano un aumento della VES o di un’alterazione della conta leucocitaria. Di norma non sono presenti altre alterazioni di laboratorio che lascino presagire l’esistenza di una neoplasia. Per un corretto inquadramento del paziente, in caso di lesione sospetta di sarcoma di Ewing, andrebbero eseguiti il dosaggio di LDH e VES, indici rispettivamente di malattia e di disseminazione sistemica [19].
L’indagine radiologica rivela come il sarcoma di Ewing assuma l’aspetto di una lesione estesa di natura osteo-distruttiva, priva di margini definiti nelle regioni, solitamente, metafisio-diafisarie e diafisarie delle ossa lunghe. Tuttavia, il sarcoma di Ewing possiede uno spettro di presentazione tra i più vari all'interno del gruppo dei sarcomi ossei, caratteristica che rende molto difficile la diagnosi radiografica. In un numero minore di casi, caratterizzati da lenta crescita del fronte di osteolisi corticale, la velocità ridotta del riassorbimento osseo consente lo sviluppo reattivo di un ispessimento corticale, quadro radiologico che rende difficile la diagnosi differenziale tra sarcoma di Ewing e
19
l’osteomielite. La neoplasia può anche mostrarsi sotto forma di una lesione litica corticale di tipo periostale, apparendo alla radiografia tradizionale come un'area di osteorarefazione concava corticale. Questa lesione “a scodella” ha origine dalla diffusione della neoplasia attraverso i canali di Havers, a cui segue lo sviluppo e la crescita di una massa tumorale che erode l'osso corticale dalla superficie esterna verso il canale midollare [29].
Le dimensioni e l’estensione del sarcoma di Ewing dipendono da diverse caratteristiche, tra le quali il tasso di crescita e l’epoca di presentazione della neoplasia. Anche la localizzazione della malattia gioca un ruolo importante nel determinare le dimensioni del tumore, infatti è tra i principali elementi che intervengono nell’influenzare il tempo di presentazione clinica e diagnosi del sarcoma di Ewing. Di conseguenza, le masse tumorali localizzate in corrispondenza di coscia, cingolo pelvico, scapola, femore prossimale, all’epoca della diagnosi, con molta probabilità avranno raggiunto dimensioni maggiori rispetto ai sarcomi di Ewing insorti in corrispondenza di segmenti anatomici più periferici. Le fratture patologiche sono relativamente frequenti, verificandosi nel 5% come primo sintomo. Le skip metastases, ben documentate nell’osteosarcoma, sono rare nel sarcoma di Ewing essendo riportati soltanto pochi casi. Tuttavia, è di comune riscontro la presenza di infiltrazione midollare la cui estensione va ben oltre la porzione ossea interessata dal tumore [19].
Anche se la radiografia tradizionale è ancora in grado di fornire informazioni utili riguardo a: diagnosi, fratture patologiche e aggressività biologica della lesione, essa non consente di ottenere informazioni affidabili riguardo all’estensione intramidollare di malattia e alla diffusione della stessa nei tessuti molli. La reale dimensione della massa tumorale può essere studiata adeguatamente esclusivamente con tecniche di imaging avanzate come TC o la RMN. Può essere difficile porre diagnosi differenziale tra sarcoma di Ewing ed osteomielite mediante radiografia tradizionale e TC. La RMN rimane quindi il gold standard per diagnosi e stadiazione in caso di sospetto sarcoma di Ewing [19, 32]. Dato che i sintomi clinici del sarcoma di Ewing non sono specifici e spesso imitano l’osteomielite o altri tumori maligni, come ad esempio la leucemia, in vista di un esame bioptico vengono sempre eseguite la radiografia tradizionale e la RMN. Questa, inoltre, è in grado di valutare la risposta della malattia alla chemioterapia analizzando la scomparsa o la permanenza della malattia extraossea, anche se non sono stati definiti dei
20
protocolli ben precisi. La scansione PET con FDG è, inoltre, raccomandata come metodica più sensibile nel rivelare i primi cambiamenti nel metabolismo tumorale a seguito di trattamento chemioterapico [33]. La biopsia rimane comunque necessaria per la conferma della diagnosi prima di qualsiasi intervento chirurgico [7].
1.3.4 – ANATOMIA PATOLOGICA
Dal punto di vista istologico, il sarcoma di Ewing è caratterizzato da strati uniformi di piccole cellule rotonde tumorali non secernenti matrice extracellulare. Queste cellule si mostrano uniformi, costituite da nuclei tondi con poche mitosi e con scarso citoplasma. Il riscontro di pseudorosette è indicativo della differenziazione neurale, caratteristica morfologica che ci permette di classificarlo come un tumore periferico primitivo neuroectodermico (PNET). All’indagine immunoistochimica, a volte può essere rivelata positività per marcatori neurali come la proteina S-100, PGP-9.5 e la vimentina. In oltre il 90% dei casi di sarcoma di Ewing, viene rivelata sulla membrana plasmatica la positività per CD99, un prodotto del gene MIC2. Tuttavia, l’espressione di CD99 non è specifica del sarcoma di Ewing, poiché può essere osservata anche in altri tumori, tra i quali: il sarcoma sinoviale, la leucemia linfoblastica, il linfoma e altre neoplasie [19, 29]. Grazie alle tecniche di indagine molecolare, come RT-PCR e la FISH, è stato riscontrato il gene di fusione EWS-FLI1, generato dalla traslocazione t(11;22) (q24;q12), che caratterizza l’85% dei sarcomi di Ewing. Un altro gene di fusione, riscontrabile nel 10% dei casi, è il gene EWS-ETV1, causato dalla traslocazione t(7;22) (q22;q12). Esistono poi delle forme rare come il gene EWS-ETV4, derivante dalla traslocazione t(17;22) (q21;q12) e il gene EWS-FEV, causato dalla traslocazione t(2;22) (q35;q12). Tutti questi prodotti di fusione codificano per proteine chimeriche che funzionano come fattori di trascrizione oncogenetici aberranti [29, 31].
Nel sarcoma di Ewing, l’accumulo di aberrazioni genetiche, a carico di più geni su diversi cromosomi, sembra avere un’influenza maggiore sul comportamento clinico e biologico del tumore rispetto ad un singolo gene di fusione alterato [7, 29, 31].
1.3.5 – FATTORI PROGNOSTICI
È stato dimostrato, in ogni studio pubblicato sul sarcoma di Ewing, che la presenza di malattia metastatica costituisce un fattore prognostico negativo, con una sopravvivenza a
21
lungo termine minore del 10% dei casi. Nei pazienti affetti da malattia localizzata, la dimensione della massa tumorale è il fattore prognostico di maggior significato [19, 34]. In diverse pubblicazioni, anche la localizzazione anatomica primaria del tumore sembra possedere un’importanza prognostica. Le lesioni centrali o pelviche hanno dimostrato una ridotta sopravvivenza rispetto alle lesioni periferiche o delle estremità. Tuttavia, negli studi in questione non è sempre stato possibile dimostrare che la valutazione del fattore “localizzazione pelvica” fosse completamente indipendente dal fattore “dimensioni della massa tumorale” [34].
Un aumento della conta leucocitaria e della VES probabilmente funziona da indicatore di malattia avanzata e sembra essere correlato ad una prognosi peggiore. L’aumento dell’LDH è stato associato a recidiva di malattia e malattia metastatica, e viene quindi incluso nell’elenco dei fattori prognostici negativi [19, 34]. Ad oggi, anche la risposta iniziale alla chemioterapia neoadiuvante è un fattore avente significato prognostico, come è stato evidenziato dall’analisi radiologica ed istologica delle lesioni tumorali nei pazienti affetti da sarcoma di Ewing sottoposti a terapia citotossica [7].
Nel sarcoma di Ewing, il fattore di rilevanza prognostica maggiormente controverso è sicuramente la resezione chirurgica del tumore primario. Numerosi studi hanno dimostrato un aumento significativo della sopravvivenza nei pazienti andati incontro a controllo locale mediante una corretta resezione chirurgica. L'intervento chirurgico sembrerebbe aumentare la sopravvivenza libera da malattia a 5 anni dal 22% al 55%. Allo stesso modo, la resezione chirurgica, se eseguita correttamente, produrrebbe un tasso di recidiva locale prossimo allo 0% ed una diminuzione del tasso di ricaduta di malattia nel sito primario dal 58% al 21%. Sfortunatamente, in molti di questi studi, la riduzione del tasso di ricorrenza locale non è statisticamente significativo [19, 34].
22
CAPITOLO 2 – ITER DIAGNOSTICO
MULTIDISCIPLINARE E TECNICHE BIOPTICHE
NEI SARCOMI DELLA PELVI
2.1 - VALUTAZIONE CLINICA E RADIOLOGICA INIZIALE
Il paziente tipico con un sarcoma pelvico lamenta per 6-12 mesi dolore all’inguine o alla schiena e presenta, o meno, una radiografia anormale. L’iter diagnostico adeguato ha inizio quando il dolore persistente o una alterazione radiografica indirizzano verso ulteriori approfondimenti diagnostici. La diagnosi iniziale e la stadiazione devono essere indagate includendo le seguenti domande:1. Si tratta di un quadro neoplastico, di una patologia degenerativa o di un’infezione? 2. In caso si tratti di tumore, la neoplasia è benigna o maligna?
3. Se maligna, si tratta di un sarcoma o di una metastasi? 4. Se è un sarcoma, è di alto o basso grado?
In alcuni casi, possono insorgere dubbi sull’origine anatomica della lesione: neoplasia primitiva dell’osso o dei tessuti molli. Determinando l’estensione del tessuto interessato (osso o tessuti molli) si può, generalmente, capire l’origine della neoplasia. Questo tipo di indagine richiede l’utilizzo di adeguate tecniche di imaging, dalla semplice radiografia a tecniche più specifiche come la scintigrafia ossea (total body), risonanza magnetica nucleare (RMN) o la tomografia computerizzata (TC) [19].
Una delle caratteristiche cliniche più significative con valore diagnostico predittivo, per i pazienti che presentano un tumore dell’osso sconosciuto, è sicuramente l’età. Se il paziente ha meno di 40 anni, molto più frequentemente si tratta di un tumore primitivo dell’osso. I pazienti con età superiore ai 40 anni sono, invece, più a rischio per metastasi da un adenocarcinoma. Le caratteristiche radiologiche, della maggior parte delle lesioni dell’osso riscontrabili con la radiografia semplice, sono differenziabili tra un processo metastatico e un sarcoma primitivo. Un osteosarcoma, generalmente, presenta un quadro
23
relativamente distinguibile alla radiografia, si presenta come una anomalia osteoblastica a margini poco definiti con possibile interessamento dei tessuti molli da entrambi i lati dell’ileo. Il condrosarcoma è il più comune tra i sarcomi diagnosticati a livello della pelvi nell’adulto, radiologicamente si presenta con una caratteristica area osteolitica policiclica con lievi calcificazioni a limiti mal definiti. Altri tumori primitivi maligni dell’osso, relativamente comuni a livello della pelvi, sono il sarcoma di Ewing, il mieloma e il linfoma. L’adenocarcinoma può essere metastatico a seguito di una neoplasia primaria della mammella, del rene, del polmone o della prostata. Alcune metastasi pelviche derivanti da adenocarcinoma sono scarsamente definite e si presentano come lesioni osteolitiche di lieve entità, a patto che non ci sia nessun coinvolgimento dei tessuti molli. Sia l’osteosarcoma sia il sarcoma di Ewing sono meno comuni nell’adulto (generalmente non si presentano dopo il 25 anno di vita), colpendo quasi esclusivamente i pazienti in età evolutiva [19, 35].
Un approccio adeguato prevede un’analisi anamnestica accurata del paziente, seguita da un approfondimento del quadro clinico che si presenta normalmente con un dolore osseo anormale a livello della pelvi. Gli esami laboratoristici che dovrebbero essere richiesti includono emocromo completo, VES, elettroliti, uremia, nitroderivani e creatininemia, fosfatasi alcalina, latticodeidrogenasi (LDH) e una completa analisi delle urine. In un uomo adulto è necessario richiedere anche il dosaggio di anticorpi specifici per la prostata, mentre nei pazienti con età superiore ai 40 anni, indipendentemente dal sesso, fondamentale è l’esecuzione di un’elettroforesi delle proteine sieriche, associata alla conta quantitativa dell’immunoglobuline, per escludere un quadro di mieloma multiplo. In questi pazienti sarebbe opportuno valutare anche il calcio sierico per individuare una possibile ipercalcemia.
Gli studi radiografici raccomandano, per tutti i pazienti con un tumore pelvico, di includere la radiografia delle pelvi, la RMN e la TC, per ottenere un quadro specifico e una stadiazione della malattia. Una scintigrafia con tecnezio dell’osso dovrebbe essere ottenuta per valutare l’estensione e l’interessamento della pelvi e la possibilità della presenza di altre lesioni ossee. Una scintigrafia delle ossa con captazione aumentata è un segno significativo di malattia attiva che richiede un approfondimento diagnostico con biopsia. Oggi in realtà, si preferisce la PET in quanto più accurata e con un rapporto costi/benefici maggiore rispetto alla tradizionale scintigrafia ossea. Tutti i pazienti con un
24
tumore osseo pelvico dovrebbero essere valutati con radiografia del torace e, se necessario, con una TC del polmone e dell’addome per escludere la possibilità di metastasi. Come indicazione generale, tutte queste indagini radiologiche dovrebbero essere effettuate prima di qualsiasi procedura bioptica. A meno che non ci sia una storia clinica di una precedente neoplasia maligna, l’iter radiografico iniziale, per il paziente pediatrico o giovane adulto, non richiede la ricerca di un carcinoma primitivo [19, 29].
2.2 - INDICAZIONI ALLA BIOPSIA E TECNICHE DI
SVOLGIMENTO
I pazienti che si presentano con dolore osseo anomalo della pelvi e che, generalmente, mostrano una lesione sospetta alla diagnostica per immagini, devono essere sottoposti a biopsia. Se l’iniziale iter radiografico del paziente, includendo anche una TC addominale e dei polmoni, dimostra un probabile adenocarcinoma primitivo, la biopsia della lesione deve essere effettuata successivamente, per confermare la diagnosi dell’apparente adenocarcinoma primitivo e la presenza di metastasi ossee, in questi pazienti, la biopsia può essere indicata allo scopo di documentare la progressione della malattia. La biopsia spesso può essere effettuata con la tecnica di aspirazione con ago sottile, piuttosto che da una biopsia a cielo aperto [3, 19].
Il successo del trattamento per qualsiasi tumore si basa su di una adeguata e accurata biopsia iniziale, in un’appropriata localizzazione. Una biopsia mal fatta può essere la prima ragione per la quale un paziente debba essere sottoposto ad una emipelvectomia esterna, senza la conservazione dell’arto. L’esecuzione della biopsia richiede esperienza ed è chiaramente meglio se attuata da un chirurgo con conoscenza nella gestione di pazienti con tumori muscoloscheletrici. Quando e dove effettuare la biopsia è una grande sfida per la maggior parte dei tumori pelvici [3, 37].
La resezione del tumore pelvico non dovrebbe essere effettuata prima di ottenere una diagnosi istologica. Per i tumori primitivi dell’osso a livello pelvico potrebbero essere richieste più informazioni di quelle fornite dalla tecnica al congelatore, tramite l’analisi della sezione permanente con formalina. Questo risulta essere particolarmente veritiero nei sarcomi ossei pelvici, quando il rischio di una contaminazione dal materiale
25
peritoneale e pelvico è elevato. Il rischio di contaminazione è parimenti importante anche per il ragionamento inverso. È necessario che il tumore non venga disseminato durante la biopsia ai tessuti circostanti, causando una successiva difficoltà nel controllo locale della neoplasia, fatto imprescindibile nella sfida per la sopravvivenza. Ecco perché, le tecniche bioptiche sono estremamente importanti per la valutazione di un tumore osseo della pelvi. Per i tumori primitivi dell’osso pelvico è fondamentale che la biopsia venga effettuata lungo la linea dell’incisione attraverso cui verrà eseguita la resezione ed è necessario garantire un campione adeguato prima di concludere la procedura bioptica. Molti sarcomi pelvici sono resecati attraverso un’incisione lungo la cresta iliaca: perciò la più sicura incisione bioptica è direttamente effettuata sopra la cresta dell’ileo [3, 19].
La maggior parte dei pazienti con un tumore pelvico sono sottoposti ad un ago biopsia. I tumori localizzati in distretti anatomici complessi, come la faccia anteriore del sacro o altri siti pelvici profondi o per tumori di piccole dimensioni, possono richiedere una biopsia con agoaspirato sotto guida TC. Ciò potrebbe richiedere l’anestesia spinale, locale o generale. Come detto, la localizzazione della puntura bioptica è determinata con le stesse modalità con la quale viene decisa la procedura in “open”, ossia, sulla linea lungo la quale viene programmata la resezione. Ogni manovra dovrebbe essere effettuata con estrema precisione per evitare la diffusione tumorale a livello retroperitoneale, sia che si tratti di una biopsia con ago aspirato, sia che si tratti di una biopsia incisionale. Un paziente, sottoposto a biopsia con agoaspirato che ottiene una diagnosi dubbia per neoplasia ossea, deve essere sottoposto alla biopsia a cielo aperto, in sala operatoria, per ottenere una diagnosi certa [37].
La decisione se utilizzare una biopsia a cielo aperto o una biopsia con ago percutaneo rimane, oggi, oggetto di controversia. Nel primo caso, si ottiene quasi sempre una quantità di tessuto sufficiente per effettuare una diagnosi preoperatoria accurata. Ciò risulta essere particolarmente importante nella pianificazione della chemioterapia neoadiuvante, in quanto esistono differenze nella durata e nella scelta degli agenti tra i diversi tumori. Tuttavia, la biopsia a cielo aperto presenta maggiori rischi di contaminazione del tessuto intrapelvico, impendendo o ostacolando la buona riuscita dell’emipelvectomia interna con conservazione dell’arto. Nel secondo caso invece, la biopsia con ago percutaneo sembra avere un rischio minore di contaminazione, ma non sempre garantisce un prelievo adeguato di materiale, vanificando di fatto l’intera esecuzione della procedura [38].
26
Indipendentemente dalla scelta, la biopsia dei tumori pelvici deve essere eseguita al termine degli studi di stadiazione, poiché l'artefatto bioptico può precludere la delimitazione accurata dei margini del tumore o la caratterizzazione del tumore su test specifici come la risonanza magnetica [38].
Oggi non si eseguono più biopsie intaroperatorie nel corso della procedura diagnostica. Al momento dell’intervento definitivo si esegue una biopsia dei margini del pezzo di resezione per valutare se sono indenni da malattia.
27
CAPITOLO 3 – INDICAZIONI PER IL
TRATTAMENTO DEI SARCOMI DELL’OSSO
Nonostante ogni tipologia di tumore osseo potrebbe colpire la pelvi, ci sono alcuni tumori maggiormente localizzati in tale area. La maggior parte dei tumori ossei della pelvi sono maligni (62% Tabella 3.1), mentre i tumori che sono localizzati al femore prossimale sono spesso benigni (68% Tabella 3.2). Nei bambini con un’età inferiore ai 14 anni, il 42% dei tumori, o lesioni similtumorali, sono le cisti ossee solitarie, le cisti aneurismatiche e l’osteoma osteoide [13]. Quando si esaminano i dati sull'incidenza, si
Table 3.1
28
dovrebbe tenere presente che i tumori comuni in sedi rare, di solito, si riscontrano più frequentemente rispetto ai tumori rari in sedi comuni. Nelle tabelle 1 e 2 viene fornita la distribuzione della frequenza del tipo di tumore in base alla sede. I tumori, e le lesioni similtumorali, riscontrati più frequentemente sono: il cordoma (25%), il tumore a cellule giganti (15%), le cisti ossee aneurismatiche (13%), il condrosarcoma (12%) nell’adulto, e l’osteosarcoma (8%), il sarcoma di Ewing (8%) nel bambino e nel giovane adulto [14]. Storicamente, la chirurgia è stata il mezzo più efficace per trattare la maggior parte dei sarcomi muscolo-scheletrici primari e l'amputazione ha avuto un ruolo preminente (nell'armamentario chirurgico). La recente evidenza che alcuni agenti chemioterapici possono avere una significativa attività anti-sarcoma, in associazione ai coincidenti progressi tecnici nella terapia con radiazioni, nelle indagini radiografiche e nella chirurgia ricostruttiva, hanno alimentato un entusiasmante interesse nei trattamenti salva-estremità. Quasi tutti questi trattamenti sono solitamente eseguiti sotto un mantello protettivo di chemioterapia neoadiuvante e adiuvante. La sola terapia con irradiazione ha dimostrato un buon controllo locale a lungo termine della massa neoplastica, ma ciononostante l'intervento chirurgico rimane, quasi sempre, un passo essenziale nella complessa gestione dei sarcomi muscoloscheletrici [11].
In questo elaborato analizzeremo nello specifico l’osteosarcoma e il sarcoma di Ewing, che rappresentano le neoplasie statisticamente più diffuse nella pelvi in età evolutiva.
3.1 – OSTEOSARCOMA
Il trattamento dei tumori maligni del bacino è uno dei problemi più impegnativi per l'oncologo ortopedico [39, 40]. L'osteosarcoma del bacino rappresenta dal 4% al 10% di tutti gli osteosarcomi [41, 42]. Questi tumori sono difficili da resecare a causa della loro localizzazione, dell’estensione locale e delle grandi dimensioni, nonché a causa della complessa anatomia e della difficoltà di stabilire ricostruzioni funzionali, durevoli e che consentano ai pazienti la massima funzionalità. L'osteosarcoma, soprattutto di tipo convenzionale, si verifica non di rado nel femore prossimale (5%) e nell'osso iliaco (3%). È molto raro nell'osso sacro, nel pubico e nell’ischiatico (<1%). Le caratteristiche radiografiche sono simili a quelle dell'osteosarcoma alle estremità. Il suo segno distintivo è la formazione di osteoidi in associazione alla distruzione asimmetrica dell'osso e
29
all'estensione asimmetrica dei tessuti molli. I tumori sono più grandi di quelli delle estremità e più spesso contengono componenti cartilaginee di grandi dimensioni (osteosarcoma di tipo convenzionale o condroblastico, a seconda della frazione di tumore costituita dalla cartilagine). Le componenti cartilaginee vengono identificate con l’RM potenziata con chelato di Gadolino (Gd) [14].
Secondo la letteratura, i pazienti affetti hanno un esito sfavorevole che va dal 4% al 32% [46, 48, 42, 43], che è in netto contrasto con il tasso di sopravvivenza a 5 anni dei pazienti con osteosarcoma delle estremità (70-75%) [44, 45]. Le ragioni di questo (scarso) risultato sono molteplici. Spesso si verifica un ritardo nella diagnosi o una diagnosi errata, che di solito è associata a una lunga durata dei sintomi e che può portare già a metastasi prima di stabilire la diagnosi. Anatomicamente, vi è una scarsa compartimentazione nel bacino a causa delle grandi dimensioni di queste lesioni che possono, quindi, attraversare l'articolazione sacroiliaca o coinvolgere le vene pelviche [41]. Ciò a sua volta rende difficile ottenere un ampio margine chirurgico [43].
Alcuni pazienti con osteosarcoma pelvico possono essere inoperabili e quindi presentano una prognosi più sfavorevole. [46, 48, 42, 43]. In questi casi, la radioterapia viene impiegata, con intento palliativo, a scopo antalgico nelle sedi ossee sia primitive che secondarie non altrimenti trattabili [46].
3.1.1– LA TERAPIA MEDICA
Negli ultimi 30 anni sono stati fatti grandi progressi nel trattamento dei pazienti con osteosarcoma non metastatico con la chemioterapia adiuvante. I chemioterapici maggiormente utilizzati sono Doxorubicina (o Adriamicina=ADM), Cisplatino (CDP), Metotrexate (MTX) ad alto dosaggio e Ifosfamide (IFO), usata singolarmente o in associazione ad Etoposide. I tassi di sopravvivenza a cinque anni sono aumentati dal 10-20% al 70% [29, 48, 49]. La chemioterapia neoadiuvante, generalmente somministrata per 8-10 settimane prima dell'intervento, offre diversi vantaggi: tratta precocemente la malattia micrometastatica riducendo il rischio di metastasi a distanza; aumenta le probabilità di ottenere margini negativi nelle procedure di resezione (degli arti;) e consente, inoltre, di pianificare la conservazione degli arti e le procedure di ricostruzione. Dopo la rimozione del tumore, la valutazione della necrosi sul tumore primario è un
30
importante indice prognostico. Infatti, una migliore necrosi istologica è associata ad una migliore sopravvivenza [50, 51]. La risposta istologica viene definita buona quando la percentuale di necrosi chemio-indotta è uguale o superiore al 90% (pazienti good
responders). È stato proposto per il trattamento dei sarcomi ossei localizzati, un
modulatore immunitario, il muramil tripeptide liposomiale (MEPACT o Mifamurtide), somministrato una volta a settimana per circa un anno, in associazione alla chemioterapia adiuvante [7]. Questo protocollo ha dimostrato un buon vantaggio in termini di sopravvivenza, in particolar modo, se utilizzato in combinazioni chemioterapiche con l’Ifosfamide [47]. In Europa è stato approvato per il trattamento dei pazienti con età inferiore a 30 anni [46].
Le maggiori dimensioni del tumore alla diagnosi, la scarsa risposta alla chemioterapia e le difficoltà nell'ottenere margini negativi (durante la resezione), sono attribuibili ad una prognosi infausta negli osteosarcomi pelvici. Attualmente, la maggior parte dei pazienti con osteosarcomi pelvici viene trattata con la stessa strategia delle lesioni agli arti, vale a dire chemioterapia neoadiuvante, seguita da intervento chirurgico ritardato e chemioterapia adiuvante. Non esistono chiare evidenze circa la migliore combinazione chemioterapica possibile. I migliori risultati in letteratura, validi soprattutto per gli osteosarcomi dell’estremità, sono ottenuti con combinazioni di MTX-CDP-ADM+/- IFO [47]. Non vi è evidenza circa la durata ottimale e la dose cumulativa dei farmaci da utilizzare. Tuttavia, i principali studi riportati in pazienti in età evolutiva hanno utilizzato dosi cumulative di Methotrexate di almeno 60 g/m2, Cisplatino 500-600 mg/m2, Adriamicina 350-450 mg/m2, Ifosfamide almeno 30 g/m2 [47], per un periodo di circa 6-10 mesi. L'intergruppo europeo per l'osteosarcoma ha dimostrato che la sopravvivenza non differiva tra i gruppi a cui erano stati assegnati 6 cicli di Doxorubicina e Cisplatino in 18 settimane, o un multidrug regime somministrato in 44 settimane. La sopravvivenza globale riferita è stata del 65% a 3 anni e del 55% a 5 anni in entrambi i gruppi [52]. Bacci e colleghi hanno aggiornato questa esperienza con un protocollo insolito. I ricercatori hanno somministrato cisplatino intra-arterioso in due cicli preoperatori [53]. I poor responder (cioè quelli con necrosi <90%), sono stati trattati dopo l'intervento con Ifosfamide ed Etoposide, aggiunti a Doxorubicina, Metotrexato e Cisplatino. La sopravvivenza libera da eventi a 10 anni è stata del 67% per i good responder e del 56% per i poor responder. Al follow-up mediano di 11,5 anni, la sopravvivenza globale è stata