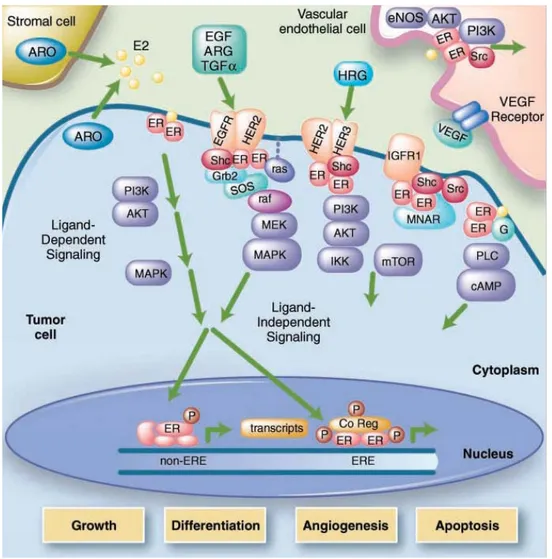
Expression of the K303R estrogen receptor alfa mulation induces hormonal resistance in breast cancer
142
0
0
Testo completo
(3)
(4)
(7)
(8)
(9)
(11)
(13)
(14)
(15)
(16)
(17)
(18)
(20)
(21)
(22)
(23)
(24)
(25)
(28)
(32)
(34)
(35)
(37)
(38)
(41)
Figura



+7






Documenti correlati
Despite these limitations, we have observed that among apparently normal sized fetuses a depressive effect of antenatal steroids is more likely if the weight percentile is in the
Lo scopo di questo sistema è quello di individuare ed identificare strutture ed oggetti sotterranei utilizzando la propagazione di onde elettromagnetiche ad alta
Mario Molteni presenta l’articolo “Organizzazioni non profit e sviluppo: il caso del Cono Norte di Lima”, pubblicato da Clara Caselli nel 2005 nella rivista Non profit, edita da
È giusto altresì tenere a mente che nei libri sul folklore di Cocchiara si valuta Corso come studioso delle tradizioni popolari e non come folklorista di guerra, d’altronde avremo
• For hemispherical domes, the ratio between the buckling loads P cr and F cr (respectively generated by a uniform external pressure and a uniform load over a
Senza un chiaro quadro delle caratteristiche dell’antecedente latino, infatti, risulta diffi- cile stabilire il confine tra l’effettiva attività di compendio del volgarizzatore,
Così come per i musei e tutti gli altri luoghi della cultura, anche le sale da concerto, i teatri o le piazze che portano in scena l’opera lirica o la musica classica
[r]
