INTRODUCTION ...2
RESVERATROL ...3
Resveratrol metabolism ...5
BIOLOGICAL ACTIVITY OF RESVERATROL...6
Resveratrol as an antioxidant...6
Resveratrol as a phytoestrogen...7
Resveratrol and cardiovascular system...9
APOPTOSIS ...13
Resveratrol and apoptosis...15
Ceramide and apoptosis...16
THE AIM OF THE THESIS ...20
NAPHTALENE ANALOGUES OF RESVERATROL...21
Results and discussions ...27
HETEROATOMS CONTAINING RESVERATROL ANALOGUES ...30
Results and discussions ...49
EXPERIMENTAL SECTION ...57
GENERAL DETAILS ...58
BIOLOGICAL ASSAY ...59
SYNTHETIC PROCEDURES...63
RESVERATROL
Resveratrol (RES) is a naturally occurring phytoalexin (3,5,4-trans-trihydroxystilbene) produced by a wide variety of plants, such as grape, peanuts, and mulberries in response to stress, injury, ultraviolet (UV) irradiation, and fungal infection as part of their defense mechanism.
Resveratrol was first identified in 1940 as a constituents of white hellebore (Veratrum
grandiflorum), and later in the dried roots of Polygonum cuspidatum, called Ko-jo-kon in
Japanese, used in traditional Chinese and Japanese medicine to treat gonorrhea favus, suppurative dermatitis and hyperlipidemia (Fig. 1).
Fig. 1. Polygonum cuspidatum.
Later, reveratrol was also detected in the leaf epidermis and the skin of grape berries but not in the flesh. Fresh grape skin contains 50 to 100 mg resveratrol per gram, and the concentration in wine may range from 0.2 to 7.7mg per liter. The epidemiologic finding of an inverse relationship between consumption of red wine and incidence of cardiovascular disease has led to the “French paradox”.
The skeleton of the molecule comprises two aromatic rings joined by a styrene double bond. The presence of the double bond facilitates trans and cis isomeric forms of RES, wich correspond to E and Z diasteromers, respectively.
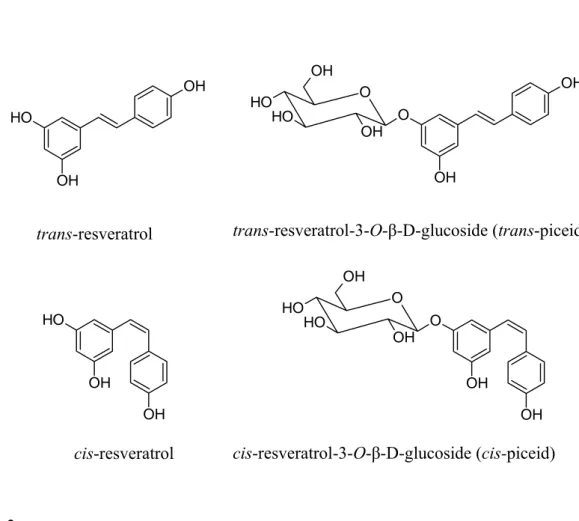
In plants, polyphenols (including resveratrol) are generally found as 3-O-β-D-glucosides, called piceids (Fig. 2).
Resveratrol is susceptible to oxidative degradation, while the glycosylated piceid form is resistant. Glycosylated resveratrol maintains its biological activity, is more stable and soluble, and therefore is more readily absorbed by the human intestine.
Fig. 2. Structural formulas of resveratrol. O H OH OH trans-resveratrolo O H OH OH cis-resveratrolo O O OH O H O H OH OH OH cis-resveratrolo-3-O-glucoside (cis-piceide) OH OH O O OH O H O H OH trans-resveratrolo-3-O-glucoside (trans-piceide) trans-resveratrol-3-O-β-D-glucoside (trans-piceid) cis-resveratrol-3-O-β-D-glucoside (cis-piceid) trans-resveratrol cis-resveratrol
Resveratrol metabolism
Resveratrol is present in dietary products as cis- and trans-resveratrol but mainly in glycosylated forms named piceids (3-O-β-D-glucosides). Plants and pathogens as well as the human digestive tract contain enzymes that oxidize polyphenols. Glycosylation inhibits enzymatic oxidation of resveratrol, thereby preserving its biological activity and increasing its stability and bioavailability. Since intestinal cells are only able to absorb aglycone resveratrol, the absorption process requires glycosidases. Therefore, the relative amounts of aglycone and glycosylated resveratrol in foods may modulate the absorption rate.
In human, the excretion time depended on the concentration of resveratrol (aglycone and conjugates) present in plasma, but there was no direct correlation between excreted and introduced amount. This observation suggests that while small amounts of resveratrol are rapidly metabolized and eliminated, retention and accumulation of the compound in tissues occur only over a certain dose of intake, thus becoming potentially available for cellular uptake and intracellular signaling. Conjugation with glycidic or sulfatidic groups is probably aimed at promoting excretion, since conjugates are not intracellularly active. Flavonoids such as quercetin inhibit the glucuronidation of resveratrol and therefore may increase its bioavailability. This observation may partly explain why low concentrations of dietary compounds have synergistic effects.
BIOLOGICAL ACTIVITY OF RESVERATROL
Resveratrol has been the focus of a number of studies investigating its biological attributes, which include antioxidant activities, antiplatelet aggregation effect, antiatherogenic property, estrogen-like growth promoting effect, immunomodulation and chemoprevention. A primary impetus for research on resveratrol has come from the paradoxical observation that a low incidence of cardiovascular diseases may coexist with intake of a high-fat diet, a phenomenon known as the “French Paradox”.1
Resveratrol as an antioxidant
Oxidative stress evoked by reactive oxygen species (ROS) is a paramount initiator, mediator and aggravator of various clinical disorders such as neurodegenerative diseases, diabetes and cardiovascular disease. The term ROS refers to molecules or parts of molecules that possess unpaired electrons in valence orbitals. This electron-deficient state makes these agents highly reactive and they can damage adjacent molecules by abstracting an electron from or donating an electron to them. They are invariably produced in aerobic environments through a variety of mechanisms.
Physiological levels of ROS participate in cellular functions and in transcriptional and posttranscriptional control. Indeed ROS have been recognized as important signal transduction intermediates that control gene expression, cell differentiation, immune activation, and apoptosis. However, excessive ROS accumulation may induce the oxidative modification of cellular macromolecules (lipids, proteins, and nucleic acids) with deleterious potential, damaging DNA and acting as tumor promoters.
Evidence has accumulated that resveratrol is both a free radical scavenger and a potent antioxidant because of its ability to promote the activities of a variety of antioxidative enzymes. The ability of poliphenolic compounds to act as antioxidants depends on the redox properties of their phenolic hydroxyl groups and the potential for electron delocalization across the chemical structure.
Resveratrol as a phytoestrogen
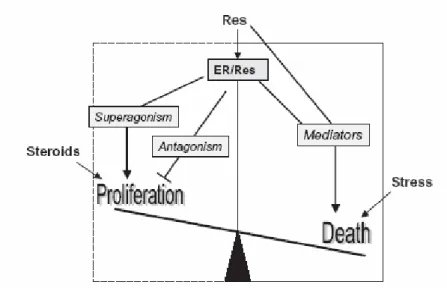
Resveratrol present a structural similarity to the synthetic estrogen diethylstilbestrol (DES) (Fig. 3). This suggested that it might be “estrogenic”, some studies demonstrated its ability to compete with labeled 17-β-estradiol (ES) for receptor binding (ER), and activate expression of estrogen-regulated genes. It binds ERs in the low micromolar range with an affinity lower than that of estradiol; therefore, it behaves as a weak competitor. Despite the lower binding affinity, resveratrol may act as a superagonist in activating hormone receptor-mediated gene transcription. The superagonistic induction of gene expression is related to the promoter context and varies according to cell type. Aside from superagonism, resveratrol also exerts an antiestrogen action by triggering parallel pathways that inhibit estrogen-induced cellular outcomes, such as proliferation, tumoral transformation and progression. In the presence of estrogen, resveratrol exerted a mixed agonistic–antagonistic action in ER-positive breast cancer cells.2
O H OH A O H OH OH A
Dietistilbestrolo (DES) trans-resveratrolo
Fig. 3. Chemical structures of RES and DES
Like other phytoestrogens, resveratrol appears to act as a SERM (a selective estrogen receptor modulator), whose effect on ER-mediated gene expression vary depending on cell type, the identity of the target gene, the presence or absence of additional estrogens, and other factors.
At the low doses provided by dietary intake, resveratrol may act as a weak estrogen competitor, according to the receptor expression and hormonal status of tissues; it counteracts the proliferative effects of hormones and provides a balancing antitumoral activity. Tissue specific expression of α and β ERs, cofactors regulating DNA binding and
different gene promoters, all modulate the cellular response to resveratrol. In the absence of endogenous hormones and according to cellular specificity, the superagonistic activity of resveratrol may act in an opposite manner and prevent tissue senescence and apoptosis. When stress signals overcome proliferative signals, or when these latter are missing (absence of hormones), the polyphenol induced pathway may switch to apoptosis (Fig. 4).
Resveratrol and cardiovascular system
Numerous studies are related to the comprehension of the “French paradox”. In fact, despite their preference for high-fat and high cholesterol diet, the French have a much lower prevalence of cardiovascular disease. This paradoxical phenomenon is referred to the vasoprotective effect of red wine. The cardioprotective mechanism of resveratrol included its role as an intracellular antioxidant and anti-inflammatory agent and the involvement of nitric oxide (NO). In kidney cells, resveratrol was also found to exert its protective action through upregulation of NO.
Resveratrol inhibits platelet activation and aggregation
Resveratrol has been shown to dose-dependently decrease platelet aggregation, which contributes to the process of atherosclerosis. Platelets stick to the endothelial surface of blood vessels where they can activate the process of thrombus formation, and their aggregation could set into motion the process of vascular occlusion. Eicosanoids synthesized from arachidonic acids have been shown to modulate platelet adhesion. The ability of resveratrol to inhibit platelet aggregation could be linked to its ability to inhibit eicosanoid synthesis.
Resveratrol and NO
Nitric oxide synthase (NOS) is a heme-containing monooxygenase with a reductase domain and an oxygenase domain. Constitutive isoforms (endothelial, neuronal and mitochondrial NOS) provide low intracellular concentrations of the short-lived free radical nitric oxide (NO). An inducible form of NOS (iNOS), activated at the transcriptional level during inflammation, provides high concentrations of NO. NO is known for its vasodilatatory properties, its inhibition of platelet adhesion and aggregation, and of adhesion molecule expression, and its suppression of cell growth and migration. It is thus apparent that NO and resveratrol share common targets. High levels of endogenously produced NO may either induce apoptosis (by generating toxic reactive intermediates) or inhibit it, depending on the intracellular redox state.
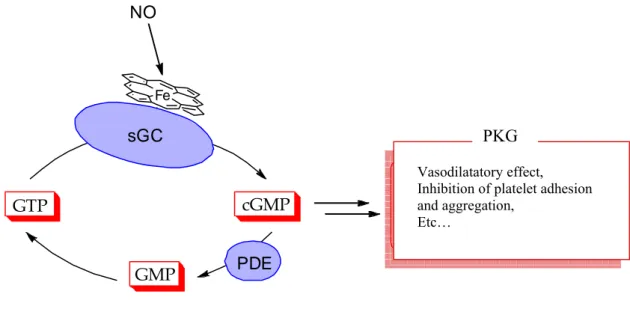
Several studies show that resveratrol upregulates NO production and potentiates NO-induced vasodilatatory effects through the NO-cGMP pathway (Fig. 4).
Cyclic GMP is produced from GTP, a guanine nucleotide triphosphate, both by soluble (sGC) and particulate (pGC) guanylyl cyclases. Nitric oxide activated the sGC. The cyclic GMP is degraded by another group of enzymes known as phosphodiesterases (PDE). After degradation to GMP (a non-cyclic monophosphate), ATP as supplier of metabolic energy is required to obtain again the initial compound: GTP. Many of the effects of cyclic GMP are mediated by the protein kinase G (PKG), an enzyme that after activation by cyclic GMP is able to phosphorylate and change the function of different proteins. The upregulation of NO from resveratrol is involved in the protection of the heart from ischemia-reperfusion injury.
Fig. 4. NO – cGMP pathway.
One of the most powerful techniques for cardioprotection is preconditioning (PC). The most generalized method of classic PC is mediated by cyclic episodes of several short durations of reversible ischemia, each followed by another short duration of reperfusion. Several regulatory pathways have been identified in different systems. Three important factors, adenosine A1 receptor, multiple kinases including protein kinase C (PKC), mitogen-activated protein kinase MAPKs, tyrosine kinases, and mitochondrial
ATP-Fe NO sGC cGMP GMP GTP PDE Vasodilatazione Inibizione piastrinica
Regolazione della muscolatura liscia Regolazione immune
ecc.
Vasodilatatory effect, Inhibition of platelet adhesion and aggregation,
Etc…
sensitive potassium (KATP) channels are known to play a crucial role in PC-mediated
cardioprotection.
Resveratrol and K+ channels
Activation of KATP channels appears to be an adaptive mechanism that protects the
myocardium against ischemia-reperfusion injury. The activation of this ion channel is at least partially responsible for the increase in outward K+ currents, shortening of action potential duration (ADP), and an increase in extracellular K+ concentration during anoxic and globally ischemic condition. It appears that delayed PC, irrespective of PC stimulus is always mediated by KATP channels.
K+ channels belong to a heterogeneous and almost ubiquitous family of proteins, wich selectively allow K+ ions to move across the cell membrane, thereby entering in the regulation of several cellular functions, such as neuronal excitability, neurotransmitter release, smooth muscle tone and hearth activity.3 Among the different factors playing an influence on the activity of different classes of K+ channels, the raise of the intracellular
concentration of free calcium ions determines the opening of a family of channels, known as calcium-activated. These channels are further subclassified into three groups on the basis of their biophysical characteristics of single channel conductance: small conductance group (SK), intermediate conductance group (IK), and big conductance group (BK).
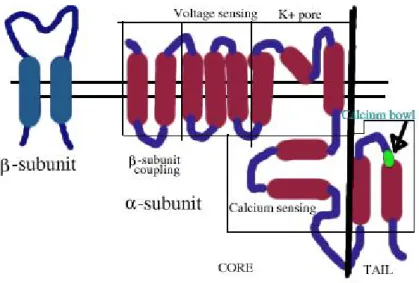
Among the three groups of K+ channels, the BK are the most studied, and they play a relevant role on the modulation of the tone of vascular smooth muscle (VSM). They are activated both by elevated cytosolic Ca2+ levels and by membrane depolarization, and consist of two distinct subunits: α and β (Fig. 5).4 Moreover, both their calcium-dependent and voltage-operated activation ensure an effective function of feed-back regulation, through a massive outward flow of K+ ions, leading to a membrane hyperpolarisation,
which counteracts the membrane depolarization and the entry of Ca2+ ions in excitable cells.
Fig. 5. Domainstructure of BK channels.
The activation of vascular BK channel can be due to intracellular cGMP, through a cGMP dependent protein kinase, to cAMP-dependent, cAMP-independent, G-proteins and GMP. They can also be activated by many endogenous agents, such as reactive oxygen species,and nitric oxide.
Agents that cause BK channel activation are called BK openers or activators. BK openers stabilize the cell by increasing efflux of potassium ions, leading to hyperpolarization, and thus decrease the cell excitability and/or cause smooth muscle relaxation. Activators of these channels have potential therapeutic implications because of the profound involvement of BK channels in conditions such as hypertension, coronary artery spasm, urinary incontinence, and several neurological disorders.
Recent studies show that resveratrol act as a BK-opener.5 In particular, resveratrol and the synthetic benzimidazolone NS 1619, a well-known BK activators, were tested to determine a possible vasodilator mechanism of action. Their vasodilator profiles, seems to be related to the release of endothelial NO. In particular, the functional effects due to the activaton of endothelial BK-channels by these drugs seem to be more pronounced and relevant than those due to the activation of BK channel expressed in the VSM cells.
APOPTOSIS
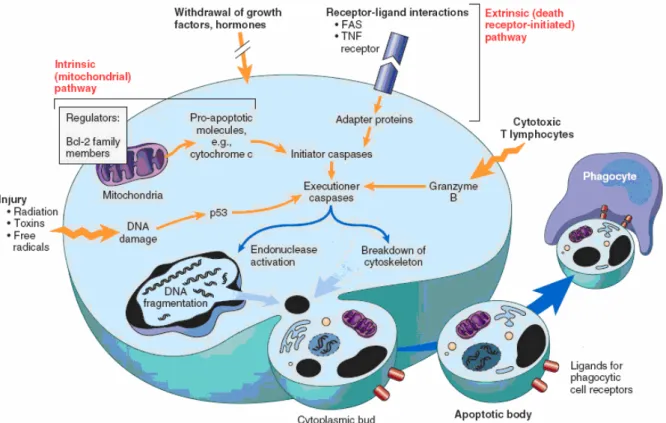
Apoptosis or programmed death cell refers to a mechanism by which cell die in an orderly manner through a genetically governed event or via the triggering of a series of parallel, cross-talking pathways that induce orderly cell death. Two central pathways lead to apoptosis: (i) positive induction by ligand binding to a plasma membrane death receptor, and (ii) negative induction by loss of a suppressor activity involving the mitochondria (Fig. 6).
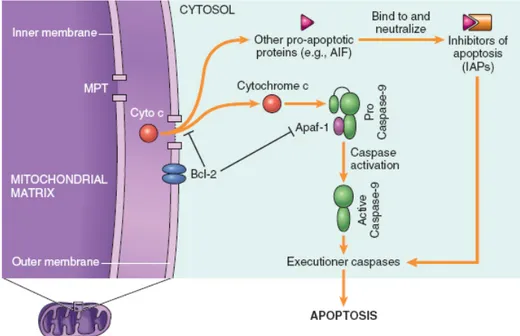
Each pathway feeds into the activation of a pivotal group of thiol aspartate-specific proteases, i.e., caspases. Caspases are activated by a wide range of stimuli from death receptors, loss of survival signals, redox stress, and ultraviolet light and irradiation, to the release of cytochrome c and other death promoting factors (such as AIF (apoptosis inducing factor) and Smac/DIABLO) from the mitochondria into the cytosol, all of wich serve as triggers to activate caspases. In the classic death receptor model of Fas receptor acivation, the Fas ligand exist as a homotrimeric membrane molecule, with each FasL trimer binding three FasR molecules on the surface of the target cell. This results in the death inducing signaling complex (DISC) that is a complex of Fas (trimer), Fas-associated death domain (FADD), and procaspase-8. Upon recruitment by FADD, procaspase 8 oligomerization drives its self-activation through autoprocessing, and active caspase-8 then activates downstream caspases, committing the cell to apoptosis. The mitochondria play an active role in apoptosis, which is regulated by a group of proteins known as the Bcl-2 family of proteins (antiapoptotic, and proapoptotic)(Fig. 7).
Whereas antiapoptotic members of the Bcl-2 family protect mitochondrial integrity by inhibiting membrane depolarization/permeabilization and translocation of death amplifying protein, the proapoptotic counterparts neutralize these effects by dislodging antiapoptotic members from the mitochondria and forming oligomers that function as channels for the release of transmembrane protein. The cytosolic cytocrome c triggers the recruitment of the protein Apaf-1(apoptotic protease activating factor-1), procaspase-9, and dATP, to bring about their assembly of the apoptosome. Apoptosome formation triggers activation of caspase-9, which accelerates apoptosis by activating other caspases.
Resveratrol and apoptosis
Antitumor agents impair procarcinogen metabolic activation and interaction with cellular targets (DNA, proteins); they inhibit cancer development by blocking tumor cell transformation and proliferation and by inducing tumor cell death. Therefore, chemoprevention and chemotherapy can be obtained by acting at multiple levels and by impairing combinatorial effects responsible for mutagenesis and mitogenesis. Among food-derived molecules that have been screened for the ability to inhibit or reverse such cellular processes, resveratrol is particularly interesting because it affects a broad range of intracellular mediators involved in the initiation, promotion and progression of cancer.
The results relating to resveratrol’s apoptotic capability are confusing and remain controversial. Resveratrol appears to induce cell death, apoptosis, through different mechanisms: (i) cell death receptors, (ii) mitochondria pathway, (iii) nuclear factors and cell cycle arrest, and (iiii) sensitization to drug-induced and free-radical-associated apoptosis, in the context of its use as a cancer chemotherapeutic agent.
Ceramide and apoptosis
Traditional anticancer agents exert their action by blocking the final events of cell cycle, such as DNA synthesis or mitosis. Therefore, these drugs often result poorly selective and quite toxic.
An alternative in cancer therapy might be the use of drugs able to act before the final events of cell cycle, controlling those mechanisms that regulate the levels of the different cell populations.
These levels in multicellular organisms depends on a complex dynamic balance between the mechanisms which regulate cell proliferation and those involved in programmed cell death, commonly termed apoptosis. Alterations and defects to these control mechanisms which determine whether a cell proliferates or dies, are the main pathogenesis of a variety of human diseases, including cancer, auto-immune disease and viral infections. It has been demonstrated that tumor progression may depend not only on an uncontrolled cell proliferation but also on dysfunction in the physiological pathways of apoptosis.
For this reason, it is nowadays believed that the possibility of pharmacological interventions able to restore the physiological apoptotic death pathway represents a concrete expectation in an anticancer therapy with a selective toxicity toward malignant but not normal cells, whose apoptotic pathways are not altered.
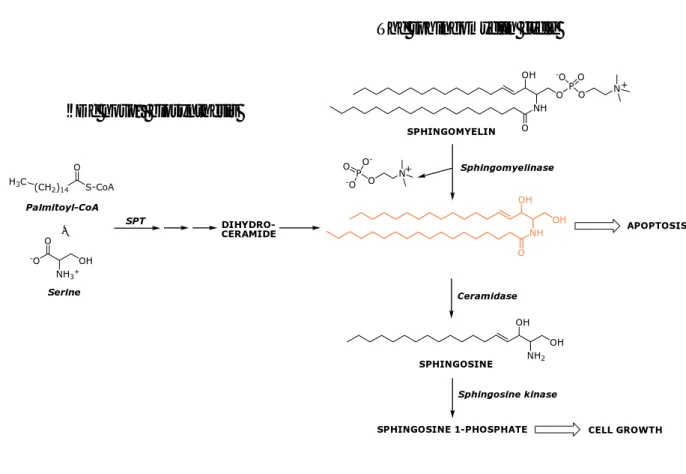
Ceramide, a sphingosine-based lipid molecule, has attracted great attention due to its emerging role as an intracellular effector molecule in apoptosis; in particular, ceramide represents the second messenger of the so-called ‘sphingomyelin pathway’ (Fig. 8).
Fig. 8. Ceramide biosynthesis.
Ceramide is generated by hydrolysis of membrane sphyngomielin by sphingomyelinases. It has been identified as a cell death effector and its generation increases in response to apoptotic stimuli including stress, radiation, chemotherapy, and death ligands. In contrast, shingosine-1-phosphate (S1P) a ceramide metabolite, has been mainly characterized as an antiapoptotic sphingolipid mediating cell proliferation and survival.6
Ceramide is also produced by de novo synthesis through ceramide synthase, and propagates the signal within the cell by the activation of stress activated protein kinases (SAPKs), leading to apoptotic death through the interaction of ceramide with caspases and other apoptotic signaling cascades.
On this basis, ceramide could be viewed as a possible therapeutic agent able to induce apoptosis in tumor cells. However, it is important to point out that natural ceramide is unable to cross the cell membrane and, therefore, cannot be used directly for therapeutic purposes. NH O O OH P O O -O N SPHINGOMYELIN Sphingomyelinase N O P O -O -O NH O OH OH OH OH NH2 APOPTOSIS Ceramidase SPHINGOSINE Sphingosine kinase
SPHINGOSINE 1-PHOSPHATE CELL GROWTH
+ + S-CoA (CH2)14 O Palmitoyl-CoA O NH3+ OH -O Serine DIHYDRO-CERAMIDE H3C SPT +
The sphingomyelin cycle
Recently, resveratrol was shown to promote intracellular accumulation of ceramide in breast and prostate cancer cells.7,8 Resveratrol enhanced the de novo synthesis of this sphingolipid by increasing the activity of the rate limiting enzyme. This observation identified a new important checkpoint in the actions of resveratrol. Although this polyphenol triggers multiple pathways, ceramide production may be a common step that drives cells toward irreversible death.
Some experiments have been done to demonstrate that ceramide mediates the antiproliferative/proapoptotic effect of resveratrol on MDA-MB-231, a highly metastatic and invasive breast cancer cell line, and to investigate the relative role of de novo biosynthesis and sphingomyelin hydrolysis in the antiproliferative/proapoptotic role of ceramide after resveratrol treatment.
These experiments showed that resveratrol were able to significantly block cell proliferation, cell viability, and colony formation and to induce apoptosis in a dose-dependent manner. The researcher tested whether accumulation of endogenous ceramide were correlated with the biological effects induced by resveratrol, and concluded that the induction of endogenous ceramide by resveratrol mirrors the induction of biological effects.
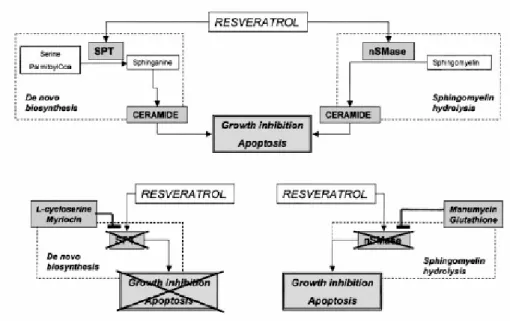
In this study they show that resveratrol has a potent antiproliferative and proapoptotic effect on MDA-MB-231, a highly invasive and metastatic cell line from human breast cancer known to be resistant to several anticancer drugs. The resveratrol-induced biological effect is dose dependent and correlates with a dramatic increase of endogenous ceramide. They further show that resveratrol induced ceramide accumulation is consequent to both activation of SPT, the key rate-limiting enzyme of the de novo synthesis of sphingolipids, and nSMase, the main enzyme responsible for the sphingomyelin/ceramide cycle (Fig. 9, upper panel). The profound biological effects of resveratrol in MDA-MB-231, an ER-negative cell line, confirms that the resveratrol cellular response is independent of ER status as observed in other breast and prostate cell lines. They demonstrated that some resveratrol-induced ceramide is indeed responsible of the resveratrol-resveratrol-induced biological effects by using ad hoc inhibitors of the enzymes involved in ceramide generation. SPT inhibitors were able to counteract resveratrol-induced inhibition of cell proliferation and induction of apoptosis (Fig. 9, lower panel, left). In contrast, nSMase inhibitors did not affect the resveratrol-induced biological responses (Fig. 9, lower panel, right).
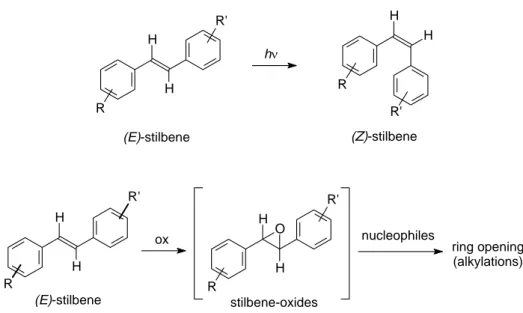
(E)-stilbene H H stilbene-oxides H H O R' R ox nucleophiles ring opening (alkylations) R' R
NAPHTALENE ANALOGUES OF RESVERATROL
Application of resveratrol as an antitumour agent is limited by several factors, some of which are related to the presence of a stilbene-type scaffold. First of all, its photosensitivity causes, under UV irradiation, its convertion into the (Z)-isomer, less stable and biologically less active.9,10 Moreover, resveratrol is metabolically unstable. For example its stilbene
double bond may be oxidized by cytochrome P450 monooxygenases into highly reactive epoxides that may act as carcinogenic metabolites (Fig 10).11
Fig. 10. Instability of stilbene derivatives
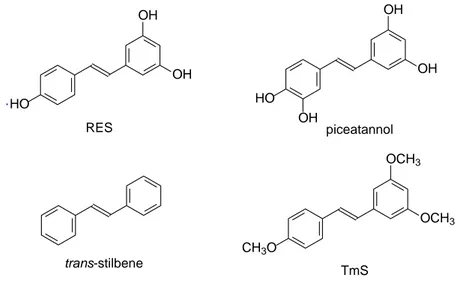
We have participated in studies demonstrating that the presence of free phenolic OH groups in the resveratrol molecule are fundamental to induce ceramide-mediated apoptosis. In these studies we, initially, compared the ability of resveratrol and three resveratrol analogues to induce both ceramide increase and growth inhibition in the androgen receptor (AR)-negative prostate cancer cell line PC3. The resveratrol analogues employed are: (i) piceatannol (3,3’,4’,5-trans-tetrahydroxystilbene), bearing an additional hydroxyl group in the 3’ position when compared to RES; (ii) trans-stilbene, the nonhydroxylated analogue;
H H R R' R H H R' hν (E)-stilbene (Z)-stilbene
and (iii) TmS (3,4’,5’-trimethoxy-trans-stilbene), with methoxy replacing the hydroxy groups (Fig. 11).12
Fig. 11. Structure of stilbenoids used in the study.
All these compounds showed significant antiproliferative effects, with the only exception of unsubstituted stilbene, as we can see from the proliferation index shown in Fig. 12.
Fig. 12. Antiproliferative activities of RES, piceatannol, (E)-stilbene and TmS. HO OH OH RES piceatannol HO OH OH OH trans-stilbene TmS CH3O OCH3 OCH3 . 0 2 4 6 0 1 2 3 4 TmS Days Vehicle 16 μM 32 μM 64 μM P rol if er a ti o n ind ex 0 2 4 6 0 1 2 3 4 TmS Days Vehicle 16 μM 32 μM 64 μM P rol if er a ti o n ind ex TmS Days Vehicle 16 μM 32 μM 64 μM P rol if er a ti o n ind ex 0 2 4 6 0 1 2 3 4 (E)-Stilbene Days P rol if er a ti o n ind ex 16 μM32 μM64 μM Vehicle 0 2 4 6 0 1 2 3 4 (E)-Stilbene Days P rol if er a ti o n ind ex 16 μM32 μM64 μM 0 2 4 6 0 1 2 3 4 (E)-Stilbene Days P rol if er a ti o n ind ex 16 μM32 μM64 μM (E)-Stilbene Days P rol if er a ti o n ind ex 16 μM32 μM64 μM Vehicle 0 2 4 6 0 1 2 3 4 Vehicle 16 μM 32 μM 64 μM Prol if erati o n ind ex Days RES 0 2 4 6 0 1 2 3 4 Vehicle 16 μM 32 μM 64 μM Prol if erati o n ind ex Days RES 0 2 4 6 0 1 2 3 4 Piceatannol Vehicle 16 μM 32 μM 64 μM Days P rol if er at io n ind ex 0 2 4 6 0 1 2 3 4 Piceatannol Vehicle 16 μM 32 μM 64 μM Days P rol if er at io n ind ex
The compounds showing an antiproliferative effects were later tested for the ceramide accumulation in the same cancer cell line, and while TmS showed an antiproliferative activity that was not associated to a significant ceramide accumulation, RES and piceatannol caused an antiproliferative effect associated with ceramide accumulation.
This is consistent with what was known in the literature, i.e. that TmS inhibits cellular growth by interfering with the mitotic process by impairing tubilin polymerization (it is an inhibitor of tubilin polymerization).13,14 Therefore, the presence of free phenolic OH groups seems to be fundamental to get this ceramide-mediated mechanism of action.
For these reasons, RES would appear as a good lead for developing new molecules endowed of better biopharmacological properties.
Most research groups working in this field have tried to synthesize and test new analogues of RES by essentially modifying the external substituents (moving OH’s to different positions, removing some of them, introducing extra substituents such as Cl, CF3,
Fig. 13. Resveratrol analogues with stilbene structure.
In the first study were synthesized a family of monohydroxylated (E)-stilbenes, with fluoro, difluoro, methoxy or dimethoxy substituents (compounds ax-jz). Evaluation of the apoptosis-inducing properties of hydroxylated (E)-stilbenes was undertaken in two human cancer cell lines, MDA MB 468 (breast) and HCT 116 (colon). In all cases, the human mammary carcinoma cell line MDA MB 468 was the most sensitive cell line examined with GI50 values in the low micromolar range, with agents jy (0.96 μM), iz (1.1 µM), gy
(1.4 µM), and iy (1.6 μM).15
In the second study a variety of substituents were introduced at positions 3’ and 4’ (OH, NH2, OCH3), and the replacement of the 3,5-hydroxy groups with methoxy functions was
k l m
n o p
also investigated. All the new synthesized compounds were assayed in vitro for cell growth inhibition and for their ability to induce apoptosis in HL60 cells, and they showed an appreciable activity.16 In no cases was ceramide accumulation evaluated.
Moreover, in all these reports the stilbene scaffold is still preserved. Unfortunately stilbene derivatives may present problems if developed as drugs, because of the high reactivity of the stilbene double bond, as previously described. For these reasons we wanted to develop more “drug-like” analogues of RES by modifying the stilbene double bond.
The simplest modification that came up to our mind was to synthesize a more rigid derivative. The resulting molecular target would be a naphthalene analogue possessing two condensed benzene rings in the place of the styrene portion of RES.17 To assess the importance of the phenolic groups in this new class of resveratrol analogues we then synthesized a series of naphthalene-based resveratrol analogues, which possess either “all-free” hydroxyls (1), or various combinations of O-methylated OH substituents (2-4) (Fig. 14).
Fig. 14. Resveratrol and its naphthalene analogues.
This simple, yet unexplored, structural modification enables preservation of the original geometrical orientation of the pharmacophore groups of resveratrol, while adding more conformational rigidity to the whole structure.
OH OH HO H H OH OH HO .MeO OH OH . HO OMe OMe . MeO OMe OMe 1 2 3 4
b Br OTBDMS OTBDMS MeO OH OH + MeO B(OH)2 . . a RO Br MeO OMe B(OH)2 + RO OR' OR' a
Compounds 1, 2 and 4 were synthesized as outlined in Scheme 1. A Suzuki Pd-catalyzed cross-coupling reaction between 3,5-dimethoxyphenylboronic acid (7) and 6-bromo-2-naphthol (5) or 6-methoxy-2-bromo-naphthalene (6) was employed to synthesize compounds 2 and 4, respectively. The completely demethylated compound 1 was obtained by treating 4 with boron tribromide at -78 °C.
Scheme 1
Reagents and conditions: (a) Pd(PPh3)4, K3PO4 (aq), dioxane, 80 °C, 16 h; (b) BBr3, dry CH2Cl2,
-78°C.
The synthesis of monomethylated compound 3 was achieved as shown in Scheme 2. In this case, 6-methoxynaphthylboronic acid (8)18 was again submitted to a Suzuki reaction with 3,5-(di-tert-butyldimethylsilyloxy)-1-bromobenzene (9)19. Acidic aqueous workup caused complete desilylation of the protected phenolic groups, affording pure compound 2.
Scheme 2
Reagents and conditions: (a) (1) Pd(PPh3)4, K3PO4 (aq), dioxane, 80 °C, 16 h. (2) HCl (aq).
5: R= H 6: R= Me 2: R= H, R’= Me 4: R= Me, R’= Me 1: R= H, R’=H 7 8 9 3
Results and discussions
These resveratrol analogues were compared for their growth inhibitory effect on MDA-MB-231, a highly metastatic breast cancer cell line. Cells were treated with compounds 1-4 at different concentrations, ranging from 0.25 μM to 64 μM for 6 days. Proliferation index was evaluated by the sulforhodamine B (SRB). The values are the mean ±SD of three independent experiments (Fig. 15).
The IC50 of the four analogues, and of resveratrol, are reported in Table 1. Compounds 1
and 3 showed an antiproliferative effect like resveratrol itself whereas the antiproliferative effects of 2 and 4 were lower.
Table 1. IC50 for resveratrol and resveratrol analogues.
Compound IC50 (µM) RES 1 2 3 4 20.5 ± 2.6 12.7 ± 1.3 28.3 ± 3.8 7.2 ± 1.0 45.1 ± 6.8
(IC50 (μM) was calculated as the mean ± SD of the
proliferation values obtained at 2, 4, and 6 days of treatment with each compound used at different concentrations in three independent experiments).
Futhermore, to test whether the antiproliferative effect of the four analogues involved an apoptotic mechanism mediated by ceramide accumulation, they were submitted to PARP cleavage and to the diacylglicerole-kinase (DGK) assay. The MDA-MB-231 cells, treated for 4 days with the four resveratrol analogues at the corresponding IC50 (Table 1), displayed
a level of ceramide significantly higher (p < 0.01) than the level induced by resveratrol only when treated with compound 1. To assess whether the antiproliferative effect could be traced to an apoptotic mechanism, we tested the induction of poly-(ADPribose)-polymerase (PARP) cleavage by the four analogues after treating MDA-MB-231 cells with the IC50
(see Table 1) for 4 days. The 86 kDa band, which is an evidence of PARP cleavage, was detected only with compound 1 (Fig. 16). Thus, only the resveratrol analogue with intact hydroxyls and an additional naphthalene ring exerts an antiproliferative effect via an apoptotic mechanism.
Fig. 16. Ceramide accumulation and proapoptotic effect from resveratrol and synthetic analogues.
In conclusion, our data shows that the introduction of a naphthalene ring into the resveratrol molecule confers an improved ceramide-mediated proapoptotic activity.
RES OH OH HO OH OH HO 1 10 N HO OH OH
HETEROATOMS CONTAINING RESVERATROL ANALOGUES
Based on these findings, we considered replacing the naphtol moiety with other heterocyclic or pseudo-heterocyclic mimics, in order to check the effect of an increase of polarity and hydrosolubility. We started from replacing the naphtol moiety with a more polar quinoline moiety (Fig. 17).20 In order to predict the hydrophilic properties of new
analogues, theoretical values of ClogP, tPSA (topological polar surface area) e ClogS (S= water solubility in mol/L) have been calculated utilizing QikProp software v2.5 (rel 21) (Schrödinger). The results are shown in Table 2.
The dissolution of an organic molecule is determined by the highly interdependent influences of aqueous solubility, ionizability (pKA), and lipophilicity (octanol/water log P).
Indeed, the polar surface area (PSA) is a very significant descriptor for drug transport properties, such as human intestinal permeation and blood-brain barrier penetration.
Table 2. ClogP, tPSA (Å2) and ClogS values for compound 1 and quinoline analogues 5 obtained with QikProp software. Compound ClogP tPSA (Å2) ClogS 1 2.21 67.41 -3.21 10 3.01 82.70 -3.48
We then synthesized quinoline 10 and its Cl-substituted precursors 14 and 15. They were obtained as reported in Scheme 3. 3,5-Dimethoxyphenylacetic acid 11 was converted to acylchloride 12 by reaction with thionyl chloride. The intermediate 12 afforded amide 13 by reaction with m-anisidine. In this reaction, PS-DIEA was used to neutralize the reaction mixture. PS-DIEA is dimethyl-isopropyl-ethyl amine which is supported on polystyrene. This resin captured HCl that was released during the reaction and remained in a solid state. Finally, we separated 13 by filtration, avoiding problems of purification. Compound 14 was obtained from intermediate 13 by Vilsmeier reaction with phosphoryl chloride in DMF, and it was demethylated with boron tribromide to give chloroquinoline 15. Finally chloroquinoline 15 afforded quinoline 10 by catalytic hydrogenation under 1 atm of H2 on
14 13 12 11 10 15 e d c b a N HO OH OH N HO Cl OH OH N MeO OMe OMe Cl MeO NH O OMe OMe COCl OMe MeO COOH OMe MeO Scheme 3
Reagents and conditions: (a) SOCl2, CH2Cl2 , 60 °C; (b) m-anisidine, CH2Cl2, DMAP, PS-DIEA, r. t.; (c)
POCl3, DMF, 75 °C; (d) BBr3, CH2Cl2, -78 °C; (e) H2/Pd-C, 24 h.
The quinoline analogue 10 together with its precursors 14, and 15 were tested for the antiproliferative activity (see in Results and Discussion, pag 49).
Furthermore, we synthesized derivatives 16, 17, 18 and 19 as novel heterocyclic analogues (Fig. 18), in which the phenolic ring of RES was replaced by a methoxy-substituted benzothiazole. In fact, similar derivatives have been reported to be good antiproliferative agents.21,22
These compounds showed a Clog P =3.36, higher than compound 1, but a better value of TPSA (82.7 Å2 with respect to 71.4 Å2), due to the presence of the nitrogen atom, that confers more hydrophilicity to the molecule.
RES OH OH HO OH OH HO 1 N S OH OH HO 16 N S OH HO OH 17 N S HO OH OH 18 N S OH OH OH 19
Fig. 18.Resveratrol, naphtalene and benzothiazolic analogues.
Derivatives 16 and 17 were obtained as reported in Scheme 4. The p-anisoyl chloride 20 afforded amides 23-24 by reaction with substituted amines 21-22. Compounds 25-26 were obtained by thionation of benzanilides with Lawesson’s reagent in refluxing chlorobenzene. Cyclization of thiobenzanilides with potassium ferricyanide afforded benzothiazoles 27-28 that were finally demethylated with boron tribromide to give the hydroxylated benzothiazoles 16-17.
Scheme 4 27-28 25-26 23-24 21-22 20 16-17 d N S OH c b a OMe COCl + NH2 NH O OMe N H S OMe N S OMe R1 R2 R1 R2 R1 R2 R1 R2 R1 R2
Reagents and conditions: (a) DMAP, PS-DIEA, CH2Cl2, r.t.; (b) Lawesson’s reagent, 129 °C; (c)
K3Fe(CN)6, NaOH30%, EtOH, H2O; (d) BBr3, CH2Cl2, -78 °C.
Benzothiazoles 18 and 19 were synthetized as respectively reported in Schemes 5 and 6, with a reaction route similar to that of compounds 16 and 17.
Nevertheless, the cyclization of thioamide 36 (Scheme 6) afforded a mixture 1:1 of the 5-methoxybenzothiazoles 37 and its 7-methoxy isomer 38. The mixture was submitted to demethylation with boron tribromide, but after cromatography we were only able to isolate pure compound 19.
N H O MeO OMe OMe N H S MeO OMe OMe N S MeO OMe OMe N S HO OH OH COOH MeO OMe COCl MeO OMe a b c d e 29 30 31 32 33 18 Scheme 5
Reagents and conditions: (a) SOCl2, CH2Cl2, 60 °C; (b) DMAP, PS-DIEA, CH2Cl2, r.t.; (c) Lawesson’s
e d c b a COCl MeO OMe COOH MeO OMe N S OH OH HO N S OMe OMe MeO N H S OMe OMe MeO N H O OMe OMe MeO + N S OMe OMe OMe N S OH OH OH + 29 34 35 36 37 38 39 19 Scheme 6
Reagents and conditions: (a) SOCl2, CH2Cl2 , 60 °C; (b) DMAP, PS-DIEA, CH2Cl2, r.t.; (c) Lawesson’s
reagent, 129°C; (d) K3Fe(CN)6, NaOH30%, EtOH, H2O; (e) BBr3, CH2Cl2, -78 °C.
We have continued the series of heterocycles derivatives, and synthesized the new analogues showed in figure 19, based on benzofuran, phthalimmide, and salicylaldoxime scaffolds.
The theoretical values of ClogP, tPSA and CLogS have been calculated with QikProp software in order to predict hydrophilic properties also of some of these new analogues. The results are shown in table 3.
OH OH HO 1 N S OH OH H2N 40 41 S N OH OH H2N N O O HO OH OH 42 OH OH O N H HO 43 O OH OH HO 44 45 O OH OH HO
Fig. 19. New heterocycles and pseudo-heterocycles analogues designed.
Table 3. CLogP, tPSA (Å2) and CLogS of heterocycles compounds (41, 43, 44), resveratrol and
compound 1 obtained with QikProp software.
Compound CLogP tPSA (Å2) CLogS
RES 1.99 67.41 -2.75
1 2.21 67.41 -3.21
44 1.08 82.97 -2.31
43 0.26 101.49 -1.96
From a theoretical point of view, all these new derivatives showed better hydrophilic properties than resveratrol. In particular compound 41 and derivative 44 showed an appreciable increase of solubility with a CLogS values of -2.66 and -2.31 respect to -3.21 of compound 1. Salicylaldoxime derivative 43 possessed a good hydrofilicity with a tPSA= 101.49 Å2 higher than those of compound 1 (tPSA=67.41 Å2) and a ClogP=0.26 lower than those of compound 1 and (ClogP=2.21).
The synthesis of benzofuran derivatives 44 and 45 was achieved as reported in Schemes 7 and 8, respectively.23
The 2-hydroxy-5-methoxybenzaldehyde 46 was reduced to alcohol 47 using sodium borohydride. Treating alcohol 47 with triphenylphosphine hydrobromide gave phosphonium salt 48. The key step for the formation of the benzofuran backbone was achieved by an intramolecular Wittig reaction between triphenylphosphonium salt and 3,5-dimethoxybenzoic acid. Wittig reaction gave the desired benzofuran 49, at a yield of 47%. Finally benzofuran 49 was demethylated with boron tribromide to give the desired product
44.
On the other hand, the synthesis of the benzofuran derivative 45 required the protection of the 2-hydroxy group of benzaldehyde 50 to obtain alcohol 53. In fact the direct reduction of 2-hydroxy-4-methoxybenzaldehyde gave a mixture not easily separable by chromatography. We have tried the MEM chloride as a protecting group but also in this case we did not detect the desired product. Finally, the protection of hydroxyl group with
tert-butyldimethylsilylchloride (TBDMSCl) and following deprotection afforded alcohol 53. The subsequent reactions were the same as those for benzofuran 44.
MeO CHO OH OH OH MeO a b PPh3Br MeO OH c O OMe OMe MeO d O OH OH HO 44 49 48 47 46 MeO OH O H MeO OTBDMS O H 51 MeO OTBDMS OH 52 MeO OH OH MeO OH PPh3Br O MeO OMe OMe O HO OH OH 53 54 55 45 a b c d e f 50 MeO OH OH 53 b Scheme 7
Reagents and conditions: (a) EtOH, NaBH4; (b) PPh3·HBr, CH3CN, reflux; (c) 1) 3,5-dimethoxybenzoic
acid, DCC, DMAP,CH2Cl2 2) Et3N, dioxane, reflux; (d) BBr3, dry CH2Cl2, -78°C.
Scheme 8
Reagents and conditions: (a) TBDMSCl, DIPEA, DMF; (b) EtOH, NaBH4; (c) TBAF, dry THF; (d)
We envisaged the possibility of bioisosterically replace the phenolic ring present in resveratrol. The salicylaldoxime moiety was identified for the structural relatedness with the phenolic group.24 It is characterized by a six-membered pseudo-ring (A'),formed by an intramolecular hydrogen bond. This ring (A’) presents several features that indicate its similarity with the phenolic A-ring: (i) both rings have approximately same size and same planar л-conjugated (at least partially, in the case of the salicylaldoximes) hexagonal geometry; (ii) the OH of the oxime group is attached to an sp2 hybridized atom (nitrogen) that is intramolecularly hydrogen-bonded to the ortho phenol and has a pKa value around
10, within the pKa range of typical phenolic OH groups (Fig. 20).
OH OH O H A O OH OH N O H H A' 1 6
Fig. 20. Structural relationship between the phenolic ring (A) and the pseudo-cycle (A').
The synthesis of compound 43 was accomplished as shown in Scheme 9. 3-Bromophenol
56 was alkylated with allyl bromide to give the intermediate allyl ether, which underwent Claisen rearrangement at 180 °C and afforded a mixture 1:1 of 58 and its isomer 59. After a difficult purification we could isolate the desired isomer 58. Rearrangement of the terminal double bond of 58 to an internal, conjugated position was achieved by treatment with potassium t-butoxide in DMSO. The bromide 60 was then submitted to a Pd-catalyzed cross-coupling in typical Suzuki conditions with 3,5-dimethoxyphenylboronic acid. Salicylaldehyde 62 was obtained by oxidative cleavage of the double bond present in 61, using sodium periodate and catalytic amounts of osmium tetroxide.
Salicylaldoxime 43 was obtained by demethylation of 62 with boron tribromide and by subsequently treating aldehyde 63 with hydroxylamine hydrochloride.
Br OH Br Br O Br OH a b Br OH OMe OMe HO OMe OMe HO O H c d e OH HO O H OH OH O N OH HO H f g 43 63 62 61 60 58 57 Br OH 56 59 Scheme 9
Reagents and conditions: (a) K2CO3, CH3CN, 80 °C; (b) N-methylaniline, 180 °C, 48h; (c) t-BuOK,
DMSO, 55 °C, 2h; (d) 3,5-dimethoxyphenylboronic acid, Pd(PPh3)4, K3PO4, dioxane; (e) OsO4 in t-BuOH,
NaIO4, dioxane, H2O, 50 °C; (f) BBr3, dry CH2Cl2; (g) NH2OH·HCl, MeOH.
Phthalimide 42 was prepared as shown in Scheme 10. 3,5-Dimethoxyaniline 65 was heated at 200 °C with 4-hydroxyphtalic acid 64 to generate the corresponding phthalimide
66.
O OH O OH HO + NH2 OMe MeO O O N OMe OMe HO O O N OH OH HO 66 42 a b 64 65 Scheme 10
Reagents and conditions: (a) heat, 200°C; (b) BBr3, dry CH2Cl2, -78°C.
Some studies reported in the literature demonstrated that the 2-aminothiazole scaffold is a good mimic, in particular for narcotic analgesics25 and dopaminergic agents26.Therefore we
thought interesting to replace the phenolic portion (A) with two isomer of 2-aminothiazole
portion (A' and A''), like in compounds 40 and 41 (Fig. 21).
N S OH OH H2N S N OH OH H2N HO OH OH 1 7a 7b A A '' A'
Fig 21. 2-Aminothiazoles derivatives.
The synthesis of compound 40 is reported in Scheme 11. 4-Bromoaniline 67 was submitted to a Suzuki reaction with 3,5-dimethoxyphenilboronic acid, affording derivative
68.27 The cyclization of product 68 with KSCN, and Br2 in AcOH afforded the bromo
substituted amminobenzothiazole 70 instead of the desired product 69. This is probably due to the activation of the aromatic ring which then easily undergoes to the electrophilic substitution. The activation may be due to the presence of the amino group in the para position of the phenyl ring togheter with the methoxy groups on the other aromatic ring.
We have then tried a non electrophilic oxidant in the place of Br2such as an hypervalent
iodine reagents, the Dess-Martin periodinane28 (DMP), in CH2Cl2 at room temperature
together with KSCN, but the reaction led to a complex mixture in which the desired product was not identified. A catalytic hydrogenation with Pd/C 10% of bromo derivative 70 did not afford the desired product 69 devoid of the bromo substituent on the phenyl ring. This was probably due to the presence of sulfur atom that poisoned the palladium catalyst.
Also the synthesis of compound 41 had no success (Scheme 12). The 2-bromoaniline 71 was submitted to a Suzuki reaction with 3,5-dimethoxyphenilboronic acid, affording derivative 72. Benzothiazole 73 was obtained by cyclization of product 72 with KSCN/Br2,
but again the demethylation with boron tribromide did not give the desired product 41.
Scheme 11
Reagents and conditions: (a) 3,5-dmethoxyphenylboronic acid, Pd(OAc)2, PPh3, EtOH/toluene 1:1,
Na2CO3, 100 °C; (b) KSCN, Br2/AcOH; (c) H2, Pd/C, EtOH; (d) BBr3, dry CH2Cl2.
H2N Br a OMe OMe H2N b S N H2N OMe OMe S N H2N OMe OMe Br c d S N H2N OH OH 7a 23 24 25 26 67 40 69 68 70
Scheme 12
Reagents and conditions: (a) 3,5-dimethoxyphenylboronic acid, Pd(OAc)2, PPh3, EtOH/toluene 1:1,
Na2CO3, 100 °C; (b) KSCN, Br2/AcOH; (c) BBr3, dry CH2Cl2. Br H2N a OMe OMe H2N b N S H2N OMe OMe c N S H2N OH OH 7b 27 28 29 71 72 73 41
We pointed our attention to the replacement of the stilbene scaffold present in resveratrol with a linear portion. We substituted the metabolically unstable stilbene double bond with a more stable amido, thioamido or sulfonamido moiety.
Initially, we designed and synthesized the amido and thioamido compounds showed in Figure 22.
Fig. 22. Amido and thioamido analogues.
We were aware that anilides and thioanilides may exist as s-cis or s-trans conformers. From 1H NMR studies of N-substituted anilides and thioanilides, there is known to be a strong preference for the s-cis isomer (N-phenyl group trans to carbonyl oxygen).29 The
s-trans isomer is detectable only when large substituents are present on the N-phenyl ring or
when R=H. The preference for s-cis conformation would make these derivatives different from RES (s-trans). Nevertheless this is a dynamic equilibrium that may be affected by many factors when these molecules are placed in a biological environment. Therefore, considering also the easy synthetic accessibility of these compounds, we thought this approach to be worthwhile (Fig. 23).
RO NH OR OR X RO NH X OR OR RO NH X OR OR NH X RO OR OR OR NH X RO OR OR OR 74: X=O, R=Me 75: X=O, R=H 76: X=S, R=Me 77: X=S, R=H 78: X=O, R=Me 79: X=O, R=H 80: X=S, R=Me 81: X=S, R=H 82: X=O, R=Me 83: X=O, R=H 84: X=S, R=Me 85: X=S, R=H 86: X=O, R=Me 87: X=O, R=H 88: X=S, R=Me 89: X=S, R=H 90: X=O, R=Me 91: X=O, R=H 92: X=S, R=Me 93: X=S, R=H
N X R' R N R' X R
Fig. 23. Equilibrium between s-cis and s-trans substituted anilides and thioanilides.
The synthesis was achieved as outlined in Scheme 12. The variously substituted benzanilides were prepared by the interaction of the appropriate benzoyl chloride with different anilines. The benzanilides were converted to their thiobenzanilides with Lawesson’s reagent in chlorobenzene. Final compounds were obtained by demethylation with boron tribromide.
s-cis s-trans
COOH COCl NH O NH O NH S NH S a b d d c R2 R1 R2 R1 R1 R2 R2 R1 NH2 R1 R2 R1 S N O O R2 R1O OR1 OR1 S N O O R2 R1O OR1 OR1 Scheme 13
Reagents and conditions: (a) SOCl2, dry CH2Cl2; (b) DMAP, DIEA, dry CH2Cl2; (c) Lawesson’s reagent,
chlorobenzene, 180 °C; (d) BBr3, dry CH2Cl2, -78°C.
We also synthesized sulfonamido analogues with different substituents (H, Me, Et, Bn) on the nitrogen atom (Fig. 24)
Fig. 24. Sulfonamidoderivatives. R1, R2= 4-OH, 4-OMe, 2,4-(OH)2, 2,4-(OMe)2, 3,5-(OH)2, 3,5-(OMe)2 94-97: R1=Me, R2=H, Me, Et, Bn. 98: R1, R2=H 99-102: R1=Me, R2=H, Me, Et, Bn. 103: R1, R2=H
SO2Cl MeO N S O O OMe H N S O O OH R2 a b R1 R1 NH2 R1 c N S O O OMe R2 c R1 N S O O OH H R1
The benzenesulfonanilde skeleton was obtained by coupling the appropriate benzenesulfonyl chlorides with the different anilines in pyridine. Compounds 94 and 99 were demethylated with boron tribromide to obtain the final compounds 98 and 103.
Additionally, N-alkylation of the benzenesulfonanilide moiety was performed with methyliodide, ethyliodide and benzylbromide affording derivatives 95-97 and 100-102. All the N-alkylated compounds were submitted to demethylation without obtaining the desired derivatives.
Scheme 13
Reagents and conditions: (a) Py; (b) NaH, R2I (or R2Br), DMF; (c) DMAP, DIEA, dry CH2Cl2.
R1= 2,4-(OH)2, 2,4-(OMe)2,
3,5-(OH)2, 3,5-(OMe)2
Results and discussions
The novel analogues were tested to assess their antiproliferative effects on MDA-MB-231, a highly metastatic breast cancer cell line, and on HGC-27, a gastric cancer cell line. The sulforhodamine B cell proliferation assay was used. Cells were treated with 0.25-64
μM resveratrol or synthetic analogues for up to 6 days. The values are the mean ( ±SD of
three independent experiments).
The IC50 for the new analogues are reported in Table 4, together with the IC50 of
Table 4. IC50 values of resveratrol and novel analogues. Compound MDA-MB-231 IC50 (μM) HCG-27 IC50 (μM) RES OH OH HO 20.5 29.3 1 OH OH HO 12.7 12.2 10 N OH OH HO 17.4 67.7 15 N OH OH HO Cl 47.4 28.1 11 N S HO OH OH >100 >100 12 S N HO OH OH 67.6 >100 13 S N HO OH OH 19.4 42.1 14 S N OH OH OH 18.1 48.2 44 O OH OH HO 46.1 >100 43 OH OH O H N HO 19.2 >100
Assays of the remaining compounds are currently underway.
All the new reveratrol analogues showed a higher antiproliferative activity on MDA-MB-231 than on HCG-27 cell line.
Salicylaldoxime derivative exhibited a good antiproliferative activity on MDA-MB-231 with an IC50=19.2 µM, whereas no activity was found on HCG-27 cancer cell line.
Quinolinic analogues showed an appreciable antiproliferative activity on MDA-MB-231 cancer cell line; in particular compound 10, which lacks the chlorine substituent, is characterized by an IC50 value comparable to that of the naphtalene analogue 1. Among
phenylbenzothiazolic derivatives, compounds 13 and 14, having only one –OH group on the benzothiazolic scaffold, are the most active of the series on both cancer cell lines.
We may conclude that the replacement of naphthalene ring of compound 1 with a more polar heterocyclic or pseudo-heterocyclic scaffold generally preserves the antiproliferative activity, as for quinoline 10, benzothiazoles 13 and 14 and salicylaldoxime 43.
As mentioned in the introduction, resveratrol acts as a BK channel opener. Hence resveratrol structural analogues could represent a promising approach in order to develop a new class of potential BK openers.
The synthesized compounds were then submitted to in vitro assays evaluating their vasorelaxing properties on rat aortic rings, in the presence or in the absence of selective potassium channels blockers, as a preliminary screening test to point out their potential activation of potassium channels. The results are shown in Table 5.
Table 5. Vasorelaxant activity of resveratrol and resveratrol analogues
Compound Emaxa at 3x10-5M pIC
50b RES OH OH HO 83.0 ± 5.0 5.16 ± 0.13 1 OH OH HO 86.5 ± 2.1 4.98 ± 0.01 10 N OH OH HO 17.2 ± 3.0 N.C 15 N OH OH HO Cl 93.1 ± 4.9 4.89 ± 0.02 11 N S HO OH OH 85.8 ± 5.3 5.11 ± 0.05 12 S N HO OH OH 73.8 ± 0.5 4.71 ± 0.01 13 S N HO OH OH 90.5 ± 2.4 4.93 ± 0.01 14 S N OH OH OH 77.7 ± 4.2 4.85 ± 0.03 44 O OH OH HO 96.9 ± 3.1 4.98 ± 0.04 43 OH OH O H N HO 47.3 ± 8.6 N.C
a Maximal vasorelaxing response expressed as percentage of contractile tension. b Vasorelaxant potency expressed as negative log of the concentration evoking a half-reduction of the contractile tone.
Assays of the remaining compounds are currently underway.
The results indicate that many of the synthesized compounds had a nearly complete vasorelaxing efficacy, with only two molecules (10 and 43) showing a low efficacy. With respect to potency, synthesized compounds had comparable pIC50 values with respect to
Like the heterocycles derivatives, amido and thioamido analogues were tested for their antiproliferative activity on the MDA-MB-231 breast cancer line and on the gastric cancer cell line HCG-27. The IC50 values are reported in Table 5.
Table 5. IC50 of resveratrol, amido and thioamido analogues.
Compound MDA-MB-231 IC50 (μM) HCG-27 IC50 (μM) RES OH OH HO 20.5 29.3 79 HO O NH OH OH 66.9 >100 81 HO S NH OH OH 88.2 >100 83 HO NH O OH OH >100 >100 85 HO NH S OH OH >100 >100
Amido compound 79 and thioamido compound 81 with two hydroxy groups on the phenyl ring directly bound to the nitrogen atom, possesses a certain antiproliferative activity, but only on MDA-MB-231 cancer cell line.
Low antiropliferative activity showed by amido and thioamido compounds is probably due, as we had mentioned before, to the fact that these compounds exist prevalently in the
s-cis configuration, while the active form of resveratrol is the s-trans.
Amido and thioamido compounds were then submitted to in vitro assays evaluating their vasorelaxing properties on rat aortic rings, in the presence or in the absence of selective potassium channels blockers. The vasorelaxing activity data, expressed as efficacy and potency (pIC50), are reported in Table 6 along with those of resveratrol.
Table 6. Vasorelaxant activity of resveatrol and amido and thioamido analogues
Compound Emaxa at 3x10-5M pIC50b
RES OH OH HO 83.0 ± 5.0 5.16 ± 0.13 79 HO O NH OH OH 37.6 ± 5.3 N.C 81 HO S NH OH OH 53.3 ± 9.5 4.52 ± 0.07 83 HO NH O OH OH 30.5 ± 6.7 N.C 85 HO NH S OH OH 46.3 ± 10.0 N.C 91 NH O OH OH OH HO 37.3 ± 11.0 N.C
a Maximal vasorelaxing response expressed as percentage of contractile tension. b Vasorelaxant potency expressed as negative log of the concentration evoking a half-reduction of the contractile tone.
The results indicate that all the synthesized compounds generally showed a low vasorelaxing efficacy.
Only thioamido compound 79 showed a significant vasorelaxing effect. Assays of the remaining compounds are currently underway.
GENERAL DETAILS
Proton NMR spectra were recorded on a Bruker AC 200 (200 MHz) spectrometer as dilute solutions in deuterochloroform or acetone deuterate. The chemical shifts are recorded relative to tetramethylsilane as internal standard and the multiplicity of the signals is designated by one of the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; br, broad; m, multiplet; with all various possible combinations (e.g. dd is double doublet). All coupling constants J, are reported in Hertz.
Flash chromatography was performed on Merck silica gel 60 or on Isolute pre-packed silica columns and the solvents employed were either of analytical grade or were distilled before the use.
All reactions were monitored by TLC using Merck silica gel 60 F24 pre-coated aluminium
backed plates which were visualised with ultraviolet light.
Dry organic solvents were stored under nitrogen and/or over sodium wire. The organic solvents were dried by distillation from the following: dichloromethane, pyridine, triethylamine, acetonitrile from calcium hydride; methanol from magnesium methoxide; benzene, toluene, THF and diethyl ether from sodium metal. N,N-dimethylformamide (DMF) Dimethylsulfoxide (DMSO) were purchased from Aldrich in SureSeal containers.
All other commercially available reagents were used as received from Fluka, Sigma-Aldrich, Merck, Lancaster.
Solvents were removed by rotary evaporation in vacuo.
Where necessary, reaction requiring anhydrous conditions, were performed in a flame or oven-dried apparatus under a nitrogen atmosphere.
BIOLOGICAL ASSAY
Antiproliferative assays have been preformed by Prof. Ghidoni research group at Laboratory of Biochemistry and Molecular Biology, San Paolo University Hospital, Medical School, University of Milan.
All drugs were dissolved in absolute ethanol (EtOH) before use. Cell Lines. Human breast cancer cell line MDA-MB-231, from American Type Culture Collection, Rockville, MD, USA, was maintained at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium,
supplemented with 5% fetal bovine serum and 100 ng/ml each of penicillin and streptomycin.
Human gastric cancer HGC-27, from Riken Gene Bank, were maintained as monolayers in RPMI medium, supplemented with 10% fetal bovine serum with 100 ng/ml each of penicillin and streptomycin at 37°C in a humidified 5% CO2 atmosphere.
Cell Proliferation by Sulforhodamine B (SRB) Assay. Cells were seeded in 96-wells tissue
culture plates at 1,3 x 103 cells/well and were allowed to adhere for 24 hours before treatment. Cells were grown in the presence of vehicle (EtOH or DMSO) (five wells) or drugs at a indicated concentrations (five wells for each treatment). Cellular growth was assessed after 2, 4 and 6 days by SRB assay. Briefly, proteins were precipitated with 10% (final concentration) trichloroacetic acid for 1 hour at 4° C and stained for 30 min with SRB dye 0.4% w/v in acetic acid 1% v/v. Finally, precipitated proteins were washed and solubilized in Tris buffer 10 mM. Absorbance (optical density, OD) was measured at 540 nm by using a microplate reader. For each treatment the proliferation index was calculated as OD TC tx / OD TC t0 where ‘OD TC tx’ is the mean optical density of treated cells at time x and ‘OD TC t0’ is the value at time zero. Data are expressed as proliferation index (fold increase of proliferation versus control at time zero).
PARP Cleavage Assay. Cells treated for 4 days with different RES analogues at
concentrations corresponding to the respective IC50 and with vehicle alone, were lysed for 20 min on ice in lysis buffer (50mM Tris-HCl pH 7.5; 250 mM NaCl; 1% Triton X-100; 10 mM sodium-pyrophosphate; 10 µl/ml of protease inhibitor cocktail; 1 mM phenylmethane - sulfonylfluoride). Cytosolic proteins were obtained by centrifugation at 15,000 x g for 15 min of extracts of cells. Proteins (25 µg) were separated on 8% SDSpolyacrylamide minigel and transferred to nitrocellulose membranes. Membranes were incubated in
blocking solution (5% nonfat dry milk in 0.05% Tween 20 in 50 mM Tris, 150 mM NaCl) and anti-PARP antibody (1:500 dilution in blocking solution). PARP, intact protein and cleaved fragment were visualized by ECLTM.
Quantification of Endogenous Ceramide. Total endogenous ceramide level was measured
using the diacylglycerol kinase assay (DGK) as previously described. Cells were treated for 4 days with different RES analogues at concentrations corresponding to the respective IC50,
and with vehicle alone. Then cells were collected and lipids were extracted according to Bligh and Dyer.
The organic phase was divided in 1/2 and 1/6 aliquots, dried and used for ceramide measurement and total phospholipids measurement, respectively. Briefly, 30 nmoles of extracted lipids were incubated at room temperature for 45 min in the presence of β- octylglucoside / dioleoylphosphatidylglycerol micelles, 2 mM dithiothreitol, 6 µg of proteins of DGK containing membranes and 1 mM ATP (mixed with [γ-32P]ATP at a final radioactivity of 13 µCi/ml) in a final volume of 0.1 ml. At the end of reaction, lipids were extracted according to Bligh and Dyer. [32P]ceramide-1-phosphate was determined by TLC separation in chloroform / acetone / methanol / acetic acid / water, (10/4/3/2/1, by vol), visualization by autoradiography, followed by scraping the radioactive ceramide-1- phosphate spots and counting in a scintillator counter. Ceramide level was referred to the level of total phospholipids.
Vasorelaxant assays have been performed by Dott. Calderone research group at Department of Pharmaceutical Sciences, University of Pisa.
All the experimental procedures were carried out following the guidelines of the European Community Council Directive 86-609. To determine a possible vasodilator mechanism of action, the compounds were tested on isolated thoracic aortic rings of male normotensive Wistar rats (250-350 g). The rats were sacrificed by cervical dislocation under light ether anaesthesia and bled. The aortae were immediately excised and freed of extraneous tissues. The endothelial layer was removed by gently rubbing the intimal surface of the vessels with a hypodermic needle. Five mm wide aortic rings were suspended, under a preload of 2 g, in 20 ml organ baths, containing Tyrode solution (composition of saline in mM: NaCl 136.8; KCl 2.95; CaCl2 1.80; MgSO4 7H2O 1.05;
NaH2PO4 0.41; NaHCO3 11.9; Glucose 5.5), thermostated at 37°C and continuously gassed
with a mixture of O2 (95%) and CO2 (5%). Changes in tension were recorded by means of
an isometric transducer (Grass FTO3), connected to a unirecord microdynamometer (Buxco Electronics). After an equilibration period of 60 minutes, the endothelial removal was confirmed by the administration of acetylcholine (ACh) (10 µM) to KCl (20 mM)-precontracted vascular rings. A relaxation < 10% of the KCl-induced contraction was considered representative of an acceptable lack of the endothelial layer, while the organs, showing a relaxation ≥ 10% (i.e. significant presence of endothelium), were discarded. From 30 to 40 minutes after confirmation of the endothelium removal, the aortic preparations were contracted by treatment with a single concentration of KCl (20 mM) and when the contraction reached a stable plateau, 3-fold increasing concentrations of the tested compounds or of the reference drug NS 1619 (a well-known BK-activator) or resveratrol were added cumulatively. Preliminary experiments showed that the KCl (20 mM)-induced contractions remained in a stable tonic state for at least 40 minutes.
The reference drug NS 1619 (Sigma) was dissolved (10 mM) in EtOH 95% and further diluted in Tyrode solution. Acetylcholine chloride (Sigma) was dissolved (100 nM) in EtOH 95% and further diluted in bidistilled water whereas KCl was dissolved in Tyrode solution. All the tested compounds were dissolved in DMSO (10 mM); they were all further diluted in Tyrode solution. All the solutions were freshly prepared immediately before pharmacological experimental procedures. Previous experiments showed a complete ineffectiveness of the vehicles. The vasorelaxing efficacy was evaluated as maximal
vasorelaxing response, expressed as a percentage (%) of the contractile tone induced by KCl 20 mM. When the limit concentration 0.1 mM (the highest concentration, which could be administered) of the tested compounds did not reach the maximal effect, the parameter of efficacy represented a vasorelaxing response, expressed as a percentage (%) of the contractile tone induced by KCl 20 mM, evoked by this limit concentration. The parameter of potency was expressed as pIC50, calculated as the negative Logarithm of the molar
concentration of the tested compounds, evoking a half reduction of the contractile tone induced by KCl 20 mM. The pIC50 could not be calculated for those compounds showing
an efficacy parameter lower than 50%. The parameters of efficacy and potency were expressed as mean ± standard error, for 5-10 experiments. Student t test was selected as statistical analysis, P<0.05 was considered representative of a significant statistical difference. Experimental data were analysed by a computer fitting procedure (software: Graph-Pad Prism 3.0).