INTRODUZIONE La genetica forense
La Genetica Forense è una branca della Genetica che si occupa di fornire una "identità" ad un campione biologico presente in qualsiasi traccia, e rappresenta uno strumento indispensabile per risolvere casi giudiziari o contenziosi in ambito Civile e Penale. Le moderne tecniche di genetica molecolare, permettono di determinare un profilo genico oltre che dai tradizionali campioni biologici come sangue, saliva, sperma, etc. anche a partire da tracce minime di materiale biologico quali sudore, impronte, tracce lavate, etc. Ciò consente il raffronto tra profilo genetico di tracce biologiche, repertate sulla scena di un crimine, con quelle di eventuali sospettati e/o vittime, al fine di escludere o attribuirne in maniera precisa la provenienza da una medesima persona. L’importanza della genetica nelle scienze forensi si è notevolmente accresciuta negli ultimi anni, grazie ai progressi nella conoscenza dei marcatori genetici polimorfici e all’evoluzione degli strumenti utilizzati per analizzarli. La genetica forense si è rivelata particolarmente utile per:
- Identificare individui che abbiano commesso dei crimini - Risolvere casi di paternità incerta
- Predire la popolazione di origine di un DNA (es. Africano, Asiatico ecc.) - Ottenere informazioni fenotipiche su un soggetto (es. colore occhi)
- Identificare e «ricostruire» cadaveri la cui identità sia ignota (vittime di catastrofi naturali, disastri aerei, guerre, genocidi)
- Investigazioni su persone scomparse - Identificare tracce di sostanze illegali
- Identificare DNA appartenente a specie a rischio di estinzione in crimini legati al loro contrabbando.
Fin dai primi studi sul genoma umano, si è capito che il DNA rappresenta una rilevante sorgente di variabilità, capace di differenziare ciascun
individuo rispetto agli altri in modo assai efficace. La possibilità di attribuire l’identità personale attraverso l’analisi del DNA è diventata realtà; questa metodologia ha contribuito a delineare un concetto nuovo di identità individuale, strettamente legato alle leggi della biologia, della genetica e dell’ereditarietà, del calcolo delle probabilità, indicata col termine di identificazione genetica.
Molte discipline hanno trovato giovamento e impulso dalle tecniche di biologia molecolare, applicate al DNA. Gli studi di antropologia molecolare, hanno permesso di datare con una certa approssimazione la nascita di un progenitore comune femminile del genere umano (idealmente Eva) intorno a 45 mila anni fa, e stabilire i flussi migratori avvenuti nel tempo a popolare il nostro pianeta.
Anche la genetica forense si è andata quindi formando come scienza autonoma.
Nel settore delle indagini di filiazione famose sono state le attribuzioni di figli a personaggi celebri. E non solo di paternità si tratta: qualche volta interi alberi familiari sono stati ricostruiti. In altri casi questi esami possono essere utilizzati per chiarire periodi storici cupi di dittatura e oscurantismo.
Oltre a questi tentativi di chiarire importanti avvenimenti storici, in altri casi si è cercato di risvegliare le memorie di antichi eroi, magari con fini apertamente speculativi.
L’impatto più impressionante si è avuto comunque nelle investigazioni criminali: alle tradizionali figure del dattiloscopista, dell’esperto balistico, si è affiancata oggi a pieno titolo quella del genetista forense. Le analisi di tracce di materiale biologico anche esigue rinvenute sulla scena del crimine e la possibilità di identificare per confronto o ricerca in un database colui dal quale le tracce derivano, è da alcuni anni una realtà oggettiva. Tra le sofisticatissime tecniche investigative forensi quella sicuramente più utilizzata è l'amplificazione enzimatica in vitro (PCR - Polymerase Chain Reaction) che consente di ottenere un profilo genetico anche a partire da tracce minime di materiale biologico (Low Copy Number).
Le indagini investigative possono oggi essere supportate da questo straordinario metodo analitico, per mezzo del quale è possibile spesso
risalire all’autore di un fatto criminoso e che permette di assolvere persone falsamente accusate di quel crimine.
La possibilità di accertare l’innocenza di un individuo è un aspetto di non poco conto che ha acquisito, in Italia, un’enorme importanza, alla luce dell’introduzione delle nuove norme sulle indagini difensive, nelle quali sono attribuite maggiori opportunità alle indagini tecniche coordinate dal difensore.
La standardizzazione dei metodi d’analisi ha creato uno sviluppo delle possibilità investigative, favorendo la creazione di banche dati, ove sono inseriti e custoditi i profili genetici di criminali (e non solo). Questi archivi sono così importanti e utili che moltissime nazioni li hanno rapidamente approntati, introducendo nel contempo adeguate leggi e regolamenti per la loro gestione.
L’utilità della genetica forense non si apprezza solo quando si tratta di attribuire un padre o una madre ad un figlio, o quando vi è un crimine da risolvere. Vi sono utilizzi sociali importantissimi come la possibilità di dare un nome ai poveri resti di un proprio congiunto scomparso.
Soprattutto nei grandi disastri o nelle grandi calamità, dovute o meno all’azione dell’uomo, quando il riconoscimento dei corpi è praticamente impossibile dal solo esame degli effetti personali o da altre caratteristiche somatiche diviene fondamentale possedere un metodo identificativo basato sul DNA.
La richiesta di conoscenza, unita alla disponibilità di strumenti e prodotti commerciali talvolta facilmente fruibili anche dal singolo cittadino (si pensi ai kit fai da te venduti all’estero in farmacia e usati per determinare la paternità), ha determinato un’ampia diffusione sul territorio dei test d’identificazione personale, per i più disparati scopi.
La genetica forense è materia di grande complessità che richiede conoscenze di carattere multidisciplinare:
- una chiara conoscenza del nostro ordinamento giudiziario, fondamentale per comprendere le attribuzioni che i codici, civile e penale, e le norme di deontologia professionale, impongono alle figure degli esperti.
- attività di tipo criminalistico come il sopralluogo giudiziario e la repertazione, sono propedeutiche e necessarie alle successive attività di
laboratorio.
- analisi di tipo medico legale volte all’identificazione del tipo di materiale biologico da sottoporre agli accertamenti risultano indispensabili.
- esami di laboratorio volti alla determinazione del profilo genetico sono inoltre particolarmente delicati e insidiosi quando un reperto biologico, per sua natura deteriorabile, debba essere esaminato.
- la corretta presentazione del dato analitico, corredata delle appropriate valutazioni probabilistiche è infine un’altra fase che richiede particolare attenzione, perché alla prova del DNA possa essere attribuito il reale valore.
In questo lungo iter analitico che costituisce parte stessa dell’esame genetico-forense, dalle fasi prodromiche all’accertamento alla presentazione dei risultati, possono verificarsi errori. Per questo motivo sia il giudice che il difensore dovrebbero conoscere almeno le potenzialità e limiti della materia, in modo da poter escutere con competenza i propri consulenti e instaurare con loro un più consapevole dialogo per l’affermazione della verità.
La variabilità del genoma umano
La variabilità genetica: mutazioni e polimorfismi
La diversità tra individuo ed individuo è generata dalla variabilità genetica 1:
differenze nel fenotipo rispecchiano differenze nel genotipo. Alcune riguardano caratteristiche fisiche molto evidenti quali i capelli, il colore degli occhi e della pelle, altre sono meno palesi ma più importanti quali il gruppo sanguigno, il sistema HLA, fattori che influiscono sulla risposta ai farmaci o sulla probabilità di contrarre malattie infettive o cardiovascolari. Alcune differenze hanno un effetto dominante, altre sono recessive. Poiché più geni influenzano un carattere (poligenia) e fattori non genetici (ambientali) possono interferire e modulare in modo diverso l'effetto dei geni (multifattorialità), il rapporto tra genotipo e fenotipo non è sempre così semplice: vi sono molte differenze tra le persone dovute completamente o in
parte a processi stocastici durante lo sviluppo, o dovute a influenze da parte dell'ambiente e non a fattori genetici; a volte diversi alleli mutanti dello stesso gene possono avere effetti diversi, e alleli di altri geni possono influenzare il fenotipo: la distinzione tra caratteri monogenici e caratteri complessi (multifattoriali) non è netta. Nonostante molte differenze tra un genoma umano e un altro, la maggior parte di queste differenze influisce molto poco o per niente sul fenotipo. La diversità genetica è dovuta a due eventi che si verificano nel processo di divisione delle cellule germinali (meiosi): l'assortimento indipendente dei cromosomi e il crossing-over, che fanno sì che le cellule figlie originatesi contengano un patrimonio genetico aploide diverso tra loro; altra importante fonte di variabilità genetica è la mutazione, definita come un qualsiasi cambiamento nella sequenza del DNA, e che ricopre un ampio spettro di eventi con differenti incidenze e meccanismi molecolari. Si parla di mutazione sia quando il cambiamento riguarda un singolo nucleotide (sostituzioni, inserzioni e delezioni), sia quando si verificano piccole inserzioni e delezioni di poche basi, ma anche nel caso di inserzioni, delezioni, duplicazioni e inversioni di regioni del DNA lunghe alcune megabasi, di espansione o contrazione nel numero di elementi di DNA ripetuti in tandem, di inserzioni di elementi trasponibili, di traslocazioni di segmenti cromosomici e qualsiasi tipo di anomalie nel numero dei cromosomi. Il termine generico mutazione spesso indica una variazione patogenica, in contrasto col termine polimorfismo, che descrive un cambiamento di sequenza nel gene che non ha alcun effetto o funzione. Ad ogni modo, vi sono ovvi problemi in questa definizione, poiché è molto difficile, se non impossibile, sapere se un cambiamento nella sequenza del DNA causa o meno un cambiamento fenotipico. Inoltre mutazioni che causano malattie sono presenti, in alcune popolazioni, con frequenze superiori all' l % e perciò possono essere classificate come polimorfismi. Si parla infatti di polimorfismo quando nella popolazione esistono almeno due forme alleliche e l'allele più raro è presente con una frequenza uguale o superiore all'l%; il termine variante, invece, indica un allele con frequenza al di sotto dell'l %. Chiaramente, poiché le frequenze alleliche spesso variano tra le popolazioni, una variante per una popolazione potrebbe essere un polimorfismo per un'altra. Non tutte le mutazioni vengono trasmesse da una
generazione all'altra e contribuiscono al cambiamento evoluzionistico: solo le mutazioni che si verificano nella cellule germinali verranno ereditate dalle generazioni successive, mentre quelle che si verificano nelle cellule somatiche potranno avere conseguenze serie, come il cancro, ma non avranno ruolo in termini evoluzionistici
I marcatori molecolari del DNA
I marcatori biallelici (inserzioni Alu, indels, SNPs)
Gli elementi Alu, le inserzioni/ delezioni (indels) ed i polimorfismi a singolo nucleotide (SNPs) sono indicati come marcatori biallelici; hanno una frequenza di mutazione estremamente bassa2, sull'ordine di 10-8, e vengono anche indicati col termine unique event polimorphisms (UEPs) oppure unique mutation events (UMEs) poiché, con tutta probabilità, ognuno di questi eventi si è verificato una volta sola nella storia.
Un primo tipo di marcatori biallelici utilizzati per gli studi sulla diversità genetica fra le popolazioni è rappresentato dagli short interspersed nuclear elements (SINEs), che consistono in brevi sequenze identificabili, inserite in particolari posizioni del genoma.
Inserzioni Alu
Nel genoma umano, una gran parte del DNA moderatamente ripetitivo si trova sotto forma di sequenze di circa 300 bp intercalate a DNA non ripetitivo. Almeno metà del materiale duplex rinaturato è tagliato dall'enzima di restrizione Alu 1 in un singolo sito, posto a 170 bp dall'inizio della sequenza: le sequenze tagliate sono tutte membri della famiglia Alu. Nel genoma aploide ci sono circa 300 000 membri di questa famiglia ampiamente dispersi. I singoli membri della famiglia Alu non sono identici, bensì correlati. Sembra che la famiglia umana si sia originata da una duplicazione in tandem di 130 bp, con una sequenza non correlata di 31 bp inserita nella metà destra del dimero. Le due ripetizioni sono a loro volta chiamate "metà destra" e "metà sinistra" della sequenza Alu. I singoli membri della famiglia
Alu hanno un'omologia media alla sequenza consenso dell'87%.
La sequenza umana è correlata all'RNA 7SL, un componente della particella di riconoscimento del segnale. L'RNA 7SL è codificato da geni che sono trascritti attivamente dalla RNA polimerasi III ed è possibile che questi geni (o geni ad essi correlati) abbiano dato origine alle sequenze Alu inattive. I membri della famiglia Alu possono essere inclusi all'interno di unità di trascrizione di geni strutturali, come dimostrato dalla loro presenza nell'RNA nucleare lungo. L'inserzione di un elemento Alu in un locus è un evento unico; infatti, una volta avvenuta, essi sono marcatori stabili, non soggetti a perdita oppure ad ulteriori riarrangiamenti.
Le Indels
Il termine Indel viene usato come abbreviazione di un evento di mutazione/ricombinazione che può far parte di due classi: un’inserzione (insertion) o una delezione (deletion) che possono essere avvenute nel corso degli anni, e che hanno prodotto delle differenze nelle sequenze di DNA in esame.
Le indels probabilmente rappresentano tra il 16% ed il 25% di tutti i polimorfismi di sequenza negli esseri umani. Pertanto, giacché il database degli SNP attualmente contiene circa 10 milioni di SNPs, nelle popolazioni umane sono presenti almeno 1,6-2.500.000 polimorfismi Indel.
Come illustrato nell'articolo dei ricercatori Ryan E. Mills 3, Christopher T. Lutting et al. (Genome Research, 2006), una nuova strategia computazionale ha consentito l'identificazione sistematica dei polimorfismi indel nel genoma di diversi esseri umani. Mills e Lutting usarono questa strategia per costruire una mappa umana iniziale di variazioni indels non ridondanti che contiene 415.436 nuovi indels distribuiti in tutto il genoma umano, la cui lunghezza varia da 1 a 9989 bp.
Circa il 36% di questi indels si trovano all'interno dei promotori, introni ed esoni di geni noti. Così, come gli SNPs, alcuni di questi indels mostrano un impatto sulla funzione del gene umano. Cinque grandi classi indel sono state identificate analizzando la loro sequenza di DNA:
(1) inserzioni e delezioni di coppie a singola base rappresentano circa un terzo (29.1%) di tutte le indels della collezione e si tratta prevalentemente di indels A:T e T :A;
(2) espansioni monomeriche di varie lunghezze ed
(3) espansioni multibase di 2-15 unità ripetute rappresentano anch’esse complessivamente un terzo (29,5%) delle indels della collezione.
Quest'ultima categoria comprende le espansioni (CA)n, le quali vengono comunemente utilizzate come marcatori genetici e le espansioni trinucleotidiche che hanno dimostrato essere responsabili di malattie umane (Warren et al. 1987).
(4) Oltre ad espansioni ripetute, sono state individuate indels lunghe da 100 fino a 30 kb causate dall'inserimento de novo di elementi trasponibili (piccola frazione pari allo 0,59%) .
(5) I restanti indels mostrano un ampio spettro di sequenze di DNA apparentemente casuali (40,8%) che vanno da 2 bp a 9989 bp di lunghezza. In fine Mills e Lutting esaminarono la distribuzione genomica di tali indels verificando che si trovavano in tutto il genoma ad una densità di un indel per 7,2 kb di DNA. Le indels generalmente sono distribuite in tutto il genoma secondo la quantità di DNA che è presente su ciascun cromosoma, ad eccezione dei cromosomi 4, 5, 8, 14 e 18.
Gli SNPs (single nucleotide polymorpshism)
La differenza più semplice tra due sequenze di DNA omologhe è la sostituzione nucleotidica, in cui una base viene cambiata con un'altra. Quando una pirimidina viene sostituita con una pirimidina o una purina con una purina, la differenza viene chiamata transizione; quando una purina viene sostituita da una pirimidina, o viceversa, abbiamo una transversione. Questi tipi di differenze sono esempi di SNPs (single nucleotide polymorphisms). Le inserzioni o delezioni (indels) di una singola base sono incluse nella categoria degli SNPs, anche se il meccanismo attraverso il quale si originano e il trattamento analitico differiscono da quelle delle sostituzioni nucleotidiche.
Come ogni polimorfismo gli SNPs sono formati da alleli diversi: poiché nell'uomo le forme trialleliche e tetraalleliche sono rarissime mentre la quasi totalità è costituita da due alleli, in bibliografia vengono spesso menzionati come "polimorfismi biallelici". Due processi fondamentali danno origine alla mutazione per sostituzione: l'errata incorporazione di nucleotidi durante la replicazione del DNA e la mutagenesi causata da modificazione chimica delle basi o da danni fisici dovuti a radiazioni ultraviolette o ionizzanti. Quando una cellula diploide si divide, tutto il suo DNA deve essere replicato affinché ogni cellula figlia contenga due copie del genoma aploide. La replicazione del DNA avviene con elevata fedeltà. Una nuova base è incorporata se si appaia con la base esistente nel DNA stampo a singola elica. L'esistenza del corretto numero di legami idrogeno tra le basi è insufficiente per assicurare che una A si leghi solo con una T e una C solo con una G: infatti la DNA polimerasi, enzima responsabile della sintesi del DNA, richiede anche la corretta geometria delle coppie di basi prima che si formi il legame con il filamento che si sta generando. A volte può capitare che venga incorporata una base sbagliata, a causa di una rara forma chimica transiente delle basi che ne altera le capacità di appaiamento. In realtà la DNA polimerasi ha anche attività di esonucleasica: in pratica esamina la base incorporata e, se non la riconosce come giusta, la elimina e prova di nuovo ad abbinare il corretto nucleotide complementare. Questo sistema di controllo permette di diminuire la probabilità di errata incorporazione di basi: errori nella replicazione si verificano con una frequenza di 109–1011 per nucleotide. L'integrità del materiale genetico è costantemente insidiata da processi chimici e fisici che alterano le basi o danneggiano la struttura fisica della molecola del DNA. Danni alla molecola di DNA possono essere causati anche da agenti mutageni chimici. Anche le radiazioni UV possono modificare la struttura del DNA formando dei legami tra timine adiacenti sullo stesso filamento, i cosiddetti dimeri di timina; le radiazioni ionizzanti possono invece rompere i legami tra le due eliche complementari o formare ioni reattivi (radicali liberi) all'interno della cellula e provocare sostituzioni nucleotidiche. Agenti mutageni chimici e fisici sono importanti cause o contribuiscono all'insorgenza di molti tumori; ad ogni modo il loro effetto sulle cellule della linea germinale può essere molto diverso da quello sulle
cellule somatiche. Non tutte le mutazioni che si verificano vengono trasmesse alle generazioni cellulari successive; le cellule hanno infatti la capacità di rilevare e riparare questi danni attraverso i sistemi di riparazione del DNA che permettono di correggere errori a livello di un singolo filamento, quali il mismatch repair e il nucleotide excision repair, e quelli che invece intervengono in caso di rottura della doppia elica, quali la ricombinazione omologa e l'end-joining non omologa. A livello genomico, le mutazioni possono verificarsi in qualsiasi regione, sia all'interno di geni sia in regioni intergeniche, con diversi effetti sul fenotipo. Sostituzioni all'interno di geni possono essere causa di malattie ed è quindi importante conoscere gli effetti di tali cambiamenti: si può passare da una completa neutralità alla mancanza totale della proteina. Una sostituzione che non altera la codifica di un aminoacido è conosciuta come "silente" o sostituzione "sinonima", mentre una mutazione che provoca cambiamento di un aminoacido è detta "non-sinonima" o "missenso". Un cambiamento di base che trasforma un codone per un aminoacido in un codone di stop è detta "non-senso". Inserzioni o delezioni di una singola base (indels) dentro la regione codificante del gene determinano lo slittamento della lettura del codice genetico (frameshift). Questo tipo di mutazione è uno dei più dannosi, in quanto la sequenza aminoacidica viene completamente alterata. Mutazioni al di fuori del gene possono influire sulla sua espressione alterando ad esempio il suo promotore o gli enhancers o i segnali di poliadenilazione; mutazioni a livello degli introni possono modificare lo splicing dell' RNA.
Frequenza e distribuzione degli SNPs nel genoma umano
L'interesse per gli SNPs è elevato in virtù del loro potenziale uso come marcatori molecolari negli studi di associazione gene-malattia. Sono stati fatti numerosi studi di risequenziamento (sequenziare lo stesso locus in diversi individui) di particolari loci e questo offre un ritratto della diversità degli SNPs in tali regioni. Complessivamente, la media della diversità nucleotidica (π, rappresenta la probabilità che una determinata posizione nucleotidica si trovi in condizione di eterozigosi quando comparata tra due cromosomi presi
a caso nella popolazione) sia negli studi sull'intero genoma che negli studi di uno specifico locus e circa 7,51 x 10-4; questo vuol dire che ci si aspetta di trovare in media 1 SNP ogni 1.331 bp circa. Dato che il DNA aploide umano e costituito da circa 3,3 x 109 bp si deduce rapidamente che gli SNPs esistenti possano essere quantificati nell'ordine di più di tre milioni. In effetti sono già stati identificati 1,42 milioni di polimorfismi di un singolo nucleotide. Ma una stima dei polimorfismi presenti nel genoma umano, considerando la frequenza minima dell' 1% per l'allele meno frequente, si spinge oltre 11 milioni di siti SNPs. L'effettivo valore di π varia significativamente tra i cromosomi, da 5,19 x 10-4 per il cromosoma 22 a 8,79 x 10-4 per il cromosoma 15. Regioni del genoma che mostrano alta densità di SNP potrebbero derivare da un'assegnazione errata tra sequenze che non sono omologhe ma paraloghe (altamente simili, con più del 97% di similarità), originate da duplicazioni segmentali e che costituiscono circa il 5% del genoma. Un recente studio ha mostrato che l'apparente densità media di SNP è elevata nelle regioni duplicate da 0.69 per Kb a 1.33 per Kb, suggerendo che questi SNPs siano varianti di sequenze paraloghe (PSVs). Il "ciclo vitale" di uno SNP può essere riassunto individuando quattro fasi principali:
1. comparsa di un nuovo allele variabile attraverso una mutazione nucleotidica;
2. sopravvivenza, contro le probabilità, del nuovo allele attraverso le prime generazioni;
3. aumento sostanziale della frequenza; 4. fissazione nella popolazione.
La durata della vita di uno SNP destinato a essere fissato da un nuovo allele è stimata 284 mila anni.
I marcatori multiallelici (VNTR, STR)
I marcatori multiallelici hanno più di due alleli, solitamente molti di più, per ogni locus, sono costituiti da ripetizioni in tandem, ovvero sequenze di
nucleotidi di varia lunghezza ripetute in successione per un certo numero di volte, localizzate solitamente in regioni non codificanti del DNA. I marcatori multiallelici comprendono i minisatelliti o VNTR (variable number tandem repeats), con lunghezza della sequenza ripetuta lunga 10- 100 bp, ed i microsatelliti o STR (short tandem repeats ), con sequenze ripetute solitamente di 2-6 bp.
I primi hanno percentuali di mutazione per generazione del 6- 11%, mentre i secondi di ~ 0,2%. In tutti i due i casi comunque sono presenti molti alleli per ogni locus. Per questi motivi i marcatori multiallelici hanno un maggior potere risolutivo dei marcatori biallelici, forniscono più informazioni e quindi sono più efficaci nel permettere la distinzione di due profili genetici ottenuti dalla loro tipizzazione.
I VNTR o minisatelliti
Con l'acronimo VNTR (Variable number of tandem repeats) si indicano i polimorfismi dovuti a variazioni nel numero di sequenze ripetute in serie. Tale denominazione è assegnata ad una eterogenea e numerosa classe di loci che presentano variazioni nel numero di ripetizioni di sequenze, organizzate ininterrottamente una dietro l'altra, senza materiale estraneo interposto. Secondo la lunghezza della sequenza ripetuta, dell'arrangiamento delle unità di ripetizione e del loro livello di variabilità, le VNTR si classificano in microsatelliti, minisatelliti e satelliti; nelle analisi di associazione genetica, il termine polimorfismo VNTR è normalmente utilizzato in relazione ai minisatelliti ipervariabili. Sebbene esse siano considerate marcatori, alcune sono localizzate nelle regioni codificanti dei geni, altre in segmenti regolatori, oppure in regioni centromeriche o telomeriche ritenute componenti funzionali importanti dei cromosomi; alcune VNTR contengono interi geni e la loro struttura ripetitiva può giocare un ruolo nella espressione del gene (ad esempio i geni del DNA ribosomiale).
I minisatelliti sono stati i primi marcatori del DNA utilizzati in genetica forense per la risoluzione dei problemi d’identificazione. Tali sequenze, scoperte nei primi anni Ottanta dal genetista inglese Alee Jeffreys, sono
altamente polimorfiche e consistono in ripetizioni in serie di sequenze lunghe tra 8 e 100 bp arrangiate in copie variabili tra 5 e anche più di 1.000 unità, organizzati in cluster prevalentemente sulle estremità dei cromosomi e con elevato contenuto di GC. Sebbene le unità ripetute varino considerevolmente per dimensioni, tutte presentano una comune sequenza centrale (core), GGGCAGGAXG; sono sistemi altamente dinamici sotto l'aspetto evolutivo, con complessa variabilità di lunghezza e di sequenza e con un tasso di mutazione che arriva fino a valori di 1,4 x 10-3. Anche se molti degli schieramenti si trovano in prossimità dei telomeri, si rintracciano sequenze minisatellite nelle parti codificanti del genoma (gene della mucina 1e del DRD4) oppure legate ai promotori dei geni con effetti sull'espressione dei geni stessi (minisatellite dell'insulina, gene della cistatina B, minisatellite dell'HRASl ).
Gli STR o microsatelliti
Weber e May nel 1989 hanno scoperto gli STR-s; in seguito molti autori hanno introdotto lo studio di questi polimorfismi nel campo medico forense, quali Edwards et al. nel 1991, Polymeropoulos et al. nel 1992, Caskey e Hammond nel 1992. La scoperta di un numero elevatissimo di polimorfismi del DNA e successivamente l'applicazione delle metodiche PCR (Polymerase Chain Reaction) al loro studio, determinarono una vera e propria rivoluzione, in particolar modo nel campo delle indagini criminalistiche : essendo il DNA presente in tutte le cellule nucleate, e molto più resistente a fenomeni di degradazione fisica rispetto ai marcatori proteici tradizionali, fu possibile estendere la ricerca ad una vasta gamma di reperti biologici: sangue, sperma, saliva, urina, formazioni pilifere, resti ossei, etc.
Nel 1989 è stata riportata l'esistenza nel genoma umano di loci ipervariabili appartenenti al DNA non codificante; le mutazioni che avvengono in questa regione, essendo meno soggette alle forze di pressione selettiva, vengono trasmesse alla discendenza con un aumento della variabilità genetica.
numero variabile di unità ripetitive presenti nei diversi alleli. In relazione alle piccole dimensioni della sequenza di base, tali loci sono stati definiti microsatelliti o Short Tandem Repeats possedendo unità di ripetizione comprese tra le due e le sei paia di basi, con lunghezza massima di circa 350 bp. A differenza dei minisatelliti che sono localizzati prevalentemente nelle regioni terminali dei cromosomi e sono presenti in numero limitato nel genoma umano, i microsatelliti sono distribuiti uniformemente su tutto il cromosoma e ricorrono ogni 10.000 nucleotidi; sono una forma elementare di DNA ripetitivo; in base alla lunghezza dell'unità di ripetizione si dividono in dinucleotidi [(es. (AG)], trinucleotidi [es. (ATG)], tetranucleotidi [es. (TAGA) ], pentanucleotidi [es. (AAAAT)], esanucleotidi [es. (CCCAAA) ], in cui l'unità di ripetizione è costituita da 2, 3, 4, 5, 6 paia di basi rispettivamente.
I microsatelliti possono essere classificati in base alla loro struttura in tre famiglie:
• Microsatelliti puri (CACACACACACACACACACA); • Microsatelliti composti (CACACACAGAGAGAGA); • Microsatelliti interrotti (CACATCACATTCATTCA ).
I microsatelliti puri sono formati da un unico tipo di unità ripetuta n volte; quelli composti sono costituiti da due o più motivi ripetuti ciascuno n volte, mentre i microsatelliti interrotti da un'unità ripetuta ed interrotta da motivi di DNA di più paia di basi. Le ripetizioni composte da due-, tre- e quattro-nucleotidi sono le più frequenti nei genomi eucariotici.
Vantaggi e svantaggi dei microsatelliti
La rapida diffusione dei polimorfismi STRs in ambito forense, sia per quanto riguarda le indagini d’identificazione criminale sia per la ricerca biologica della paternità, è legata a molte caratteristiche che presentano:
(1) sono sistemi facilmente amplificabili tramite PCR che possono essere tipizzati con un alto grado di specificità in tempi relativamente brevi utilizzando tecniche non isotopiche altamente sensibili e riproducibili quali l'elettroforesi in gel di poliacrilamide nativo e denaturante.
(2) la loro moderata variabilità consente una corretta tipizzazione degli alleli, la quale avviene mediante confronto con un marcatore di peso molecolare noto. Inoltre la presenza di poche forme alleliche permette una valutazione semplice di parametri statistici quali frequenze alleliche, indice di eterozigosità ed equilibrio di Hardy-Weinberg.
(3) il ristretto range molecolare dei loci STR consente un'amplificazione omogenea prevenendo il cosiddetto fenomeno "allele drop-out", ossia l'amplificazione preferenziale dell'allele a più basso peso molecolare negli eterozigoti con formazione di falsi omozigoti.
(4) infine le ridotte dimensioni molecolari delle relative regioni polimorfe permettono l'amplificazione di DNA stampo altamente degradato riscontrabile in tessuti in via di decomposizione o putrefatti in resti ossei molto antichi o in tessuti inclusi in paraffina.
Tuttavia non tutti i microsatelliti conosciuti sono ugualmente adatti a scopi forensi; le principali considerazioni per la selezione dei loci sono il potere discriminativo (PD), l'assenza di linkage, la presenza di frequenze geniche ben bilanciate, l'accordo con l'equilibrio di Hardy- Weinberg e l'assenza di piccole bande shadows (bande accessorie derivate da un allineamento non corretto delle basi durante il processo di duplicazione tramite PCR).
Occorre dapprima valutare il potere discriminativo, che indica la probabilità, a priori, che due individui all'interno di una popolazione possiedano genotipi differenti; maggiore è questo valore, più basso è il livello di condivisione casuale delle forme alleliche. In genere questo valore deve essere superiore a 0.9. Il potere discriminativo è strettamente legato a due principali parametri: il numero degli alleli, cioè il grado di polimorfismo e l'indice di eterozigosità, cioè la loro distribuzione all'interno della popolazione. E' preferibile impiegare sistemi con eterozigosità maggiore di 0,7. Recenti sviluppi nel campo della genetica permettono ogni anno di scoprire centinaia ed anche migliaia di loci STR, che sono conservati in specifici database. Il GDB (Human Genoma Database) dell'Università di Baltimora, creato allo scopo di mappare tutto il genoma, all'inizio del 1995 conteneva più di 7000 loci STR all'interno dei quali più di 2000 erano costituiti da tetranucleotidi ma soltanto circa 200 presentavano un grado di eterozigosità elevato (superiore a 0.85).
Il cromosoma Y
Il cromosoma Y4 rappresenta uno dei due cromosomi sessuali, una delle 23 coppie di cromosomi omologhi umani. I cromosomi sessuali vengono indicati come X ed Y, mentre gli altri 44 sono definiti autosomi e sono presenti sia negli individui di sesso maschile che in quelli di sesso femminile.
Il ruolo principale del cromosoma Y nell'uomo, è determinare il sesso maschile e la fertilità, tramite una serie di geni che controllano lo sviluppo dei testicoli e la spermatogenesi .
Poiché lo possiedono solo gli individui di sesso maschile ed in singola copia, ha delle caratteristiche singolari all'interno del genoma umano:
- è costitutivamente aploide, cioè non esiste un suo omologo come invece avviene per gli autosomi o per il cromosoma X nella donna,
- si trasmette esclusivamente con modalità patrilineare, cioè da un padre ai suoi figli maschi,
- la sua sequenza è costituita per metà da DNA satellite ripetuto in tandem,
- evita in gran parte la ricombinazione alla meiosi, ad eccezione di due piccole porzioni che ricombinano con il cromosoma X,
- l'unico processo che agisce incisivamente nella sua diversificazione è la mutazione e
- in virtù di queste due ultime proprietà, la sua sequenza rimane altamente conservata lungo una linea di discendenza maschile (Jobling 5 and Tyler-Smith 2003).
Il cromosoma Y è uno dei più piccoli cromosomi umani, è lungo circa 60 Mb (57.701.691 bp) di cui sono note informazioni di sequenza soltanto per circa 23 Mb, e rappresenta uno dci più piccoli cromosomi umani, maggiore solamente ai cromosomi 21 (47 Mb) e 22 (49 Mb). Costituisce circa lo 0,38% del DNA nelle cellule del maschio ed è ovviamente assente nelle cellule della femmina.
Vi sono stati individuati appena 140 geni, nessuno dei quali contiene informazioni essenziali per la vita di un individuo, e la stragrande
maggioranza del suo DNA sembrerebbe non avere alcuna funzione.
Il cromosoma Y è un piccolo cromosoma acrocentrico, caratterizzato da una ridotta parte di eucromatina che comprende il braccio corto del cromosoma (Yp) e la parte prossimale del braccio lungo (Yq), in cui si trovano i pochi geni deputati al differenziamento delle caratteristiche sessuali maschili ed al controllo della spermatogenesi, mentre la restante parte dell'Yq è costituita da un'ampia regione eterocromatinica , dove si trovano sequenze non codificanti altamente ripetute. Per la maggior parte della sua lunghezza non è soggetto a ricombinazione, non è in grado di procedere al crossing-over con l'omologo cromosoma X, eccetto che per le due estremità dei bracci del cromosoma Y, le cosiddette regioni pseudo-autosomiche (PAR): all'estremità del braccio corto (Yp) si trova la PAR l, lunga circa 2,5 Mb, mentre all'estremità dcl braccio lungo (Yq) c'è la PAR2, lunga meno di 1Mb. La presenza di una tale regione di appaiamento è critica per la segregazione dei cromosomi X e Y durante la gametogenesi maschile. La restante parte, circa il 95%, tuttavia non si appaia né ricombina con il cromosoma X. Di conseguenza, originariamente fu denominata regione non ricombinante dell'Y (non-recomblning region of the Y chromosome, NRY - Butler 6 2005). Nella pubblicazione di Skaletsky 7 et al. (2003) questa regione venne rinominata regione dell'Y specifica per il sesso maschile (male specific region of the Y chromosome, MSY), in quanto sono state trovate prove che al suo interno avviene ricombinazione intracromosomica con una frequenza pari a quella con cui si verifica il crossing-over alla meiosi fra due autosomi. La regione MSY è suddivisa all'incirca equamente tra regioni eucromatiche contenenti geni funzionali e regioni eterocromatiche prive di geni. Infatti, all'interno dell'MSY ci sono due blocchi di eterocromatina, ovvero la regione centromerica ed una sequenza di circa 30 Mb situata sul braccio lungo del cromosoma, ed una porzione eucromatinica di circa 23 Mb, 8 Mb sul braccio corto e 14,5 Mb su quello lungo. Nella pubblicazione di Skaletsky 7 et al.
(2003), viene descritta la sequenza della porzione eucromatinica. Nell'MSY sono state trovate 156 unità di trascrizione, 78 delle quali codificanti proteine, tutte localizzate nelle due regioni di eucromatina. L'eucromatina, localizzata negli intervalli di delezione da 1 a 6, contiene almeno 30 geni differenti che possono essere suddivisi in due categorie: i geni che sono
esclusivamente o prevalentemente espressi nel testicolo ed geni che sono espressi ubiquitariamente nell'organismo e presentano degli omologhi sul cromosoma X.
I geni testicolo-specifici, tendono ad essere presenti in copie multiple e sono organizzati in cluster, circoscritti in una data regione oppure dispersi lungo il cromosoma.
Più precisamente nella porzione eucromatica, adiacente alla regione PAR del braccio corto del cromosoma Y, c'è un gene critico che controlla lo sviluppo sessuale in senso maschile, chiamato regione di determinazione del sesso (sex-determining region Y, SRY).
Nell'uomo, l'assenza del cromosoma Y quasi sempre porta allo sviluppo di una femmina, perciò questo gene non è presente sul cromosoma X. SRY codifica per un prodotto genico che induce il tessuto gonadico indifferenziato dell'embrione a formare testicoli. Questo prodotto è chiamato fattore di determinazione testicolare (testis-determining factor- TDF). SRY (o una sua versione correlata) è presente in tutti i mammiferi finora esaminati, ciò è indice della sua funzione essenziale nell'ambito di questo eterogeneo gruppo di animali.
Alcuni recenti risultati ottenuti da David Page et al. hanno fornito una visione verosimilmente completa della regione MSY del cromosoma Y umano. Questo lavoro, completato nel 2003, è basato sulle informazioni ottenute dal Progetto Genoma Umano, in cui è stato ormai sequenziato il DNA di tutti i cromosomi. La regione MSY consiste di circa 23 milioni di coppie di basi (23 Mb) e può essere suddivisa in tre classi.
La prima regione è denominata regione trasposta dell'X (X-ransposed region) e contiene circa il 15% della regione MSY. Le X-trasposte costituiscono due blocchi della lunghezza totale di 3,4 Mb che si trovano sul braccio corto (Yp), presentano un'identità del 99% con le sequenze nucleotidiche della banda Xq21, non danno crossing-over col cromosoma X alla meiosi e comprendono solamente due geni codificanti, ambedue con omologhi sul cromosoma X. Il loro nome è dovuto al fatto si sono originate a causa di una massiccia trasposizione di materiale genetico dal cromosoma X al cromosoma Y, avvenuta successiva mente alla divergenza dell'uomo dallo scimpanzé durante l'evoluzione umana (dai 3 ai 4 milioni di anni fa), la quale
è stata seguita da un'inversione sul braccio corto (Yp) che ha separato la sequenza nei due blocchi.
La seconda regione è denominata regione degenerativa dell'X e contiene circa il 20°/o della regione MSY. Le X-degenerate si trovano in otto distinti segmenti, distribuiti sia sul braccio lungo che su quello corto, che hanno una lunghezza totale di 8,6 Mb. Presentano un’identità di sequenza con le loro omologhe sul cromosoma X che va dal 60 al 96%. Esse sono probabilmente ciò che rimane delle sequenze di qualche autosoma antico, dal quale sono evoluti i due cromosomi del sesso. Nelle X-degenerate si ritrovano tutti i dodici geni espressi ubiquitariamente nel corpo umano ed uno solo degli undici espressi solo nei testicoli. Due di questi geni sono omologhi a dei geni X -linked.
La terza area, la regione ampliconica, contiene circa il 30% della regione MSY. Gli ampliconi sono costituiti da sette segmenti sparsi per tutta la MSY che hanno una lunghezza totale di 10,2 Mb. La maggior parte di queste sequenze presenta identità intracromosomiche del 99,9%. Molte di queste sequenze inoltre sono palindromi, ovvero ci sono coppie di segmenti di DNA duplicati nei quali una delle due copie è invertita ed il grado di identità fra le sequenze nucleotidiche è molto elevato. All'interno di queste sequenze c'è la più alta densità di unità di trascrizione del cromosoma Y (circa 60 unità di trascrizione), fra le quali ci sono nove famiglie di geni ( TSPY, VCY, XKRY , CDY, HSFY, RBMY, PRY, BPY2, DAZ) codificanti proteine per lo sviluppo e la funzionalità testicolare, tutti espressi esclusivamente o prevalentemente nei testicoli. Queste sequenze si sono originate per eventi di ricombinazione intracromosomica.
I marcatori genetici del cromosoma Y e la loro utilità in ambito forense La specificità del cromosoma Y per il sesso maschile e la modalità di trasmissione patrilineare, hanno suscitato notevole interesse riguardo le possibili applicazioni nell’ambito della genetica forense. Se consideriamo che circa il 93% dei crimini ed il 99% dei delitti a sfondo sessuale è commesso da uomini, troviamo anche delle ragioni statistiche a questo accresciuto
interesse.
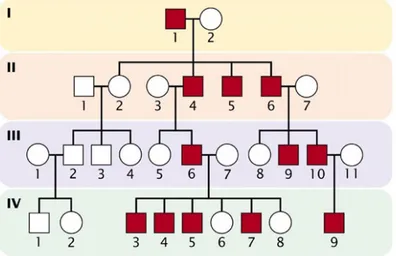
L'utilizzo in ambito forense dell'analisi e della tipizzazione del cromosoma Y richiede alcune considerazioni: prima fra tutte la trasmissione patrilineare e la conseguente assenza di ricombinazione, per cui l'intero cromosoma Y è condiviso anche dai fratelli e da tutti i discendenti per linea paterna (Fig. 1).
Fig. 1. Il cromosoma Y osserva una modalità di trasmissione patrilineare. In particolare i soggetti IV-7 e IV-9 condividono lo stesso cromosoma Y pur essendo parenti di sesto grado.
Posseduto solo dai maschi e costitutivamente aploide, si trasmette di padre in figlio ed evita in gran parte la ricombinazione meiotica, ad eccezione dei due segmenti PARl e PAR2 che ricombinano con il cromosoma X. L'importanza di avere una NRY così ampia consiste nel fatto che le combinazioni di stati allelici di marcatori situati lungo il cromosoma, solitamente passano inalterate da generazione a generazione, come un unico assetto aploide (Jobling 5 and Tyler-Smith 2003). L'unico evento che agisce nella
diversificazione di queste combinazioni è la mutazione, dunque è più facile che determinati assetti aploidi si fissino in una popolazione o che la loro distribuzione risenta di particolari eventi legati alla storia di questo gruppo di individui, costituendone al tempo stesso una sorta di testimonianza.
Come esemplificato nella pubblicazione di Jobling e Tyler-Smith (2003), se si assume un rapporto di 1:1 fra uomini e donne nelle popolazioni, visto che la donna ha due cromosomi X e l'uomo un cromosoma X ed un cromosoma Y, la proporzione totale di cromosomi Y sarà 1/4 rispetto ad ogni tipo di
autosoma ed 1/3 dei cromosomi X. Se si ipotizza che su tutti i cromosomi agiscano gli stessi processi di mutazione, ci si aspetta che la diversificazione della sequenza riguardi molto meno il cromosoma Y degli altri cromosomi, come in effetti avviene.
Si ritiene inoltre che il cromosoma Y sia più soggetto alla deriva genetica, per cui si verificano cambiamenti nelle frequenze geniche di un aplotipo.
Per questi motivi marcatori opportunamente individuati sul cromosoma Y umano rappresentano marcatori di linee di discendenza paterne chiamati lineage markers. La loro utilità sta nel fatto che consentono di ricostruire discendenze andando a ritroso anche di molte generazioni. Questo elemento è di fondamentale importanza per gli studi antropologici ed evoluzionistici. Lo studio delle distribuzioni dei marcatori del cromosoma Y nelle varie popolazioni in ambito forense, può consentire di individuare la provenienza geografica di un individuo dato un campione di materiale genetico. Sempre nel campo forense, l'analisi del cromosoma Y assume un ruolo di primaria importanza in determinati casi:
- nei casi di paternità "deficitari": qualora non sia disponibile il padre, è possibile fare degli accertamenti indiretti attraverso fratelli del padre o altri soggetti imparentati in linea paterna;
- nei casi di mixing biologici che coinvolgono più di due soggetti maschili: in questi casi l'analisi può stabilire il numero di tali soggetti;
- nei casi di violenza sessuale: in tali casi, infatti, può essere difficile discriminare la componente genetica derivante dalla vittima (cellule mucosali) rispetto a quella derivante dal colpevole (seme); il cromosoma Y è l'unico materiale genetico posseduto esclusivamente dall'individuo di sesso maschile (colui che ha compiuto il reato in questo caso specifico) e non è quindi soggetto alle contaminazioni da parte del DNA della vittima. Chiaramente è possibile anche rilevare l'eventualità in cui più di un individuo abbia partecipato alla violenza;
- nei casi di identificazione di persone scomparse, mediante la comparazione con il profilo ottenuto da parenti di linea paterna.
Per le caratteristiche biologiche osservate, il cromosoma Y ha il più basso livello di polimorfismi dei 24 diversi cromosomi umani. In particolare, il cromosoma Y mostra una variazione di sequenza ogni circa 10.000
nucleotidi, mentre gli altri cromosomi in media mostrano una variazione di più di un nucleotide ogni 1000.
Per tipizzare il cromosoma Y si analizzano marcatori polimorfi: essendo il cromosoma Y presente in singola copia nel genoma di un individuo di sesso maschile, la sua analisi genererà, per ogni marcatore, un genotipo costituito da un singolo allele. L’insieme ordinato degli alleli, osservati su uno specifico cromosoma, è definito aplotipo. Se analizziamo un numero di marcatori superiore o uguale a 18, parliamo di aplogruppo, se analizziamo un numero minore di marcatori parliamo di aplotipo: un aplotipo di 18 marcatori costituisce un aplogruppo. Nel 1997 è stata definita una serie di marcatori necessari per garantire “l’aplotipo minimo” per la tipizzazione del cromosoma Y (Kayser 8 et al. 1997). L’aplotipo minimo è definito dai loci: DYS19; DYS389I; DYS389II, DYS390; DYS391; DYS392; DYS393 e DYS385a/b. Nel 2003, è stato raccomandato l’utilizzo di due marcatori addizionali (DYS438 e DYS439) per definire in modo più preciso l’aplotipo minimo. Se l’aplotipo ottenuto dall’analisi del cromosoma Y di un sospettato non coincide con quello del colpevole la diagnosi di esclusione è certa. Il discorso cambia radicalmente quando si tratti di effettuare una conferma sulla base dell’aplotipo del cromosoma Y. Il problema chiave è la condivisione dello stesso aplogruppo (aplotipo) da parte di tutti i discendenti per linea paterna: stabilire l’identità biologica di una persona esclusivamente sulla base dei risultati forniti dall’analisi dell’aplotipo del cromosoma Y è impresa piuttosto avventata, in quanto lo stesso aplotipo può essere condiviso da tutti i parenti in linea paterna, e suscettibile di errori di false attribuzioni. La tipizzazione del cromosoma Y non è un sistema identificativo e, sebbene particolarmente dirimente in particolari condizioni, dovrebbe essere sempre accompagnata dall’analisi del DNA autosomico. Il suo utilizzo appare quindi un elemento di prova addizionale, piuttosto che una valida alternativa.
Le inserzioni Alu
Il polimorfismo YAP (Y-Alu polymorphism), è stato il primo marcatore binario del cromosoma Y umano ad essere scoperto (Butler 6 2005). Lo YAP
appartiene alla famiglia Alu ed è costituito da un'inserzione di 280 bp nel sito nucleotidico DYS287, situato sul braccio lungo del cromosoma Y. Le due forme alleliche, YAP- (assenza di inserzione) e YAP+ (presenza di inserzione), indicano rispettivamente la forma ancestrale e la forma derivata. Gli individui che presentano l'inserzione YAP sul proprio cromosoma Y possono appartenere a due soli aplogruppi: O o E. Per questo motivo tale polimorfismo viene generalmente testato sull'intero campione come screening iniziale per indirizzare le analisi successive in quanto permette di distinguere due grossi gruppi di linee del cromosoma Y (O e E/ tutti gli altri aplogruppi).
Gli Y-SNPs
Con il termine Y-SNPs ci si riferisce agli SNPs del cromosoma Y. La maggior parte degli SNPs sono biallelici, cioè hanno una forma ancestrale ed una forma mutante. Visto che questi polimorfismi interessano una sola base della sequenza, gli eventi che li originano sono mutazioni per sostituzione di base. Ogni individuo può possedere anche milioni di SNPs ed è per questo che la loro tipizzazione viene sfruttata in molti campi della ricerca genetica. Una volta avvenute, queste mutazioni possono fissarsi in una popolazione nel corso delle generazioni a causa della bassa frequenza con cui avvengono, divenendo quindi popolazione- specifiche. Questo è particolarmente vero per gli Y-SNPs, i quali possiedono un'utilità unica nello studio di eventi demografici anche molto antichi della storia della specie umana.
La tipizzazione di questi marcatori, in campo forense, può consentire l'attribuzione della provenienza geografica ad un determinato campione o l'appartenenza ad un gruppo etnico, dato che costituisce un utile integrazione alle informazioni ottenibili dai marcatori biallelici. Può rivelarsi utile anche nell'attribuzione di campioni ai loro donatori in caso di tracce biologiche estremamente deteriorate.
Gli STRs, o Microsatelliti o Short tandem Repeats, rappresentano i marcatori del cromosoma Y più utilizzati nella pratica forense; sono una classe di markers multiallelici costituiti da brevi (da 2 a 6 nucleotidi e con lunghezza massima di 350 bp) sequenze ripetute in serie diffuse lungo il genoma. Oggigiorno si conoscono circa 219 STRs sul cromosoma Y.
A differenza degli STRs autosomici, nei quali la variabilità è data da tre fattori diversi quali riassortimento cromosomico, ricombinazione e mutazione, gli STRs del cromosoma Y vedono implicata nella loro variabilità aplotipica soltanto la mutazione. Di conseguenza, gli studi di identificazione genetica, implicano l’analisi di un numero maggiore di marcatori per ottenere un potere discriminativo paragonabile a quello degli STRs autosomici; si deve inoltre considerare che gli Y-STRs vengono ereditati come un unico aplotipo dal padre, e questo complica la distinzione dei soggetti in linea maschile di una famiglia i quali membri condivideranno tutti uno stesso aplotipo, a meno di una rara evenienza di una mutazione. Per queste ragioni, gli Y-STRs, sono più comunemente utilizzati in ambito forense per individuare componenti maschili in miscele di DNA nel caso in cui sia presente un elevato background femminile, o per ricostruzioni di paternità tra individui di sesso maschile.
A tali scopi sono disponibili, ad oggi, pannelli Y-STR tra cui alcuni kit commerciali fino a 23 marcatori; esistono anche database di riferimento di grandi dimensioni ed in crescita continua per stimare le frequenze Y-STR aplotipiche tra diverse popolazioni umane in base alle distribuzioni geografiche. Esistono tuttavia alcune limitazioni all’utilizzo di questi pannelli in ambito forense, poiché, nonostante la diversità degli aplotipi Y23, il potere discriminativo rispetto agli STR autosomici rimane notevolmente inferiore. Mediante studi è stato dimostrato che la diversità aplotipica Y-STR, misurata con gli attuali set Y-STR, può essere incrementata e la differenziazione della linea maschile può essere migliorata aggiungendo ulteriori Y-STR debitamente selezionati.
Il deficit più grande nell’utilizzo di tali marcatori del cromosoma Y nella pratica forense è rappresentato dall’incapacità dell’esclusione di parenti, per parte paterna, vicini o lontani dell’imputato, nel caso in cui sia stato
depositato il materiale biologico al posto dell’imputato stesso: questo trova spiegazione nel relativamente basso tasso di mutazione, di sole alcune mutazioni per locus ogni mille generazioni.
Gli attuali set Y-STR generalmente non consentono di differenziare un profilo ottenuto dalla corretta identificazione del sospetto da un profilo ottenuto da diversi maschi correlati per parte paterna con il sospetto.
Rapidly mutating Y-STR
Nel Settembre 2010 Kaye N. Ballantyne, Miriam Goedbloed et al., in un lavoro intitolato " Mutability of Y-chromosomal microsatellites: Rates, Characteristics, Molecular bases and ForensicImplications'', studiarono i tassi di mutazione di 186 Y-STRs, analizzando il DNA di 1966 coppie europee padre-figlio per ciascun marker e dando osservazione diretta di 352,999 trasferimenti meiotici, fino ad oggi il più grande studio nel suo genere. Nel complesso sono state identificate 924 mutazioni in 120 (64.5%) dei 186 marcatori Y-STRs studiati. Più precisamente, 91 marcatori Y-STRs (48.9%) hanno mostrato tassi di mutazione nell'ordine di 10-3, altri 82
marcatori (44%) nell'ordine di 10-4 e 13 marcatori (6.9%) nell'ordine di 10-2.
Attraverso tutti i 186 marcatori Y-STRs, il tasso di mutazione medio era di 3.35 x 10-3 (intervallo di credibilità del 95%, da 1.79 x 10-3 a 6.38 x 10-3), simile al tasso di mutazione ottenuto da Kayser et al. e Hohoff et al. per un piccolo numero di Y-STRs. In particolar modo, i 13 marcatori Y-STRs con tassi di mutazione superiori a 1 x 10-2, i quali rappresentano solo il 7% dei marcatori studiati, sono stati caratterizzati dal più elevato numero di mutazioni osservate in questo studio (circa 462 delle 924 mutazioni totali, ossia il 50%).
Questi 13 marcatori sono stati così definiti "rapidly mutating (RM) Y- STR" (tutti con tassi di mutazione superiori a 1x10-2) e sono i seguenti: DYF387Sl, DYF399Sl, DYF403Sl, DYF404Sl, DYS449, DYS518, DYS526, DYS547, DYS570, DYS576, DYS612, DYS626 e DYS627.
)
(DYF387Sl, DYF404Sl e DYS526 con due copie, DYF399Sl con tre copie e DYF403Sl con quattro copie) mentre otto sono marcatori singola copia (sebbene sei di questi marcatori contengano diversi loci Y-STRs all'interno del singolo amplicone, e solo due, DYS570 e DYS576, sono ripetizioni semplici, con un solo locus Y-STR rispettivamente).
I 13 RM Y-STRs sono stati combinati in un unico set in base all'ipotesi che maschi strettamente correlati (coppie padre-figlio oppure coppie di fratelli) possano essere differenziati da mutazioni degli STR solo se gli RM Y-STRs sono tra loro combinati. In linea di principio, una mutazione ad uno solo dei 13 RM Y-STRs potrebbe bastare per la differenziazione individuale. Per ipotizzare un'analisi statistica della capacità degli RM Y-STRs di differenziare tra parenti maschi ed al fine di confrontare la loro potenzialità con quella del set Yfiler comunemente utilizzato, è stato calcolato in primo luogo il tasso di mutazione osservato per ciascuno dei due set per mezzo di un approccio bayesiano. Il numero di mutazioni osservate in ogni coppia padre-figlio per ciascun set Y-STR è stato analizzato mediante una distribuzione di Poisson. Per la distribuzione degli RM Y-STR è stato stimato a posteriori un tasso di mutazione medio di 1.97 x 10-2 (intervallo di
credibilità del 95% da 1.8 x 10-2 a 2.2 x 10-2), il quale è risultato essere 6.5 volte più elevato di quello stimato per il set Yfiler (tasso di mutazione medio di 3.0 x 10-3. Successivamente è stata stimata la probabilità di osservare almeno una mutazione in ciascuno dei due set Y-STR per una data coppia padre-figlio, un'analisi fondamentale per soddisfare i criteri minimi di differenziazione tra parenti maschi. Utilizzando le stime a posteriori appena citate, la probabilità di osservare almeno una mutazione nel set RM Y-STR è di 0.1952. Tale valore è risultato essere 4 volte superiore rispetto al valore stimato per il set Yfiler pari a 0.047; quindi tale probabilità è significativamente superiore rispetto al set Yfiler (p < 5.0 x 10-7).
Infine Ballantyne et al. fornirono una prova teorica così come empirica della capacità degli RM Y-STRs di differenziare individui maschi strettamente correlati. A tale scopo, sono state genotipizzate ulteriori 103 coppie di parenti maschi da 80 pedigree maschili che erano tra loro collegate da 1 a 20 generazioni, confrontando poi i risultati con quelli ottenuti mediante l'utilizzo del set Yfiler per gli stessi campioni.
Nel complesso è stato dimostrato che il set RM Y-STR distingue il 70.9% di combinazioni di parenti maschi con almeno una mutazione, un aumento di 5 volte superiore del livello di differenziazione maschile ottenuto con il set Yfiler (solo il 13% di combinazioni di parenti maschi); in particolare, la differenza significativa (t=6.389, p<0.0001) è simile alle aspettative statistiche riguardanti le coppie padre-figlio inizialmente utilizzate per stabilire i tassi di mutazione STR. All'interno del pedigree, il set RM Y-STR ha distinto il 70% delle coppie padre-figlio, il 56% dei fratelli e il 67% dei cugini.
In contrasto, il set Yfiler non è stato in grado di differenziare nessuna delle coppie padre-figlio e dei cugini, ma solo Il 6% dei fratelli. Inoltre, in tutti i parenti separati da più di 11 generazioni sono state individuate una o più mutazioni con il set RM Y-STR, mentre solo il 33% sono stati differenziati con Yfiler.
L'esame di questi 13 RM Y-STR è stato inoltre fondamentale per dimostrare che la compresenza di livelli ottimali dei tre parametri che influenzano la mutabilità di una dato Y-STR (lunghezza dì ripetizione, numero totale di ripetizioni e complessità totale) sembrerebbe aumentare di un ordine di grandezza o più il tasso di mutazione degli Y-STRs stessi. Infatti il numero medio di ripetizione totale per gli RM Y-STR, pari a 32.8, è più di due volte superiore rispetto ai restanti 173 Y-STR utilizzati in tale studio, così come la complessità totale della sequenza ripetuta, che risulta essere 2.7 volte superiore rispetto ai restanti Y-STR.
In un secondo fondamentale studio, pubblicato nel Maggio del 2012 da Kaye N. Ballantyne, Victoria Keerl et ai. ed intitolato "A new future of forensic Y-chromosome analysis: Rapidly mutatlng Y-STRs for differentiating male relatives and paternal lineages", il potere degli RM Y-STRs di differenziare tra individui non imparentati (di separare linee paterne), è stata testato con lo Human Genome Diversity Panel (HGDP),fornito dal Centre d'Etude du Polymorphisme Humain (CEPH). Il set completo comprende 668 maschi provenienti da 59 popolazioni dislocate in 8 regioni geografiche distinte. Per misurare la capacità di risoluzione della stirpe paterna, è stato utilizzato il set ridotto H592, in cui tutti i parenti di primo e secondo grado sono stati rimossi. Ciò ha determinato un gruppo finale di studio di 604 maschi non
strettamente correlati provenienti da 51 popolazioni localizzate in 8 regioni geografiche: 81 dall'Africa sub- sahariana (6 popolazioni), 20 dal nord Africa (1popolazione), 50 dal Medio Oriente (3 popolazioni), 163 dall'Asia sud-occidentale (8 popolazioni), 17 dall'Oceania (2 popolazioni), 83 dall'Europa (8 popolazioni), 22 dall'America (5 popolazioni) e 169 dall'Asia orientale (18 popolazioni).
Mentre la capacità degli RM Y -STRs di distinguere tra parenti di sesso maschile è stata testata con 305 persone provenienti da 127 pedigree separati o da piccole famiglie (156 coppie di parenti). Tali campioni provenivano dalle aree germaniche di Greifswald, Kiel e Berlìno, dall'area di Leuven del Belgio e da Varsavia, oltre che dal Canada e dalla Germania centrale. Inoltre, tutti i parenti di primo e secondo grado inclusi nel pannello HGDP-CEPH sono stati esclusi dall'analisi dei parenti maschi.
Le relazioni familiari tra campioni inclusi nell'HGDP-CEPH sono state precedentemente individuate da 783 loci STR autosomici, mentre la natura della relazione familiare (ad es. padre-figlio, cugini, zio- nipote) è stata calcolata sulla base della condivisione allelica. Anche in tal caso, per illustrare i valori del set RM Y-STR, sono stati confrontati i risultati con quelli ottenuti con il set Yfiler, il più grande set Y-STR ad ora disponibile in commercio.
Tra i 604 maschi indagati in tale studio, 595 aplotipi unici sono stati osservati con il set RM Y-STR (capacità di discriminazione del 98.3%) e 511 con il set Yfiler (capacità di discriminazione del 90.4%), ossia un aumento statisticamente significativo dell'8% su scala globale (t-test: t=4 .334, p=0.002). Sei delle 8 regioni geografiche in tutto il mondo (Africa sub-sahariana, nord-Africa, Medio Oriente, sud/ovest Asia,Oceania ed Europa) sono state pienamente risolte con il set RM Y-STR, mentre solo l'Africa del nord con il set Yfiler; ciò si traduce in un aumento della capacità di discriminazione dei 13 RM Y- STR in tutte le regioni geografiche del mondo. Il maggior incremento è stato osservato in Medio Oriente, dall'84% di capacità discriminativa con Yfiler al 100% con RM Y-STR, mentre le altre regioni geografiche hanno visto un aumento dell' 11% (Africa sub- sahariana e sud/ovest Asia), dell'8% (est asiatico), del 6% (Oceania), del 5% (sud America) e dell'1% (Europa) rispetto al set Yfiler.
Con il set RM Y-STR solamente tre aplotipi sono stati ripartiti tra 8 individui maschi dei 604 uomini indagati in tale studio, mentre con il set Yfìler è stata ottenuta la condivisione di 33 aplotipi tra 85 individui maschi. I tre aplotipi sopracitati riguardano, rispettivamente, una coppia di individui appartenenti alla popolazione Kalash dell'Asia del sud e due gruppi di tre individui, uno dalla popolazione Yakut dell'Asia orientale e l'altro dalla popolazione Karitfana del sud America.
Oltre ad una maggior capacità di discriminazione, il set RM Y-STR ha mostrato un aumento sostanziale della diversità aplotipica rispetto al set Yfiler. Anche se la differenza tra la diversità aplotipica osservata con i due set di marcatori non è statisticamente significativa (t-test: t =2.074, p=0.072), il pattern osservato è molto simile a quello ottenuto per la capacità di discriminazione tra i due set. Solo due regioni geografiche non hanno mostrato alcun aumento della diversità aplotipica con il set RM Y-STR rispetto al Yfìler, ossia il nord Africa (diversità aplotipica pari a 1 per entrambi i set) ed il sud America (diversità aplotipica pari a 0.9870 per entrambi i set). Si può quindi assumere che, a livello globale, la diversità aplotipica è aumentata da un valore di 0.9994 con il set Yfiler ad un valore di 0.99996 con il set RM Y-STR .
Un aspetto molto importante è dato dal fatto che per raggiungere la massima capacità discriminativa nonché la massima diversità aplotipica all'interno di ciascuna regione geografica rappresentata nel pannello HGDP CEPH, il set RM Y-STR ha richiesto un numero notevolmente più basso di marcatori rispetto al set Yfiler. Solo 2 marcatori RM Y-STR (DYF399Sl e DUF403S1) sono stati sufficienti a risolvere tutti gli individui in quattro regioni geografiche (regione sub-sahariana, nord Africa, Europa e Asia orientale), mentre a livello globale, sono stati necessari 7 RM Y-STR per ottenere la massima capacità discriminativa nonché diversità aplotipica, rispetto ai 12 del set Yfiler.
Il confronto tra le coppie di individui facenti parte del pannello HGDP-CEPH ha dimostrato che gli RM Y-STR consentono l'individuazione di un numero maggiore di differenze alleliche rispetto al set Yfiler, sia in assoluto che in termini relativi . Dei 222.000 confronti allelici effettuati in tale studio, il numero medio di differenze alleliche individuato tra le coppie
è pari a 11.81 con il set Yfiler (69 .4% dei possibili alleli) mentre è pari a 17.99 con il set RM Y-STR (85.7% dei possibili alleli). Ciò si traduce anche in un numero relativamente elevato di differenze tra i loci RM Y-STR, più precisamente 11.66 dei 13 loci sono differenti (89.7%).
Gli alti tassi di mutazione degli RM Y-STR hanno consentito di ipotizzare che i maschi, anche se strettamente correlati, possono essere differenziati con tale set dato che una singola mutazione in un unico locus può essere sufficiente per separare individui imparentati. Come già anticipato, nello studio effettuato da Ballantyne et al. nel 2010 era già stato individuato un tasso medio di differenziazione, tra individui tra loro collegati da 1 a 20 generazioni, pari al 70%. Combinando i risultati ottenuti da questo precedente studio con i nuovi dati, è stato possibile genotipizzare con il set RM Y-STR ben 156 coppie di parenti maschi di varia ascendenza biogeografica; mentre solo il 15% dei parenti maschi è stato differenziato con il set Yfiler.
Tuttavia, nelle indagini forensi, risultano essere più rilevanti ai fini investigativi le strette relazioni parentali quali padre-figlio, fratelli e cugini rispetto alle relazioni di parentela meno strettamente correlate (generalmente superiori alle 10 generazioni). Considerando rapporti di parentela più stretti, il set Yfiler è stato in grado di differenziare solo il 7.7% delle coppie padre-figlio, l'8% dei fratelli ed il 25% dei cugini (separati da 4 eventi meiotici). Per contro, il set RM Y-STR è stato in grado di differenziare il 66% di tutti i parenti maschi analizzati, ben 4.4 volte in più rispetto ad Yfiler; più precisamente, questa percentuale comprendeva il 48.7% delle coppie padre-figlio (6 volte di più rispetto a Yfiler), il 60% dei fratelli (7.5 volte di più rispetto a Yfiler) e il 75% dei cugini (3 volte di più rispetto a Yfiler).
Tutte le coppie separate da 9 o più eventi meiotici si distinguevano per almeno 1 mutazione con il set RM Y-STR contro il solo 47% delle coppie con Yfiler. È stata quindi rilevata una chiara tendenza di aumento del grado di differenziazione con il crescere del numero di eventi meiotici.
Dato il maggior numero di mutazioni osservato nel set RM Y-STR, è stato possibile rilevare anche un aumento del numero medio di mutazioni ad ogni ulteriore evento meiotico che si verifica tra le coppie esaminate. Infatti con una sola separazione meiotica è stata individuata una media di mutazioni di
0.1 con Yfiler e di 0.65 con RM Y-STR. Questo valore è aumentato rispettivamente fino a 0.7 e 2.0 dopo 10 meiosi, e fino a 4 mutazioni, con il set RM Y-STR, dopo 20 meiosi.
Possiamo quindi concludere affermando che i marcatori RM Y-STR presentati in questo studio mostrano sostanzialmente una più elevata capacità di discriminazione e diversità aplotipica rispetto al set commerciale Yfiler composto da 17 marcatori Y-STR. Ciò è dovuto ad un più alto tasso di mutazione di tutti gli RM Y-STR (nell'ordine di 1x10-2) che consente non solo di generare maggiori range allelici per ciascun marcatore RM Y-STR, ma anche di individuare un maggior numero di aplotipi. Inoltre, il più alto tasso di mutazione elimina qualsiasi effetto del fondatore o deriva genetica a cui è stata sottoposta una determinata popolazione, consentendo così di ottenere una più omogenea distribuzione di aplotipi tra le popolazioni di tutto il mondo.
Anche se l'aumento della capacità di discriminazione tra individui maschi non imparentati è senza dubbio molto utile, soprattutto in popolazioni con complessiva riduzione della diversità del cromosoma Y causata da effetti demografici o culturali, è per lo più l'elevata capacità di distinguere tra parenti maschi strettamente correlati e non, che favorirà notevolmente l'analisi del cromosoma Y in biologia forense. Tuttavia, il maggior numero di mutazioni osservato nei marcatori RM Y-STR evidenzia, al contempo, la loro incapacità nello stabilire il tipo di relazione familiare, come è generalmente richiesto nei casi di paternità e nei test familiari. In questi casi, di conseguenza, è consigliabile l'utilizzo dei marcatori Yfiler o di qualsiasi altro marcatore Y-STR caratterizzato da un basso tasso di mutazione.
Occorre infine ricordare che vi è una limitazione nell'applicazione del set RM Y-STR a scopi forensi, riguardanti le grandi dimensioni delle sequenze ripetute di molti dei loci RM Y-STR (sono state osservate fino a 59 ripetizioni per il locus DYF40351). Da un lato, questo elevato numero di ripetizioni è essenziale per creare alti tassi di mutazione, essendo questo uno dei tre principali fattori molecolari che influenzano la mutabilità degli Y-STR. Dall'altro, il gran numero di ripetizioni porta alla formazione, nell'analisi PCR, di un amplicone di notevoli dimensioni che può causare problemi durante l'analisi di sequenze di DNA fortemente degradato.