UNIVERSITÀ DI PISA
FACOLTÀ DI SCIENZE MATEMATICHE, FISICHE E NATURALI
Corso di Laurea in Scienze BiologicheTESI DI LAUREA
MUTAZIONI PROTROMBOTICHE COME FATTORI DI
RISCHIO PER EVENTI ISCHEMICI CEREBROVASCOLARI
CRIPTOGENICI IN SOGGETTI CON
FORAME OVALE PERVIO
Candidata: Relatori:
Francesca Antoniazzi Dr.ssa Maria Grazia Andreassi
INDICE
RIASSUNTO pag. 4
ABSTRACT pag. 7
INTRODUZIONE pag. 10
TROMBOSI: UNA MALATTIA MULTIFATTORIALE pag. 13
VARIANTE G1691A DI LEIDEN DEL FATTORE V pag. 17
VARIANTE G20210A DELLA PROTROMBINA pag. 20
VARIANTE C677T DELLA METILENETETRAIDROFOLATO REDUTTASI pag. 22
FATTORE V LEIDEN, PROTROMBINA G20210A, MTHFR C677T E RISCHIO DI TROMBOSI pag. 26
FORAME OVALE PERVIO pag. 28
SCOPO DELLA TESI pag. 33
MATERIALI E METODI pag. 34
POPOLAZIONE DI STUDIO pag. 34
ANALISI GENETICA pag. 37
ANALISI STATISTICA pag. 42
CARATTERISTICHE DEMOGRAFICHE E PRESENZA DEI MAGGIORI FATTORI DI RISCHIO PER MALATTIE CARDIOVASCOLARI pag. 43
DISTRIBUZIONE DEI GENOTIPI pag. 44
RISCHIO ASSOCIATO ALLA PRESENZA DI CIASCUN POLIMORFISMO PER ISCHEMIA CEREBRALE NEI PAZIENTI CON FOP pag. 47
ANALISI MULTIVARIATA pag. 48
ANALISI DELLA RICORRENZA DI EVENTI pag. 49
DISCUSSIONE pag. 51
CONCLUSIONI pag. 57
LETTERATURA CITATA pag. 59
RIASSUNTO
Il Forame Ovale Pervio (FOP) è una anomalia cardiaca che è presente circa nel 25% della popolazione generale e nella maggior parte dei casi non è associata ad alcuna manifestazione clinica. Numerosi studi hanno, tuttavia, evidenziato una forte associazione tra la presenza di FOP ed il rischio di eventi cerebrovascolari ischemici, presumibilmente di origine embolica, soprattutto in soggetti giovani. In questi pazienti la presenza di uno stato di ipercoagulabilità, dovuto a trombofilia ereditaria, potrebbe determinare un aumento del rischio di episodi ischemici cerebrali.
Vi sono, infatti, varianti genetiche relativamente frequenti nella popolazione generale riconosciute come importanti fattori di rischio per malattia tromboembolica, tra queste la variante G1691A di Leiden del fattore V (FVL) e la variante G20210A del gene della protrombina (PT). Recentemente, entrambe queste mutazioni sono state dimostrate essere importanti fattori di rischio per eventi cerebrovascolari ischemici, soprattutto in soggetti giovani ed in assenza di altri concomitanti fattori di rischio. Inoltre, la presenza in omozigosi della mutazione C677T della metilenetetraidrofolato reduttasi (MTHFR), enzima chiave nel metabolismo dell'omocisteina, potrebbe svolgere un ruolo importante
Scopo della tesi è stato quello di valutare l’incidenza delle varianti FVL, G20210A PT e C677T MTHFR in una popolazione di pazienti con eventi ischemici cerebrali e diagnosi di FOP, afferenti all'ospedale “G.Pasquinucci” di Massa per la chiusura transcatetere del difetto cardiaco.
Sono stati reclutati 220 pazienti (102 uomini; età media: 45.1±12 anni) e 182 soggetti di controllo appaiati per età e sesso (71 uomini; età media: 41.8±10.3 anni). Per ciascun soggetto è stata effettuata l'analisi genetica tramite un kit diagnostico commerciale che consente l’amplificazione multipla dei tre geni mediante PCR e una ibridazione allele-specifica.
I risultati hanno mostrato che la prevalenza di soggetti portatori di FVL (1.8% vs 0.5%;χ2=1.3, p=ns) e della variante 20210A PT (6.4% vs 1.1%;χ2=7.7, p=0.02) era più alta nel gruppo dei pazienti con FOP rispetto al gruppo dei controlli. La frequenza della variante 677T MTHFR era simile tra pazienti e controlli (26.8% vs 23.6%; χ2
=0.7, p=ns).
Dopo correzione per i principali fattori di rischio cardiovascolari, è risultato che la presenza del FVL o della variante 20210A PT in pazienti con FOP determina un rischio aumentato di 3.4 volte (95% CI=1.2-9.1; p=0.02) di eventi ischemici cerebrali.
Inoltre, la presenza del FVL o della variante 20210A PT era più frequente nei soggetti che hanno manifestato più di un evento ischemico rispetto a quelli che ne hanno manifestato solo uno (18.5% vs 7.2%; χ2=3.8, p=0.05).
I risultati ottenuti indicano che le mutazioni protrombotiche prese in considerazione in questo studio sono fattori di rischio importanti per ischemia cerebrale in pazienti con FOP.
ABSTRACT
Patency of the foramen ovale (PFO) is present in 25% of the general population and in most cases never causes a health disturbance. However, the presence of a PFO is considered a possible cause of unexplained, presumably embolic, ischemic neurological events in young patients.
In these patients, the procoagulant conditions, caused by congenital thrombophilia, may increase the risk for recurrent ischemic cerebral events.
In particular, genetic polymorphisms, relatively frequent in Caucasian population, are identified as risk factors for venous thrombosis: factor V Leiden (G1691A) and prothrombin (PT) G20210A.
Recently, prothrombotic mutations have been suggested as genetic risk factors for “cryptogenic” ischemic cerebrovascular disease in young adults and a relation between the prothrombotic mutation and cerebral ischemia risk in younger PFO patients has also been reported.
Several studies on genetic variant 667 of methylenetetrahydrofolate reductase (MTHFR), which plays a crucial role in regulation of plasma homocysteine concentration, reported an association between gene polymorphism and stroke.
The aim of this study was to evaluate the prevalence of the factor V Leiden, prothrombin G20210A and C677T MTHFR in patients with cerebral ischemia associated with PFO, who were referred for percutaneous transcatheter closure of their cardiac defect.
We examined 220 patients (102 males; age: 45.1±12 years) and 182, matched for age and sex, healthy subjects (71 males; age: 41.8±10.3 years).
We performed genetic analysis for each subjects by using a multiplex allele-specific polymerase chain reaction assay.
The prevalence of subjects carrying FVL (1.8% vs 0.5%;χ2=1.3, p=ns) and 20210A PT (6.4% vs 1.1%;χ2=7.7, p=0.02) was significantly higher in the group of PFO patients than in the controls. No difference was observed for C677T MTHFR genotype distribution (26.8% vs 23.6%; χ2=0.7, p=ns)
After adjustment for other vascular risk factors, the combination of either factor V Leiden or prothrombin 20210A and PFO was associated with a 3.4-fold (95% CI=1.2-9.1; p=0.02) increased risk of cerebral ischemia.
The prevalence of subjects carrying at least 1 prothrombotic genotype (FVL or 20210A PT) was significantly higher in the PFO patients with recurrent ischemic events than in patients with one ischemic event (18.5% vs 7.2%; χ2
Our results indicate that prothrombotic mutations are important risk factors for cerebral ischemia in patients with PFO.
INTRODUZIONE
Negli ultimi anni, i progressi nell’ambito della ricerca genetica e la completa conoscenza del genoma umano hanno consentito di poter identificare molti dei geni coinvolti nell’eziopatogenesi di malattie multifattoriali.
Le recenti conoscenze scientifiche hanno, inoltre, dimostrato l’importanza della suscettibilità genetica individuale nell’insorgenza e nella progressione di gran parte delle patologie.
Sappiamo, infatti, come ogni persona dimostri il 99.9% d’identità genetica rispetto ad una qualsiasi altra, mentre il restante 0.1% del nostro DNA rappresenta la variabilità individuale, che è sostanzialmente dovuta a piccole variazioni di sequenza, molto spesso sostituzioni di singoli nucleotidi chiamate SNP (mutazioni puntiformi ad un singolo nucleotide). Ad oggi, sono stati identificati circa 3 milioni di SNP, ma si calcola che ve ne siano almeno 10 milioni.
È ormai riconosciuto che gruppi di queste piccole variazioni, interagendo tra loro, siano in grado di determinare la suscettibilità o la resistenza a molte delle malattie umane più comuni.
Questo può risultare determinante nello sviluppo di nuovi modelli di stratificazione del rischio e nella scelta di trattamenti terapeutici adeguati basati
sulla conoscenza del profilo genetico di ciascun individuo (Housman e Ledley 1998).
L’identificazione della componente genetica delle malattie complesse viene unanimemente riconosciuta essere una delle principali sfide della medicina dei prossimi anni.
La possibilità di avere test genetici che predicano la suscettibilità al cancro, all’infarto o al morbo di Alzheimer, avrebbe un enorme impatto in termini di salute pubblica. Un’altra delle applicazioni più interessanti delle scoperte di genetica e genomica riguarda la possibilità di identificare la relazione tra la costituzione genetica di un individuo e la sua risposta a farmaci somministrati, sia in termini di efficacia sia in termini di tossicità (Zhou et al. 2008).
La prospettiva futura è quella di fornire prevenzione e diagnosi attraverso programmi individualizzati basati sul rischio personale (medicina predittiva) e trattamenti più mirati su base individuale (farmacogenetica). Con le parole di McKusick: “la terapia giusta per il paziente giusto” (McKusick 2001).
Recentemente, anche per le malattie cardiovascolari sono state studiate ed identificate numerose varianti genetiche di suscettibilità in grado di conferire al paziente una certa vulnerabilità ai diversi aspetti della malattia.
trombotico hanno, già, trovato ampio impiego nella pratica clinica cardiovascolare con lo scopo di individuare i soggetti a rischio e migliorare la prevenzione dei quadri clinici.
TROMBOSI: UNA MALATTIA MULTIFATTORIALE
Nel mondo occidentale la patologia trombotica è la maggior causa di mortalità. La definizione clinica di trombosi è quella di una presenza patologica di un coagulo (trombo) in un vaso sanguigno o a livello del cuore che causa l’ostruzione del flusso attraverso il sistema circolatorio. La formazione di un coagulo ostruttivo nelle vene e nelle arterie è il prodotto finale di un disequilibrio tra fattori pro coagulanti, anticoagulanti e fibrinolitici.
In base alla localizzazione in cui si forma il trombo (ad esempio nella parte venosa o arteriosa del sistema circolatorio), la patologia trombotica può essere classificata in trombosi venosa o trombosi arteriosa. Entrambi i tipi di trombosi sono considerati come patologie distinte, caratterizzate da diversi meccanismi patogenetici e da fondamentali fattori di rischio ( Federici et al. 2006, Boekholdt e Kramer 2007, Prandoni 2007, Lowe 2008).
Le manifestazioni più comuni della trombosi arteriosa sono ictus ischemico, infarto miocardico acuto ed arteriopatia periferica. I trombi arteriosi sono costituiti principalmente da piastrine, comprendono relativamente poca fibrina e si sviluppano in siti di danno alla parete vasale in presenza di un flusso di sangue ad
alta velocità (Federici et al. 2006, Boekholdt e Kramer 2007, Prandoni 2007, Lowe 2008).
Le manifestazioni più comuni della trombosi venosa sono embolia polmonare e trombosi venosa profonda. I trombi venosi sono costituiti primariamente da fibrina ed eritrociti intrappolati ed includono poche piastrine. Hanno la tendenza a svilupparsi in aree di stasi in seguito ad attivazione del sistema di coagulazione del sangue e, quindi, stati di ipercoagulabilità sono fattori di rischio importanti (Federici et al. 2006, Prandoni 2007, Lowe 2008).
La trombofilia è una tendenza alla coagulazione eccessiva del sangue, con predisposizione a sviluppare trombosi ed embolie, sia venose che arteriose, in età giovanile (sotto i 45 anni), che spesso possono andare incontro a recidive. Essa può essere determinata da condizioni acquisite o da fattori ereditati geneticamente.
Condizioni acquisite che predispongono a trombofilia possono essere: immobilità, interventi chirurgici, infiammazione, neoplasie, terapie con contraccettivi orali (Rosendaal 1997, Heit et al. 2002).
L’importanza della componente genetica nei processi di tromboembolismo venoso e arterioso è stata ampiamente studiata perché la conoscenza dei fattori di rischio genetici e di quelli ambientali può contribuire in modo significativo a
prevedere un episodio di trombosi (Kottke-Marchant 2002, Endler e Mannhalter 2003).
Tuttavia i fattori di rischio genetici non hanno lo stesso ruolo nella trombosi venosa e arteriosa.
I fattori di rischio genetici sono conosciuti fin dal 1965 quando è stata descritta la prima famiglia con tendenza ereditaria alla trombosi, causata da una deficienza di antitrombina (Egeberg 1965). Negli anni 80 sono state descritte le deficienze di proteina C e proteina S in casi di trombofilia familiare (Griffin et al. 1981, Schwartz et al. 1984).
I tests funzionali della coagulazione per ricercare la deficienza degli anticoagulanti naturali, proteina C, proteina S ed antitrombina, sono molto richiesti ed utilizzati nella pratica di laboratorio.
Al contrario, difetti genetici che causano deficienza di proteina C, proteina S e antitrombina, sembrano essere piuttosto rari ed eterogenei nella popolazione generale, per cui l’indagine molecolare risulta molto complessa e non applicabile nella routine di laboratorio (Federici et al. 2006).
Esistono, tuttavia, alcune varianti genetiche, relativamente frequenti nella popolazione generale, il cui impiego può essere estremamente utile nella
In particolare, la variante di Leiden del gene del fattore V, la variante G20210A del gene della protrombina e la variante C677T della metilenetetraidrofolato reduttasi sono stati identificati come importanti fattori di rischio tromboembolici (Endler e Mannhalter 2003, Federici et al. 2006) (Tabella 1).
Tabella 1. Difetti genetici nel tromboembolismo venoso
Difetti ereditari Pazienti con trombosi (%)
Popolazione generale (%)
Rischio Relativo per tromboembolismo venoso
Antitrombina III ~1 0.02-0.04 ~20-50
Proteina C ~2-5 0.2-0.5 7-10
Proteina S ~1-3 0.1-1 ~2
Fattore V Leiden ~20(eterozigoti) 2-3(eterozigoti) ~3-7(eterozigoti) 50-100(omozigoti) Protrombina
G20210A 20 (eterozigoti) ~2-3(eterozigoti) ~2-5(eterozigoti) MTHFR C677T 15-20(omozigoti) 5-15(omozigoti) 2-3(omozigoti)
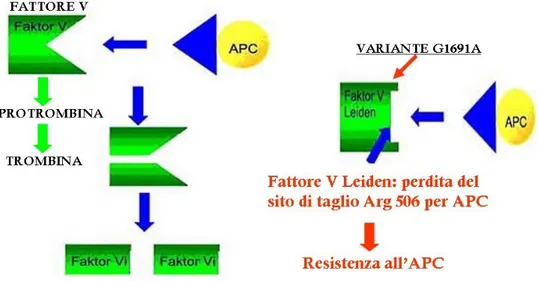
VARIANTE G1691A DI LEIDEN DEL FATTORE V
Il fattore V è uno dei numerosi fattori che intervengono nella cascata della coagulazione sia con funzioni procoagulanti che anticoagulanti; i livelli di fattore V sono controllati attraverso un’attivazione trombina-dipendente ed una inattivazione dipendente dalla proteina C attivata (APC). La trombina taglia il fattore V, generando una catena pesante amino-terminale ed una leggera carbossi-terminale, le quali formano un dimero (fattoreVa); questo assieme a ioni calcio, al fattore Xa ed a fosfolipidi anionici forma un complesso protrombinasico che promuove la conversione della protrombina (fattore II) in trombina sulla superficie piastrinica (Rosendorff e Dorfman 2007).
La trombina agisce sul fibrinogeno come un enzima proteolitico determinando la formazione di monomeri di fibrina, i quali polimerizzano e formano filamenti di fibrina, proteina fondamentale per la costituzione del coagulo.
In condizioni fisiologiche, il fattore Va viene inattivato dalla proteina C attivata che opera un taglio su di esso a livello di tre siti.
Nel 1994 è stata descritta una variante genetica comune nel gene che codifica per il fattore V, denominata variante di Leiden (FVL) come la cittadina olandese nella
adenina a livello del nucleotide 1691. A livello aminoacidico, il risultato è la sostituzione di un’arginina con una glutammina in posizione 506, a livello del primo sito riconosciuto dalla APC. Il taglio a livello dell’arginina 506 è necessario affinché vengano esposti in modo corretto gli altri due siti di taglio (Arg 306 e Arg 679) (Rosendorff e Dorfman 2007) (Figura 1).
Figura 1. Fattore V Leiden
Nel 1993 lo svedese Dahlbäck aveva osservato che il tempo di tromboplastina parziale attivato (PTT) eseguito sul plasma di alcuni individui con trombosi venosa appartenenti ad una stessa famiglia non veniva prolungato dall’aggiunta di proteina C attivata (Dahlbäck et al. 1993). Normalmente, l’aggiunta di APC causa
un prolungamento del PTT, dovuto alla inibizione dei fattori V e VIII attivati, i substrati su cui la proteina C attivata svolge la sua azione inibitoria.
L’anomalia osservata è stata di conseguenza denominata “resistenza alla proteina C attivata” ad indicare il comportamento del test funzionale.
Pazienti con il fattore V Leiden sono resistenti all’inattivazione del fattore Va, hanno pertanto un’emivita plasmatica del fattore Va più lunga e di conseguenza un aumento nella produzione di protrombina (Bertina et al. 1994, Rosendaal et al. 1995).
Molti studi identificano il fattore V Leiden come un importante fattore di rischio per trombosi venosa nella popolazione caucasica, con un rischio relativo di 3-7 volte nei soggetti portatori della variante mutata e di 50-100 volte nei soggetti omozigoti mutati (Federici et al 2006, Rosendorff e Dorfman 2007).
VARIANTE G20210A DELLA PROTROMBINA
La protrombina o fattore II (PT) è una glicoproteina che circola nel sangue in forma inattiva ed è un precursore della serin proteasi trombina. Quest’ultima agisce come pro-coagulante, attraverso l’attivazione piastrinica e la generazione di fibrina e fattore Va, VIIIa, XIIIa, e, successivamente, come anticoagulante, attraverso l’attivazione della proteina C circolante.
La regolazione dell’attività della trombina risulta, perciò, cruciale per il mantenimento dell’equilibrio emostatico (Russo et al. 2001).
Nel 1996 Poort e collaboratori hanno descritto una variante del gene della protrombina, caratterizzata da una sostituzione di una guanina con una adenina nel nucleotide 20210, che si trova nella regione non tradotta al 3’ del gene.
Questa regione è coinvolta nella regolazione genica post-trascrizionale, quale la stabilità dell’RNA messaggero o una maggior efficienza di trascrizione del messaggero stesso.
A livello fenotipico, i portatori della variante mutata presentano un aumento circa del 30% dei livelli plasmatici di protrombina, che comporta una maggiore predisposizione alla trombosi.
La prevalenza dell’allele 20210A varia da 1 a 4% nella popolazione normale con una distribuzione geografica variabile. La prevalenza della mutazione è del 3-7% nell’Europa del sud contro 2-5% nell’Europa del nord (Rosendaal et al. 1998).
VARIANTE C677T DELLA METILENETETRAIDROFOLATO REDUTTASI
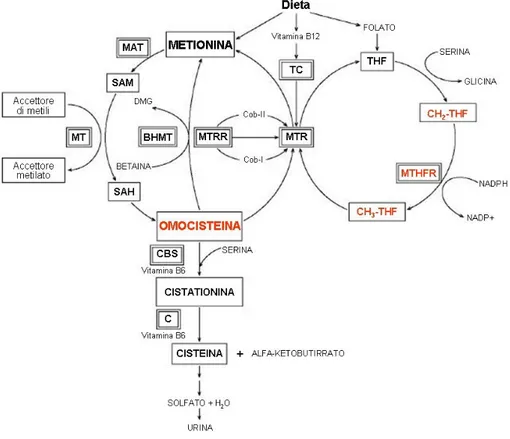
La metilenetetraidrofolato reduttasi (MTHFR) è un enzima coinvolto nel metabolismo dell’omocisteina e catalizza la riduzione del 5-10 metilenetetraidrofolato a 5-metiltetraidrofolato, necessario come donatore di gruppi metile nella reazione di rimetilazione dell’omocisteina a metionina, reazione quest’ultima catalizzata dalla metionina sintasi (MTR) in presenza di metilcobalamina (forma attiva della vitamina B12) (Trabetti 2008) (Figura 2).
Nel 1988 Kang et al. hanno identificato una variante termolabile dell’MTHFR associata con una diminuzione dell’attività dell’enzima e con un conseguente aumento delle concentrazioni plasmatiche di omocisteina.
Frosst et al. (1995) hanno identificato nel gene MTHFR una mutazione puntiforme che conferisce termolabilità all’enzima. La mutazione comporta la sostituzione di una citosina con una timina al nucleotide 677, la quale si traduce a livello aminoacidico con la sostituzione di una alanina con una valina in posizione 222.
La variante 677T è comune nella popolazione generale con una prevalenza nella popolazione caucasica di circa 15-20% nella condizione di omozigosi (Schneider et al. 1998).
La conseguenza fenotipica di tale variante è una riduzione dell’attività enzimatica dell’MTHFR pari al 50%, sino ad arrivare al 30% in condizioni di esposizione al calore. Tale variante, soprattutto in condizioni di omozigosi, comporta un moderato aumento dei livelli plasmatici di omocisteina, specialmente dopo un carico orale di metionina.
Una normale concentrazione plasmatica di omocisteina può variare da 5 fino a 15 µmol/L, l’aumento può essere moderato se la concentrazione varia da 15 a
un aumento elevato quando la concentrazione supera la soglia di 100µmol/L (Kang et al. 1992).
Numerosi studi hanno indicato, in modo definitivo, l’iperomocisteinemia come un fattore di rischio indipendente nello sviluppo di malattia aterosclerotica coronarica, di malattia cerebrovascolare e nell’insorgenza di trombosi venosa (Kang et al. 1991, Fermo et al. 1995, den Heijer 1996, Homocysteine Studies Collaboration 2002).
Ancora non è noto il meccanismo fisiopatologico tramite il quale l’omocisteina induce danno vascolare (Figura 3).
Figura 3. Iperomocisteinemia e danno cardiovascolare (modificata da Thambyrajah e Townend 2000)
E’ stato proposto che concentrazioni moderate o alte di omocisteina nel plasma possano agire determinando:
− alterazione del processo coagulativo attraverso un incremento dell’attività del fattore V ed una diminuzione dell’attività della proteina C attivata. Inoltre, l’omocisteina diminuirebbe il rilascio di ossido nitrico da parte delle cellule endoteliali con conseguente disfunzione endoteliale ed aumento del potenziale pro-trombotico (Thambyrajah e Townend 2000);
− disfunzione del tessuto endoteliale, dovuta ad una ridotta bio-disponibilità di ossido nitrico (NO), prodotto dalle cellule endoteliali (Thambyrajah e Townend 2000). La diminuzione della disponibilità di NO potrebbe, inoltre, modulare il potenziale pro-trombotico dell’omocisteina;
− proliferazione di cellule muscolari lisce, evento importante nello sviluppo della placca aterosclerotica (Schachinger et al. 1999);
− ossidazione di lipoproteine attraverso l’aumento della produzione di radicali liberi e, quindi, dello stress ossidativo (Chen et al. 2000).
FATTORE V LEIDEN, PROTROMBINA G20210A, MTHFR C677T E RISCHIO DI TROMBOSI
E’ ormai noto il ruolo che queste tre varianti hanno nell’aumentare il rischio di tromboembolismo venoso (Rosendaal 1997, De Stefano et al. 1999, Kottke Marchant 2002, Endler e Mannhalter 2003, Almawi et al. 2005, Miñano et al. 2008).
Soggetti eterozigoti per il fattore V Leiden hanno un rischio aumentato da 3 a 7 volte di andare incontro a tromboembolismo venoso, mentre soggetti omozigoti per lo stesso fattore mostrano un aumento del rischio da 50 a 100 volte (Federici et al. 2006). Tale evento trombotico è favorito in presenza di altre condizioni predisponenti quali la gravidanza, l’assunzione di contraccettivi orali ed interventi chirurgici.
Soggetti portatori della la variante 20210A della protrombina hanno un rischio aumentato da 2 a 5 volte di andare incontro a tromboembolismo venoso.
Soggetti omozigoti per la variante 677T della MTHFR hanno un rischio aumentato da 2 a 3 volte di andare incontro a tromboembolismo venoso (Federici et al. 2006).
Numerosi studi hanno valutato il rischio di trombosi in soggetti doppi eterozigoti per il fattore V Leiden e la protrombina 20210A; il risultato è stato un rischio molto più alto rispetto alla semplice somma dei rischi di ognuna delle due varianti prse singolarmente (Emmerich et al. 2001).
E’, invece, ancora dibattuto il ruolo che possono avere queste tre varianti genetiche nell’insorgenza della trombosi arteriosa (Kim e Becker 2003).
Tuttavia, sono stati pubblicati numerosi studi che mettono in relazione la presenza della variante di Leiden del fattore V e della variante 20210A della protrombina con ictus ischemici ed attacchi ischemici transitori (Lalouschek et al. 2005).
In particolar modo, il ruolo di queste due varianti protrombotiche sembra essere piuttosto rilevante in pazienti giovani con ictus criptogenetico, ovvero in assenza di possibile cause note (Grossmann et al. 2002, Aznar et al. 2004).
Recentemente, alcune evidenze apparse in letteratura hanno suggerito il possibile ruolo di queste varianti protrombotiche nell’identificazione di soggetti a rischio di eventi trombotici in un particolare gruppo di pazienti con forame ovale pervio, un difetto cardiaco che sembra essere associato ad maggiore incidenza di eventi ischemici cerebrovascolari (Pezzini et al. 2003, Botto et al. 2007).
FORAME OVALE PERVIO
Il forame ovale pervio è una anomalia cardiaca che è stata riscontrata a livello autoptico nel 25% circa della popolazione generale (Hagen et al. 1984, Lechat et al. 1988, Wu et al. 2004, Hara et al. 2005). Esso ha l’aspetto di una fessura allungata, simile ad un tunnel, situata a livello del setto interatriale e mette in comunicazione l’atrio destro del cuore con l’atrio sinistro (Figura 4).
Figura 4. Forame ovale pervio: schema
che mette in comunicazione i due atri cardiaci chiamata forame ovale (Embriologia medica, Langman) (Figura 5).
Figura 5. Sviluppo embrionale del forame ovale
A) La prima porzione del septum primum è una cresta falciforme che inizia a crescere, dalla porzione media del tetto della camera atriale comune, verso le cellule mesenchimali che formano cuscinetti endocardici; permane tra di essi una apertura detta ostium primum (OP).
B) Il septum primum si fonde con i cuscinetti endocardici ed occlude l’ostium primum.
C) Si inizia a formare una seconda parete, il septum secundum, alla destra del septum primum; si forma, inoltre, un secondo orifizio, l’ostium secundum (OS) nella porzione superiore del septum primum. Il septum secundum, infine, ricopre l’ostium secundum.
D) Vista laterale della parete interatriale con il forame ovale.
E) Vista frontale della parete interatriale (Cruz-Gonzáles et al. 2008).
ombelicale della madre, di giungere all’atrio destro, passare direttamente nell’atrio sinistro ed entrare così direttamente nel circolo sistemico .
Alla nascita, quando il circolo polmonare diviene pienamente funzionante, si modificano le resistenze vascolari e la pressione nell’atrio sinistro del cuore diviene leggermente superiore rispetto a quella dell’atrio destro.
Questa differenza di pressione fa accollare al forame ovale una piccola membrana, chiamata septum primum, che si è formata durante lo sviluppo del setto interatriale.
In condizioni normali, entro il primo anno di vita, il septum primum si salda completamente alla parete del setto. Quando ciò non accade, il forame ovale è detto pervio (FOP) ed il septum primum viene mantenuto in sede solamente dalla differenza pressoria tra i due atri. Perciò, in determinate situazioni che comportano un aumento di pressione nell’atrio destro, si può verificare un passaggio di sangue dall’atrio destro al sinistro. Un aumento cronico della pressione nell’atrio destro può verificarsi a causa di ipertensione polmonare, embolia polmonare, tosse, manovra di Valsava, immersioni subacquee (Kedia et al. 2008)
Nelle normali condizioni di vita, il FOP non comporta nessun problema e molti individui che presentano questa anomalia cardiaca non hanno nessun sintomo e possono non accorgersi della sua presenza.
Tuttavia, sebbene attualmente non esista ancora alcuna prova sicura di un rapporto causa-effetto, numerosi studi hanno confermato una forte associazione tra la presenza di un FOP ed il rischio di ischemia cerebrale (Lechat et al. 1988, Overell et al. 2000). Quando confrontati con un gruppo di soggetti di controllo senza FOP, i pazienti con FOP avevano un rischio di soffrire di un evento trombo-embolico che è quattro volte più alto; tale rischio era 33 volte maggiore nei pazienti che avevano sia il PFO che un aneurisma del setto atriale (Cabanes et al. 1993).
In alcuni casi, infatti, il FOP può essere associato con altre anomalie anatomiche quali, appunto, l’aneurisma del setto atriale (ASA), presente nel 50-80% dei casi di FOP (Hara et al. 2005). In presenza di ASA, una parte del setto interatriale mostra una dilatazione aneurismale che può sporgere all’interno di ciascuno dei due atri durante il ciclo cardiorespiratorio (Drighil et al. 2007).
A questo proposito, uno studio recente ha preso in considerazione la morfologia del forame ovale dimostrando come in pazienti con eventi cerebrovascolari i FOP
siano più larghi e più frequentemente associati con aneurisma del setto atriale (Goel et al. 2009).
Il meccanismo attraverso il quale la presenza di un forame ovale pervio possa predisporre ad eventi ischemici, tuttavia, non è stato ancora del tutto chiarito. Sono stati ipotizzati diversi possibili meccanismi che potrebbero essere responsabili di tali eventi.
Il più accreditato è il fenomeno dell’embolia paradossa dal sistema venoso periferico: in condizioni normali, un piccolo embolo che si forma a livello del sistema venoso profondo, giunge all’atrio destro e successivamente si ferma a livello dei capillari polmonari; la presenza di un forame pervio consentirebbe, invece, un possibile passaggio di sangue e, quindi, anche dell’embolo, dall’atrio destro a quello sinistro e da questo alla circolazione arteriosa cerebrale causando ictus ischemico (Ranoux et al. 1993).
Un altro possibile meccanismo prevede la formazione di trombi all’interno del tunnel del FOP, dovuti alla stasi del sangue in questo tratto (Schneider et al. 1990). In ultimo, vi è la possibile formazione di un trombo come conseguenza di aritmie atriali transitorie (Berthet et al. 2000).
SCOPO DELLA TESI
L’incertezza riguardo al processo fisiopatologico che correla il forame ovale pervio all’ischemia cerebrale, non sempre permette al medico di condurre una strategia terapeutica ottimale per il paziente.
Recenti studi, sulla base dei meccanismi supposti, hanno indicato come la presenza di condizioni procoagulanti in pazienti con FOP possa modulare il rischio di formazione di un trombo instabile in grado di causare ischemia cerebrale.
Nonostante ciò, la prevalenza di varianti genetiche protrombotiche nei pazienti con FOP non è stata sufficientemente studiata e i dati finora pubblicati mostrano, in alcuni casi, risultati contraddittori.
Pertanto, scopo della tesi è stato quello di analizzare la prevalenza dei polimorfismi del fattore V di Leiden G1691A, della protrombina G20210A e della MTHFR C677T in un’ampia coorte di pazienti con FOP ed eventi ischemici cerebrovascolari.
Questo al fine di identificare i pazienti ad aumentato rischio di eventi che potrebbero beneficiare di uno screening genetico ed essere indirizzati alle più
MATERIALI E METODI
POPOLAZIONE DI STUDIO
La nostra popolazione, in un disegno di studio caso-controllo, era costituita da 220 pazienti (102 uomini; età media: 45.1±12.0 anni) afferenti all’ospedale “G. Pasquinucci”, Fondazione G. Monasterio ,di Massa nel periodo Gennaio 2006-Dicembre 2008.
I pazienti giungevano alla nostra osservazione con diagnosi di forame ovale pervio ed indicazione ad effettuare un intervento di chiusura transcatetere per via percutanea del difetto cardiaco.
I pazienti erano selezionati, per essere inclusi nello studio, secondo i seguenti criteri di inclusione:
1. storia di ictus ischemico cerebrale, documentato clinicamente da un neurologo e da esami radiologici quali Tomografia Computerizzata Craniale o Risonanza Magnetica, o di un episodio di attacco ischemico transitorio (TIA), definito come un deficit neurologico transitorio con pieno recupero nell’arco delle 24 ore e confermato clinicamente da un neurologo;
2. presenza di un forame ovale pervio, con o senza aneurisma del setto atriale, diagnosticato tramite ecocardiografia transesofagea (TEE) o Doppler transcranico;
3. età di insorgenza dell’evento cerebrovascolare inferiore ai 55 anni per gli uomini e ai 65 anni per le donne.
La popolazione di controllo era costituita da 182 soggetti sani (71 uomini; età media: 41.8±10.3 anni), reclutati nell’ambito del nostro personale ospedaliero, che non riferivano alcuna storia precedente di eventi cerebro e/o cardiovascolari. Tutti i soggetti inclusi nello studio erano stati sottoposti ad un accurato esame anamnestico e clinico al momento del loro reclutamento.
In particolare, la popolazione di studio è stata caratterizzata per i fattori di rischio tradizionali delle malattie cerebro e cardiovascolari secondo i seguenti criteri: ipertensione, una pressione arteriosa >140/90 mmHg oppure l’uso di farmaci anti-ipertensivi; dislipidemia, concentrazioni di LDL ≥130 mg/dL, HDL<35mg/dL, trigliceridi ≥ 200mg/dL o terapia con anti-lipidemici; diabete mellito, livello di glucosio plasmatico ≥7.0 mmol/L oppure uso di farmaci ipoglicemizzanti.
Sono stati classificati come fumatori i soggetti che fumavano almeno tre sigarette al giorno al momento dello studio; come ex fumatori coloro che avevano smesso di fumare da almeno sei mesi e come non-fumatori coloro che non avevano mai fumato. I fumatori e gli ex fumatori sono stati riuniti in un unico gruppo. Ciascun fattore di rischio è stato classificato come presente o assente.
Tutti i partecipanti allo studio hanno preso visione e firmato il consenso informato e la commissione etica ha approvato il protocollo di studio.
ANALISI GENETICA
Tutti i soggetti partecipanti allo studio sono stati genotipizzati per identificare i diversi polimorfismi dei geni del fattore V, della protrombina e della MTHFR. L’analisi genetica è stata effettuata mediante l’utilizzo di un kit commerciale che permette l’identificazione simultanea dei tre genotipi su di un’unica striscia (Figura 6).
Figura 6. Analisi genetica simultanea dei tre polimorfismi mediante kit commerciale
La procedura del kit ( Nuclear Laser Medicine SRL ) si articola in tre passaggi: ISOLAMENTO DEL DNA. Da ciascun soggetto si prelevavano 100µl di sangue, fresco o congelato in provette con EDTA o citrato come anticoagulante. Si
Marker Rosso
Marker Verde Controllo Positivo
Fattore V Leiden mutato Protrombina G20210A mutato MTHFR C677T mutato Fattore V Leiden wild-type
Protrombina G20210A wild-type MTHFR C677T wild-type DNA
Genomico
miscelando per diverse volte, si lasciava riposare per 15 minuti a temperatura ambiente e si centrifugava per 5 minuti a 3000 rpm in una micro centrifuga.
Dopo la rimozione del sopranatante, si aggiungeva 1ml di Lysis Solution e si mescolava nuovamente la miscela. Il campione veniva centrifugato per 5 minuti a 12000 rpm. Dopo aver rimosso ed eliminato il sopranatante, si risospendeva il pellet con una resina (GENXTRACT Resin). La provetta veniva, poi, incubata per
20 minuti a 56°C, e per 10 minuti a 98°C. Infine, si centrifugava la miscela per 5 minuti a 12000 rpm e si raffreddava in ghiaccio.
Il sopranatante ottenuto conteneva DNA idoneo per un uso immediato in PCR. AMPLIFICAZIONE TRAMITE PCR CON PRIMERS BIOTINILATI
Si preparava una diluizione fresca (1:25 concentrazione finale 0.2U/µl) di Taq polimerasi in un buffer di diluizione. Per ogni campione si preparava una mix di reazione per PCR in ghiaccio:
− 15µl di mix di amplificazione, contenente le coppie di primer specifiche per l’amplificazione di ciascuno dei tre geni analizzati;
− 5µl di Taq DNA polimerasi precedentemente diluita; − 5µl di DNA template.
Prima di essere utilizzato il termociclatore veniva preriscaldato a 94°C. Il numero di cicli di amplificazione e la temperatura di annealing erano ottimizzati,
sottoponendo la miscela di reazione a 32 cicli di reazione, programmati secondo il seguente schema :
Pre PCR 94°C 2 minuti 30 cicli:
Denaturazione 94°C 15 secondi “Annealing “ primers 58°C 30 secondi Estensione 72°C 30 secondi Ultimo ciclo:
Estensione 72°C 3 minuti IBRIDAZIONE
La terza fase prevedeva l’ibridazione dei prodotti amplificati su una striscia contenente sonde oligonucleotidiche allele-specifiche immobilizzate secondo uno schema di bande parallele.
Le sequenze biotinilate legate alle sonde venivano rilevate utilizzando fosfatasi alcalina coniugata con streptavidina e, in seguito, il relativo substrato.
In dettaglio, regolata la temperatura di 45°C del bagno termostatato basculante, si pipettavano 10µl del prodotto amplificato nella corrispondente goccia di 10µl di DNAT nella specifica corsia dell’apposita vaschetta. Dopo aver mescolato bene,
ibridazione (preriscaldato a 45°C) in ogni corsia, si agitava delicatamente il vassoio e si inseriva le strisce (Teststrip) nelle rispettive corsie. Si incubava per 30 minuti a 45°C nel bagno basculante.
Alla fine dell’ibridazione si rimuoveva la soluzione di ibridazione tramite aspirazione a vuoto. A questo punto si eseguivano due lavaggi stringenti, di 15 minuti ciascuno, a 45°C con una soluzione di lavaggio preriscaldata anch’essa a 45°C. Veniva, poi, effettuata la fase della coniugazione a temperatura ambiente su un agitatore per 20 minuti seguita da 2 lavaggi di 5 minuti ciascuno. In ultimo, veniva effettuato lo sviluppo del colore a temperatura ambiente su agitatore coprendo le strisce dalla luce. Infine, dopo aver lavato le Teststrip diverse volte con acqua distillata, si lasciavano asciugare al buio su carta assorbente.
INTERPRETAZIONE DEI RISULTATI
Il genotipo di un campione si determinava usando il “Collector” incluso nel kit allineandola allo schema disegnato utilizzando la linea rossa (in alto) e la linea verde (in basso) come riferimento (figura 7).
Una reazione positiva nella banda di Controllo in alto indica il corretto funzionamento della Conjugate Solution e del Color Developer. Questa banda deve sempre risultare positiva.
Figura 7. Esempi di test su striscia:
a) paziente con una doppia eterozigosità per la variante G20210A della protrombina e C677T dell’MTHFR.
b) paziente eterozigote per la variante Leiden ed omozigote per la variante 677T dell’MTHFR
c) paziente omozigote per la variante 677T dell’MTHFR
ANALISI STATISTICA
L’analisi statistica è stata eseguita tramite l’uso del software per analisi statistiche Statview versione 5.0.1 (SAS Institute).
I dati sono stati espressi come media±deviazione standard. La differenza fra le medie di due variabili continue è stata valutata mediante il test di Student, mentre le differenze tra variabili non continue e la distribuzione dei genotipi sono state valutate mediante il test del χ2.
E’ stata effettuata l’analisi di regressione logistica per calcolare gli odds ratios (ORs) e gli intervalli di confidenza (95%CI) riferiti all’associazione tra varianti genetiche protrombotiche e rischio di ischemia cerebrale .
Attraverso l’analisi multivariata sono stati corretti gli ORs per gli altri fattori di rischio cardiovascolari da noi considerati.
RISULTATI
CARATTERISTICHE DEMOGRAFICHE E PRESENZA DEI MAGGIORI FATTORI DI RISCHIO PER MALATTIE CARDIOVASCOLARI
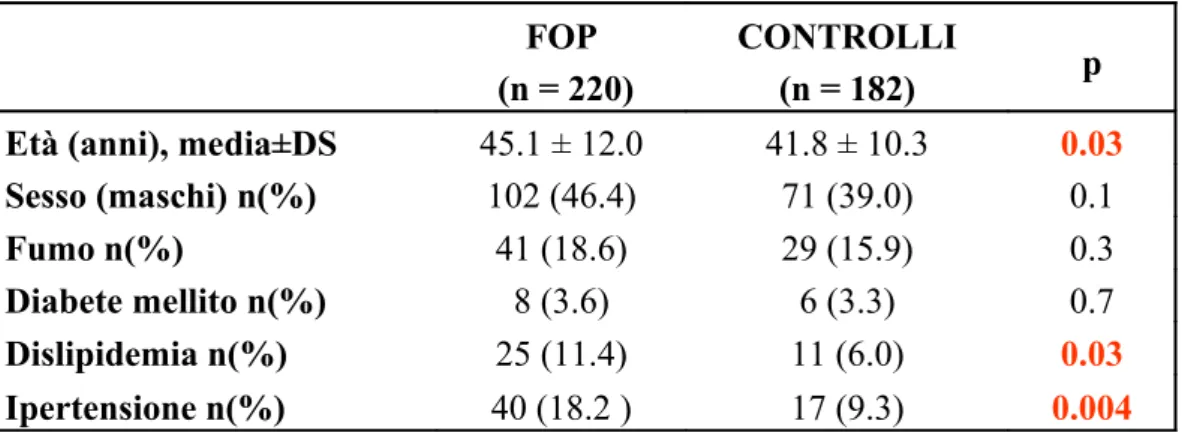
Nella tabella (2) sono riportate le caratteristiche cliniche e demografiche del gruppo dei pazienti e del gruppo dei controlli e sono elencati i più importanti fattori di rischio per malattie cardiovascolari.
Come atteso, la prevalenza di alcuni noti fattori di rischio, come dislipidemia (11.4% vs 6.0%; p=0.03) ed ipertensione (18.2% vs 9.3%; p=0.004), è significativamente più alta nel gruppo dei pazienti rispetto a quello dei controlli. Inoltre, i pazienti hanno età media maggiore rispetto ai controlli (45.1±12 vs 41.8±10.3 anni; p=0.03).
Il sesso, l’abitudine al fumo e la presenza di diabete mellito sono paragonabili tra i due gruppi.
Tabella 2. Caratteristiche cliniche e demografiche della popolazione FOP
(n = 220)
CONTROLLI
(n = 182) p
Età (anni), media±DS 45.1 ± 12.0 41.8 ± 10.3 0.03
Sesso (maschi) n(%) 102 (46.4) 71 (39.0) 0.1
DISTRIBUZIONE DEI GENOTIPI
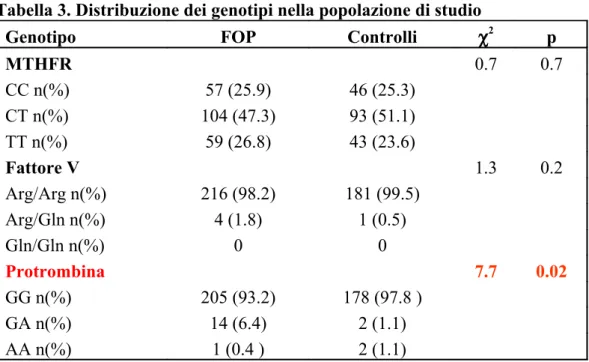
Nella tabella (3) si riporta la distribuzione dei genotipi associati a ciascun polimorfismo. La distribuzione dei genotipi nella nostra popolazione era simile a quella normalmente osservata nella popolazione caucasica
Per quanto riguarda il polimorfismo C677T del gene MTHFR, non abbiamo riscontrato differenze significative nella distribuzione dei genotipi fra il gruppo dei pazienti e quello dei controlli (χ2= 0.7, p=0.7).
Abbiamo osservato, invece, una frequenza maggiore di portatori della variante di Leiden del fattore V nel gruppo dei pazienti rispetto a quello dei controlli (1.8% vs 0.5%; χ2=1.3, p=0.2), sebbene tale differenza non raggiunga la significatività statistica. Nella nostra popolazione di studio, non sono stati trovati soggetti omozigoti mutati per il fattore V Leiden.
Per quanto riguarda il polimorfismo G20210A del gene della protrombina, abbiamo osservato una frequenza significativamente più elevata di soggetti eterozigoti mutati nel gruppo dei pazienti rispetto a quello dei controlli (6.4% vs 1.1%; χ2
Tabella 3. Distribuzione dei genotipi nella popolazione di studio
Genotipo FOP Controlli χ2 p
MTHFR 0.7 0.7 CC n(%) 57 (25.9) 46 (25.3) CT n(%) 104 (47.3) 93 (51.1) TT n(%) 59 (26.8) 43 (23.6) Fattore V 1.3 0.2 Arg/Arg n(%) 216 (98.2) 181 (99.5) Arg/Gln n(%) 4 (1.8) 1 (0.5) Gln/Gln n(%) 0 0 Protrombina 7.7 0.02 GG n(%) 205 (93.2) 178 (97.8 ) GA n(%) 14 (6.4) 2 (1.1) AA n(%) 1 (0.4 ) 2 (1.1)
Inoltre, prendendo in considerazione le combinazioni fra i vari genotipi, abbiamo osservato come la presenza di almeno uno dei due polimorfismi protrombotici, fattore V Leiden o PT 20210A, fosse presente con maggior frequenza nel gruppo dei pazienti rispetto al gruppo dei controlli (8.6% vs 2.7%; χ2=6.1, p=0.01).
Le combinazioni dei genotipi 677TT+FV Leiden e 677TT+PT 20210A non risultano significativamente differenti tra pazienti e controlli (tabella 4).
Non sono stati riscontrati soggetti con la contemporanea presenza delle due varianti mutate del fattore V della PT G20210A (tabella 4)
Tabella 4. Combinazioni genotipiche
Genotipo FOP Controlli χ2 p
MTHFR 677TT + FV Leiden 1(0.5) 1(0.5) 0.02 0.9
MTHFR 677TT + PT 20210A 2(0.9) 0 1.6 0.2
FV Leiden o PT 20210A 19(8.6) 5(2.7) 6.1 0.01
FV Leiden o PT 20210A
RISCHIO ASSOCIATO ALLA PRESENZA DI CIASCUN POLIMORFISMO PER ISCHEMIA CEREBRALE NEI PAZIENTI CON FOP
L’analisi di regressione logistica ha evidenziato che nella nostra popolazione la presenza dell’allele mutato per la protrombina è associata ad un rischio 3.3 volte maggiore di eventi ischemici cerebrali nei pazienti con FOP (95%CI=1.1-9.9; p=0.04).
Per quanto riguarda il fattore V, i pazienti FOP portatori dell’allele mutato avevano un rischio aumentato di 3.4 volte di andare incontro ad ischemia cerebrale, sebbene non statisticamente significativo (95%CI=0.4-30.3; p=0.2). Infine, abbiamo osservato come la presenza di almeno un genotipo protrombotico, FV Leiden o PT 20210A, fosse associata nei pazienti con FOP ad un rischio 3.4 volte maggiore di andare incontro ad un evento ischemico (95%CI=1.2-9.1; p=0.02) (Tabella 5).
Tabella 5. Rischio associato alle varianti genetiche protrombotiche
OR (95% CI) P
MTHFR 677TT 1.2 (0.7-1.8) 0.5
ANALISI MULTIVARIATA
Dopo aver corretto i dati per i noti fattori di rischio aterogeno (età, sesso, fumo, dislipidemia, ipertensione, diabete), la presenza di almeno un genotipo protrombotico rimaneva un fattore di rischio indipendente per ischemia cerebrale, con un rischio superiore a 4, nei pazienti con FOP (OR=4.4, 95%CI=1.4-13.3; p=0.009) (tabella 6)
Tabella 6. Fattori di rischio indipendenti di ischemia cerebrale nei pazienti con FOP OR 95% CI P Età 1.0 1.0-1.1 0.045 Sesso 1.4 0.9-2.1 0.15 Fumo 1.2 0.7-2.1 0.5 Diabete mellito 0.7 0.2-2.3 0.5 Dislipidemia 1.6 0.7-3.5 0.2 Ipertensione 1.9 0.9-3.6 0.07 FV Leiden o PT 20210A 4.4 1.4-13.3 0.009
ANALISI DELLA RICORRENZA DI EVENTI
Successivamente, in base ai dati clinici ed anamnestici, abbiamo suddiviso la popolazione dei pazienti in due gruppi in base al numero di eventi ischemici precedenti al reclutamento.
In questo modo abbiamo selezionato 193 pazienti che avevano manifestato un solo evento ischemico cerebrale prima della chiusura del forame ovale pervio, e 27 pazienti che avevano avuto più di un evento.
Come possiamo osservare nella tabella 7, i due gruppi di pazienti sono paragonabili per quanto riguarda l’età e la presenza dei principali fattori di rischio per malattie cardiovascolari.
Tabella 7. Principali fattori di rischio cardiovascolari nei due gruppi di pazienti. 1 evento (n=193) >1 evento (n=27) χ2 P Età(anni), media±DS 44.9±11.7 46.2±14.0 0.6 Sesso (maschi) n(%) 89 (46.1) 13 (48.1) 0.04 0.8 Fumo n(%) 38 (19.7) 3 (11.1) 1.3 0.3 Diabete n(%) 6 (3.1) 2 (7.4) 1.2 0.3 Dislipidemia n(%) 21 (10.9) 4 (14.8) 0.3 0.6 Ipertensione n(%) 35 (18.1) 5 (18.5) 0 0.97
più di un evento ischemico rispetto a quelli che ne hanno manifestato solo uno (18.5% vs 7.2%; χ2=3.8, p=0.05) (tabella 8)
Tabella 8. ricorrenza di eventi e varianti protrombotiche. 1 evento (n=193) >1 evento (n=27) χ2 p FV Leiden 3 (1.5%) 1 (3.7%) 0.6 0.4 PT 20210 11 (5.7%) 4 (14.8%) 3.1 0.08 MTHFR 677TT 49 (25.4%) 10 (37.0%) 1.6 0.2 FV Leiden o PT 20210A 14 (7.2%) 5 (18.5%) 3.8 0.05 FV Leiden o PT 20210A o MTHFR 677TT 62 (32.1%) 13 (48.1%) 2.7 0.09
DISCUSSIONE
Nella nostra popolazione di studio, abbiamo osservato come la frequenza di mutazioni protrombotiche sia relativamente elevata in pazienti con forame ovale pervio ed eventi cerebrovascolari ischemici avvenuti in età giovanile. In particolare, i risultati del presente studio dimostrano come la presenza della variante di Leiden del fattore V o della protrombina G20210A determini, in questi pazienti, un aumento di 3.4 volte del rischio ischemico.
Al contrario, la presenza della variante C677T della MTHFR non sembra essere direttamente associata con un aumento del rischio di andare incontro ad ischemia cerebrale nei pazienti con FOP.
In questi anni, numerose evidenze scientifiche hanno mostrato come il forame ovale pervio possa essere coinvolto nell’eziologia di eventi cerebrovascolari ischemici (Di Tullio et al. 1992, Mas 1994, Chant e McCollum 2001). Infatti, la presenza di FOP è stata associata ad una maggiore incidenza di ictus ed attacchi ischemici transitori, presumibilmente di origine embolica, soprattutto in soggetti giovani ed in mancanza di fattori di rischio concomitanti (Lechat et al. 1988, Wu et al. 2004, Hara 2005).
Circa il 40% degli ictus ischemici, infatti, sono considerati criptogenici, ovvero senza una causa apparente certa (Wu et al. 2004, Hara 2005). In questi pazienti, uno stato di ipercoagulabilità dovuto a trombofilia ereditaria potrebbe aumentare il rischio di episodi ischemici cerebrali, soprattutto in presenza di una sorgente trombogenica importante quale un forame ovale pervio.
Recentemente, alcuni studi hanno confermato il ruolo di mutazioni protrombotiche, riconosciuti fattori di rischio tromboembolico, nell’insorgenza di episodi ischemici in pazienti con FOP, suggerendo l’embolia paradossa come possibile meccanismo causale sottostante (Karttunen et al. 2003, Lichy et al. 2003, Pezzini et al. 2003, Belvis et al. 2006, Botto et al. 2007, Offelli et al. 2007) (tabella 9). In particolare, la presenza della variante 20210A della protrombina ed, in misura minore, la variante di Leiden del fattore V, sono associati ad un rischio 3-4 volte maggiore di episodi ischemici, come confermato nel presente studio su una casistica di popolazione molto ampia.
Tabella 9. Mutazioni protrombotiche e rischio di eventi cerebrovascolari in pazienti FOP: studi condotti.
Referenza
Mutazioni
OR
95% CI
Karttunen et al. 2003 FV Leiden 7.77 0.85-71.30 PT 20210A 9.41 0.44-199.54 FV Leiden o PT 20210A 2.8 1.2-6.5
Lichy et al. 2003 FV Leiden 1.02 0.50-2.08 PT 20210A 3.76 1.29-10.96 FV Leiden o PT 20210A 1.58 0.88-2.83
Pezzini et al. 2003 FV Leiden 3.60 0.92-14.16 PT 20210A 6.08 1.30-28.52 FV Leiden o PT 20210A 4.25 1.43-12.66
Belvis et al. 2006 FV Leiden 0.64 0.03-11.76 PT 20210A 3.87 0.69-21.68 FV Leiden o PT 20210A 1.68 0.34-8.33
Botto et al. 2007 FV Leiden 3.35 0.03-37.41 PT 20210A 4.70 1.22-18.18 FV Leiden o PT 20210A 4.48 1.37-14.72
Offelli et al. 2007 FV Leiden 0.49 0.04-5.56 PT 20210A 0.49 0.04-5.56 FV Leiden o PT 20210A 0.49 0.09-2.74
Le evidenze qui presentate mettono in luce come una condizione di ipercoagulabilità, dovuta a fattori ereditati geneticamente, sia importante da identificare nei pazienti con FOP per poter individuare coloro che sono a maggior rischio per embolia paradossa.
annuo di avere una recidiva di ischemia cerebrale transitoria (TIA) è dell’1.2%, e del 3.4% di avere una recidiva di ictus cerebrale (Mas e Zuber 1995).
Nel presente studio, la presenza di almeno una mutazione protrombotica, FVL o PT 20210A, era associata ad un rischio superiore a 3 di andare incontro a recidive, senza un adeguato supporto terapeutico.
Ad oggi, infatti, non vi è ancora consenso sulla strategia di trattamento ottimale, terapia farmacologica o intervento di chiusura, da adottare nei pazienti con FOP (Ranoux et al. 1993, Meissner et al. 2006, Mareedu et al. 2007, Onorato et al. 2008).
La terapia farmacologica prevede l’assunzione di anticoagulanti orali o di antiaggreganti piastrinici, ma ancora non vi è un consenso su quale sia il trattamento farmacologico più efficace o per quanto tempo esso debba essere protratto. Inoltre, l’utilizzo di farmaci, pur prevenendo la formazione di coaguli, non elimina il difetto cardiaco e, quindi la possibile fonte trombogenica; inoltre, costringe i pazienti a modificare alcune abitudini di vita che possono aumentare il rischio di emorragia.
L’intervento di chiusura prevede l’utilizzo di un catetere il quale, inserito in un vaso sanguigno attraverso una piccola incisione, viene fatto avanzare fino a
raggiungere il cuore e attraverso di esso viene introdotto un dispositivo permanente in grado di chiudere il difetto cardiaco.
L’assenza di trials randomizzati che mettano a confronto la terapia medica con l’intervento di chiusura per via percutanea porta alla mancanza di linee guida precise da seguire.
L’American College of Cardiology, l’American Heart Association e l’American Society of Echocardiography non danno raccomandazioni in proposito, mentre
l’American College of Neurology sostiene che non vi sono prove sufficienti per valutare l’efficacia della chiusura del FOP (Mareedu et al. 2007, Pinto Slottow et al. 2007, Balbi et al. 2008, Onorato et al. 2008).
In questo contesto, l’impiego di uno screening genetico in grado di identificare soggetti ad alto rischio di eventi tromboembolici può essere importante per ottimizzare la scelta delle opzioni terapeutiche da parte del medico.
Infatti, difetti ereditari trombofilici possono, ad esempio, influire sui rischi potenziali dell’intervento di chiusura per via percutanea e ridurne i potenziali benefici (Giardini et al. 2004, Wu et al. 2004).
Allo stesso modo, pazienti portatori di tali mutazioni che abbiano avuto un precedente evento ischemico, soprattutto in giovane età, sono probabilmente ad
cardiaco, senza un opportuno trattamento terapeutico mirato, diminuendo i benefici attesi di tale intervento.
Sicuramente, quindi, la possibilità di eseguire uno screening per mutazioni protrombotiche potrebbe avere sicuri vantaggi in termini di rapporto costo-beneficio, migliorando la stratificazione del rischio in pazienti con FOP ed ottimizzando la profilassi secondaria allo scopo di ridurre la probabilità di eventi ricorrenti e di indirizzare il medico alla decisione terapeutica più adeguata a ciascun paziente.
CONCLUSIONI
È certo che nei prossimi anni la pratica clinica d’ogni giorno sarà sempre maggiormente influenzata dalle scoperte della genetica.
Tuttavia, un atteggiamento responsabile induce a limitare l’impiego della genetica molecolare nella pratica clinica a sottogruppi di pazienti in cui esista un forte rischio di eventi trombotici e l’applicazione del test diagnostico possa migliorare la diagnosi e le scelte terapeutiche.
Infatti, malgrado non vi sia alcun dubbio sulla possibilità che lo screening genetico troverà una larga applicazione nella pratica clinica contribuendo ad aumentare la prevenzione e l’approccio terapeutico di molte malattie multifattoriali, è opportuno procedere con cautela alla sua applicazione nella popolazione generale ed evitare il rischio di una “chiromanzia genetica” senza valore clinico.
Bisogna ricordare che uno degli aspetti più complessi della genetica delle malattie multifattoriali è rappresentato dal fatto che esse sono il risultato dell’interazione tra fattori genetici, spesso multipli, e fattori ambientali e non sono trasmesse
suscettibilità che conferiscono al paziente una certa vulnerabilità nei confronti di queste patologie e, quindi, rappresentano fattori di rischio quantificabili in termini di probabilità di ammalarsi.
La distribuzione combinata dei fattori genetici e la loro interazione con fattori ambientali nelle persone e nelle popolazioni diverse determina la variabilità del rischio di malattia. Soltanto la capacità del medico di comprendere appieno le implicazioni dei test genetici e di interpretarne i risultati nel quadro clinico complessivo consentirà di fornire al paziente informazioni adeguate, giocando un ruolo fondamentale per un corretto uso di questi nuovi strumenti
LETTERATURA CITATA
Almawi WY, Tamim H, Kreidy R, Timson G, Rahal E, Nabulsi M, Finan RR, Irani-Hakime NA. Case control study on the contribution of factor V-Leiden, prothrombin G20210A, and MTHFR C677T mutations to the genetic susceptibility of deep venous thrombosis. Journal of Thrombosis and Thrombolysis 2005;19:189-196
Aznar J, Mira Y, Vaya A, Crella D, Ferrando F, Villa P, Estellés A. Factor Leiden and prothrombin G20210A mutations in young adults with cryptogenic ischemic stroke. Thromb Haemost 2004; 91:1031-4
Balbi M, Casalino L, Gnecco G, Bezante GP, Pongiglione G, Marasini M, Del Sette M, Barsotti A. Percutaneous closure of patent foramen ovale in patients with presumed paradoxical embolism: periprocedural results and midterm risk of recurrent neurologic events. Am Heart J. 2008;156:356-60
Belvís R, Santamaría A, Martí-Fàbregas J, Cocho D, Borrell M, Fontcuberta J, Martí-Vilalta JL. Diagnostic yield of prothrombotic state studies in cryptogenic stroke. Acta Neurol Scand 2006;114:250-253
Berthet K, Lavergne T, Cohen A, Guize L, Bousser MG, Le Heuzey JY, Amarenco P. Significant association of atrial vulnerability with atrial septal abnormalities in young patients with ischemic stroke of unknouwn cause. Stroke 2000;31:398-403
Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64-7
Boekholdt SM, Kramer MH. Arterial thrombosis and the role of thrombophilia. Semin Thromb Hemost 2007;33:588-96
Botto N, Spadoni I, Giusti S, Ait-Ali L,Sicari R, Andreassi MG. Prothrombotic mutations as risk factors for cryptogenic ischemic cerebrovascular events in young subjects with patent foramen ovale . Stroke 2007;38:2070-2073
Cabanes L, Mas JL, Cohen A, Amarenco P, Cabanes PA, Oubary P, Chedru F, Guérin F, Bousser MG, de Recondo J. Atrial septal aneurysm and patent foramen ovale as risk factors for cryptogenic stroke in patients less than 55 years of age. A study using transesophageal echocardiography. Stroke. 1993;24:1865-73
Chant H, McCollum C. Stroke in young adults: the role of paradoxical embolism. Thromb Haemost. 2001;85:22-9
Chen N, Liu Y, Greiner CD, Holtzman JL. Physiologic concentrations of homocysteine inhibit the human plasma GSH peroxidase that reduces organic hydroperoxides. J Lab Clin Med. 2000;136:58-65
Cruz-Gonzáles I, Solis J, Inglessis-Azuaje I, Palocios IF. Patent Foramen Ovale: Current State of the Art. Rev Esp Cardiol 2008;61:738-51
Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant
response to activated protein C :prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993;90:1004-8
De Stefano V, Martinelli I, Mannucci PM, Paciaroni K, Chiusolo P, Casorelli I, Rossi E, Leone G. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation . N Engl J Med 1999;341:801-6
den Heijer M, Koster T, Blom HJ, Bos GM, Briet E, Reitsma PH, Vandenbroucke JP, Rosendaal FR. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759-62
Di Tullio M, Sacco RL, Gopal A, Mohr JP, Homma S. Patent foramen ovale as a risk factor for cryptogenic stroke. Ann Intern Med. 1992;117:461-5
Drighil A, El Mosalami H, Elbadaoui N, Chraibi S, Bennis A. Patent foramen ovale: a new disease? Int J Cardiol 2007;122:1-9
Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965;13:516-30
Emmerich J, Rosendaal FR, Cattaneo M, Margaglione M, De Stefano V, Cumming T, Arruda V, Hillarp A, Reny JL. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism. Thromb Haemost 2001;86:809–16
Endler G, Mannhalter C. Polymorphisms in coagulation factor genes and their impact on arterial and venous thrombosis. Clin Chim Acta 2003;330:31-55
Federici C, Gianetti J, Andreassi MG. Genomic medicine and thrombotic risk: Who, when, how and why? International Journal of Cardiology 2006;106:3-9
Fermo I, D’Angelo SV, Paroni R, Mazzola G, Calori G, D’Angelo A. Prevalence of moderate hyperomocysteinemia in patients with early –onset venous and arterial occlusive disease. Ann Intern Med 1995;123:747-53
Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Mattews RG, Boers GJH, den Heijer M, Kluijtmans LAJ, van den Heuve LP, Rozen R. A candidate gene risk factor for vascular disease: a common mutation at the metylenetetrahydrofolate reductase locus. Nat Genet 1995;10:111-113
Giardini A, Donti A, Formigari R, Bronzetti G, Prandstraller D, Bonvicini M, Palareti G, Guidetti D, Gaddi O, Picchio FM. Comparison of Results of Percutaneous Closure of Patent Foramen Ovale for Paradoxical Embolism i Patients With Versus Without Thrombophilia. Am J Cardiol 2004;94:1012-1016
Goel SS, Tuzcu EM, Shishehbor MH, de Oliveira EI, Borek PP, Krasuski RA, Rodriguez LL, Kapadia SR. Morphology of the patent foramen ovale in asymptomatic versus symptomatic (stroke or transient ischemic attack) patients. Am J Cardiol 2009;103:124-9
Griffin JH, Evatt B, Zimmermann TS, Kleiss AJ, Wideman C. Deficency of protein C in congenital thrombotic disease. J Clin Invest 1981;68:1370-3
Grossman R, Geisen U, Merati G, Müllges W, Schambeck M, Walter U, Schwender S. Genetic risk factors in young adults with ‘cryptogenic’ ischemic cerebrovascular disease. Blood Coagulation and fibrinolysis 2002;13:583-590
Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: .an autopsy study of 965 normal hearts. Mayo Clin Proc 1984;59:17-20
Hara H, Virmani R, Ladich E, Mackey-Bojack S, Titus J, Reisman M, Gray W, Nakamura M, Mooney M, Poulose A, Schwartz RS. Patent foramen ovale :current pathology, pathophysiology, and clinical status. J Am Coll Cardiol 2005;46:1768-1776
Heit JA, O'Fallon WM, Petterson TM, Lohse CM, Silverstein MD, Mohr DN, Melton LJ 3rd. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med 2002;162:1245-8
Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA 2002;288:2015-22
Housman D, Ledley FD. Why pharmacogenomics? Why now? Nat Biotechnol 1998;16:492-3
Kang SS, Wong PW, Malinow MR. Hyperomocysteinemia as a risk factor for occlusive vascular disease. Annu Rev Nutr 1992;12:279-298
Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am J Hum Genet 1991;48:536-545
Kang SS, Zhou J, Wong PW, Kowalisyn J, Strokosch G. Intermediate Homocysteinemia: a thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet 1988;43:414-421
Karttunen V, Hiltunen L, Rasi V, Vahtera E, Hillbom M. Factor V Leiden and prothrombin gene mutation may predispose to paradoxical embolism in subjects with patent foramen ovale. Blood Coagul Fibrinolysis 2003;14:261-8
Kedia G, Tobis J, Lee MS. Patent foramen ovale: clinical manifestations and treatment. Rev Cardiovasc Med 2008;9:168-73
Kim RJ, Becker RC. Association between factor V Leiden , prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations and events of the arterial circulatory system: a meta-analysis of published studies. Am Heart J 2003;146:948-57
Kottke-Marchant K. Genetic polymorphisms associated with venous and arterial thrombosis: an overview. Arch Pathol Lab Med 2002;126:295-304
Lagman J. Embriologia Medica. 3a edizione Piccin editore, Padova
Prothrombin G20210A Mutat,ion in Patients With Ischemic Stroke/Transient Ischemic Attack Up to the Age of 60 Years. Stroke 2005;36:1405-9
Lechat P, Mas JL, Lascault G, Loron P, Theard M, Klimczac M, Drobinski G, Thomas D, Grosgogeat Y. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med 1988;318:1148-1152
Lichy C, Reuner KH, Buggle F, Litfin F, Rickmann H, Kunze A, Brandt T, Grau A. Prothrombin G20210A mutation, but not factor V Leiden, is a risk factor in patients with persistent foramen ovale and otherwise unexplained cerebral ischemia. Cerebrovasc Dis 2003;16:83-7
Lowe GD. Common risk factors for both arterial and venous thrombosis. Br J Haematol 2008;140:488-95
Mareedu RK, Shah MS, Mesa JE, McCauley CS. Percutaneous Closure of Patent Foramen Ovale: A Case Series and Literature Review. Clin Med Res 2007;5:218-226
Mas JL, Zuber M Recurrent cerebrovascular events in patients with patent foramen ovale, atrial septal aneurysm, or both and cryptogenic stroke or transient ischemic attack. French Study Group on Patent Foramen Ovale and Atrial Septal Aneurysm. Am Heart J 1995;130:1083-8
Mas JL. Patent foramen ovale, atrial septal aneurysm and ischaemic stroke in young adults. Eur Heart J 1994;15:446-9
McKusick VA. The anatomy of the human genome. A neo-vesalian basis for medicine in the 21st century. JAMA 2001;286:2289-95
Meissner I, Khandheria BK, Heit JA, Petty GW, Sheps SG, Schwartz GL, Whisnant JP, Wiebers DO, Covalt JL, Petterson TM, Christianson TJ, Agmon Y. Patent formen ovale : innocent or guilty? Evidence form a prospective population-based study. J Am Coll Cardiol 2006;47:440-5
Miñano A, Ordóñez A, España F, González-Porras JR, Lecumberri R, Fontcuberta J, Llamas P, Marín F, Estellés A, Alberca I, Vicente V,Corral J.
Leiden or prothrombin G20210A polymorphisms. Haematologica 2008;93:729-34
Offelli P, Zanchetta M, Pedon L, Marzot F, Cucchini U, Pegoraro C, Iliceto S, Pengo V. Thrombophilia in young patients with cryptogenic stroke and patent foramen ovale(PFO). Thromb Haemost 2007;98:906-7
Onorato E, Casilli F, Berti M, Anzola GP. Patent foramen ovale closure. Pro and cons. Neurol Sci 2008;29:S28-S32
Overell JR, Bone I, Lees KR. Interatrial septal abnormalities and stroke: a meta analysis of case-control studies. Neurology 2000;55:1172-9
Pezzini A, Del Zotto E, Magoni M, Costa A, Archetti S, Grassi M, Akkawi NM, Albertini A, Assanelli D, Vignolo LA, Padovani A. Inherited thrombofilic disorders in young adults with ischemic stroke and patent foramen ovale. Stroke 2003;34:28-33