UNIVERSIT ´A DEGLI STUDI DI ROMA LA SAPIENZA
Facolt´a di scienze matematiche, fisiche e naturali
TESI DI LAUREA TRIENNALE IN FISICA A.A. 2004/2005
Diffusione depolarizzata della luce
in colloidi anisotropi
Relatore:
Prof. Giancarlo Ruocco Dr. Tullio Scopigno
Candidata: Chiara Vitelli
Indice
1 Introduzione 5
1.1 Diffusione della luce . . . 5
1.2 Fluttuazioni e funzioni di correlazione temporale . . . 7
1.3 Densit´a spettrale . . . 10
1.4 Realizzazioni di un esperimento di scattering . . . 12
2 Teoria base della diffusione della luce 15 2.1 Risultati dalla teoria dell’elettromagnetismo . . . 15
2.2 Approccio molecolare alla diffusione della luce . . . 19
2.3 Geometrie di scattering . . . 20
3 Esperimenti di diffusione della luce 25 3.1 Metodo omodino . . . 26
3.2 Metodo eterodino . . . 27
3.3 Area di coerenza . . . 28
3.4 Considerazioni sperimentali: il contrasto dinamico . . . 29
4 Sistemi di molecole sferiche 33 4.1 Molecole sferiche . . . 33
4.2 Soluzioni diluite . . . 34
4.2.1 Funzione di correlazione eterodina . . . 35
4.2.2 Funzione di correlazione omodina . . . 37
5 Sistemi contenenti molecole anisotrope 39 5.1 Scattering di molecole a simmetria cilindrica . . . 40
5.2 Diffusione rotazionale di molecole lineari . . . 42
6 Risultati sperimentali 45 6.1 L’apparato sperimentale . . . 45
6.2 L’acquisizione dei dati . . . 46
6.3 Conclusioni . . . 50
Capitolo 1
Introduzione
Attraverso questo lavoro ci proponiamo di indagare la dinamica di un sistema colloidale, mediante una tecnica sperimentale di diffusione del-la luce detta fotocorredel-lazione. Ci soffermeremo sul comportamento dei gradi di libert´a rotazionali del sistema, analizzando la radiazione de-polarizzata diffusa dal campione, i.e. la luce con polarizzazione lineare ortogonale a quella della luce incidente. Verificheremo che la dinami-ca di tali sistemi presenta una sdinami-cala di tempo veloce, relativa alle vi-brazioni delle particelle attorno alle posizioni istantanee di equilibrio della struttura, e una lenta associata alle modificazioni della struttura, causate da processi cooperativi che coinvolgono gruppi di particelle. La funzione di correlazione dell’intensit´a della luce diffusa dal sistema ´e quindi caratterizzata da un doppio decadimento: il primo ´e associato alla dinamica vibrazionale, il secondo avviene su scale di tempo tipiche della diffusione delle particelle. Ci proponiamo infine di indagare come la dinamica descritta dipenda dall’invecchiamento del campione. Riportiamo di seguito i principi su cui si basa la tecnica di fotocorre-lazione utilizzata; illustriamo poi i fondamenti della teoria della dif-fusione della luce e le tecniche sperimentali ad essa legate; presen-tiamo quindi due modelli di sistemi contenenti molecole isotrope e anisotrope con le rispettive funzioni di correlazione caratteristiche; ed infine riportiamo i risultati sperimentali ottenuti.
1.1
Diffusione della luce
La radiazione elettromagnetica permette di indagare la struttura e la dinamica della materia: i fotoni, incidendo su di essa, possono essere assorbiti o diffusi. L’assorbimento di ultravioletto, visibile, infrarosso ecc. fornisce informazioni dettagliate sui livelli elettronici rotaziona-li e vibrazionarotaziona-li delle molecole e permette quindi di determinare la
dinamica delle molecole complesse. La diffusione elastica di raggi X o neutroni, per esempio, permette di ricavare informazioni sulla struttura microscopica di un sistema atomico/molecolare attraverso lo studio del fattore di struttura. Incidendo sulla materia, i fotoni possono inoltre essere diffusi, cedendo o guadagnando energia dai gradi di libert´a elet-tronici, traslazionali, rotazionali e vibrazionali delle molecole, si parla in questo caso di diffusione anelastica, e l’informazione che si ottiene riguarda la dinamica del sistema.
Ci concentreremo essenzialmente sulle propriet´a della luce diffusa dei gradi di libert´a rotazionali e traslazionali, ovvero di quello che ´e co-munemente chiamato scattering di Rayleigh ([1]).
Classicamente si pu´o interpretare il fenomeno considerando che, in-cidendo sulla materia, il campo elettrico della luce induce un dipolo oscillante nella materia tramite la polarizzabilit´a, le molecole diven-gono quindi una sorgente di luce secondaria e conseguentemente dif-fondono luce. Le dimensioni, la forma, e le interazioni molecolari del materiale che diffonde determinano, attraverso la polarizzabilit´a, la dis-tribuzione in frequenza, la polarizzazione e l’intensit´a della luce diffusa. Cos´ı dalle caratteristiche della luce diffusa da un dato sistema, ´e pos-sibile, con l’aiuto dell’elettrodinamica e della teoria della meccanica statistica dipendente dal tempo, ottenere informazioni circa la strut-tura e la dinamica molecolare del mezzo diffusore.
1.2. FLUTTUAZIONI E FUNZIONI DI CORRELAZIONE TEMPORALE 7
1.2
Fluttuazioni e funzioni di correlazione
tempo-rale
Nell’ambito della teoria della risposta lineare, ´e possibile descrivere come la materia risponde alla radiazione conoscendo i due sistemi sep-aratamente, ipotizzando quindi che radiazione e materia siano debol-mente accoppiate.
L’agitazione termica delle molecole di un sistema colpito da un’onda elettromagnetica provoca delle fluttuazioni nel tempo del campo elet-trico diffuso, tali fluttuazioni sono ben descritte dalle funzioni di corre-lazione temporale, che esprimono quanto sono correlate due grandezze per un periodo di tempo.
In generale, ogni osservabile misurata in un sistema all’equilibrio ´e una media sul tempo T :
¯ A(t0, T ) = 1 T Z t0+T t0 dtA(t) (1.1)
La media ha significato solo se il tempo T su cui ´e fatta ´e grande rispetto al periodo delle fluttuazioni di A. Idealmente A andrebbe me-diando su un tempo infinito:
¯ A(t0, T ) = lim T →∞ 1 T Z t0+T t0 dtA(t) (1.2)
Assumendo che A sia una propriet´a stazionaria del sistema, si perde la dipendenza da t0. Immaginando poi che la variabile A dipenda
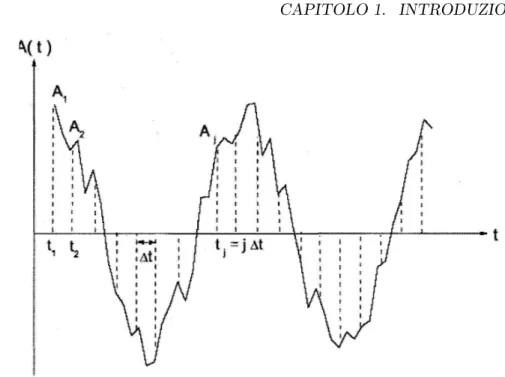
dal-la posizione e dal momento di tutte le particelle del sistema, il suo andamento nel tempo presenter´a, a causa dell’agitazione termica, un certo grado di rumore. In generale allora il valore di A sar´a diverso per due tempi t e t+τ , i due valori differiranno al crescere di τ quanto pi´u questo diventer´a grande rispetto alle fluttuazioni del sistema, mentre saranno correlati per τ piccolo. Una misura di tale correlazione ´e data dalla funzione di autocorrelazione di A:
< A(0)A(τ ) >= lim T →∞ 1 T Z T 0 dtA(t)A(t + τ ) (1.3) Si trova che 1 < A(0)2 > ≥ < A(0)A(τ ) >
1Dovendo calcolare la (1.3) a passi discreti, considerando l’asse dei tempi diviso in N
Figura 1.2: Andamento della variabile A che fluttua nel tempo, campionata su intervalli temporali discreti
ovvero il valore della funzione di correlazione al tempo zero ´e massi-mo, mentre, per τ molto grandi, i valori di A(t) e A(t+τ ) si decorrelano:
lim
τ →∞< A(0)A(τ ) >=< A(0) >< A(τ ) >=< A > 2
La funzione di autocorrelazione decade allora dal valore iniziale <A2> al valore <A>2 tanto pi´u velocemente quanto pi´u rapide sono
le fluttuazioni della variabile A. In molti casi di interesse pratico l’an-damento ´e quello di un decadimento esponenziale del tipo:
intervalli discreti sar´a allora:
< A >≈ lim N →∞ 1 N N X j=1 A(j∆t) < A(0)A(τ ) >≈ lim N →∞ 1 N N X j=1 A(j∆t)A((j + n)∆t)
Nell’ultima sommatoria si possono avere termini negativi che andranno a cancellare termini positivi; considerando invece < A(0)A(0) > si ha che tutti i termini sono positivi, infatti: PNj=1A(j∆t)A(j∆t) =PNj=1A2(j∆t) con A2(j∆t) ≥ 0
quindi:
1.2. FLUTTUAZIONI E FUNZIONI DI CORRELAZIONE TEMPORALE 9
< A(0)A(τ ) >=< A >2 +(< A2 > − < A >2) exp[−τ /τ
r] (1.4)
dove τr ´e il tempo di rilassamento o tempo di correlazione della
variabile A.
Introducendo la deviazione di A(t) dal suo valore medio come: δA(t) ≡ A(t)− < A >
ci si pu´o riferire alla funzione di correlazione delle fluttuazioni, e tenendo conto del fatto che <δA(t)>=0, si ottiene l’espressione sem-plificata:
< δA(0)δA(τ ) >=< A(0)A(τ ) > − < A >2 (1.5) da cui:
< δA2 >=< A2 > − < A >2 (1.6)
Facendo riferimento alle equazioni (1.4), (1.5) e (1.6) si ottiene allora:
< δA(0)δA(τ ) >=< δA2 > exp[−τ /τ
r] (1.7)
Si ottiene quindi che la funzione di correlazione delle fluttuazioni ha una forma pi´u semplice rispetto alla funzione di correlazione della propriet´a stessa, in quanto ´e rimossa la parte <A2>, invariante rispetto
al tempo.
In generale si pu´o introdurre un parametro che caratterizzi la scala di tempo di decadimento delle correlazioni definendo il tempo di corre-lazione τc: τc= Z ∞ 0 dτ < δA(0)δA(τ ) > < δA2 >
che per un decadimento esponenziale singolo coincide ovviamente con τr.
1.3
Densit´
a spettrale
Si definisce densit´a spettrale IA(ω) della funzione di correlazione <A∗(0)A(t)>
come la sua trasformata di Fourier nel dominio delle frequenze:
IA(ω) ≡ 1 2π Z +∞ −∞ dt exp[−iωt] < A ∗(0)A(t) > (1.8)
Negli esperimenti di diffusione di luce spesso viene misurata proprio la densit´a spettrale del campo elettrico diffuso, ci si pu´o allora ricon-durre alla funzione di correlazione temporale semplicemente trasfor-mando secondo Fourier:
< A∗(0)A(t) >=Z +∞
−∞ dω exp[iωt]IA(ω) (1.9)
da cui:
<| A |2>=<| A(0) |2>= Z +∞
−∞ dωIA(ω) (1.10)
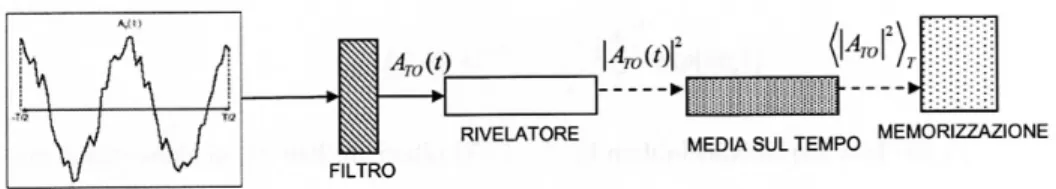
Immaginiamo di voler misurare la variabile A in un periodo T: un filtro seleziona solo un intervallo di frequenze della quantit´a AT(t),
tale segnale uscente dal filtro arriva al rivelatore il cui output ´e pro-porzionale a | AT O(t)| 2 che viene mediato sul tempo, per dare, come
risultato finale, <| AT O(t)|2>t.
Nell’intervallo (-T/2,T/2) posso espandere la A(t) in serie di Fourier:
AT(t) = 1 √ T X n Anexp[iωnt] (1.11)
Il filtro selezioner´a solo alcune delle frequenze An, questo equivale a
moltiplicare i termini della sommatoria per dei fattori Fn che possono
essere 1 o 0 a seconda che la frequenza venga filtrata o meno, ottenendo in uscita AT O.
Facendo la media temporale tra (-T/2,T/2) del modulo quadro di AT O
si ha: <| AT O |>T= 1 T X n F2 n | An|2 (1.12)
1.3. DENSIT ´A SPETTRALE 11
Si pu´o allora esprimere la funzione di autocorrelazione della vari-abile A come: < A∗(t)A(t + τ ) >= 1 T X n | An |2 exp[iωnt] (1.13)
Ora integrando τ tra [-T/2,T/2] e moltiplicando per exp[-iωmτ ] si
ottiene:
| Am |2= 2πIAT(ωm) (1.14)
dove IAT(ωm) ´e per definizione la densit´a spettrale della funzione di
correlazione <A∗(t)A(t+τ )>. La (1.12) diviene allora:
<| AT O |>T= 2π T X n Fn2IAT(ωn) (1.15) dove 2π
T pu´o essere scritto come ∆ω=ωn+1-ωn.
Nel limite T→∞ la sommatoria diviene un integrale in dω, e | F(ω)|2
´e la funzione di filtro, che ´e diversa da zero solo in un intervallo ∆ ω attorno alla frequenza ω0 se ho un filtro a banda stretta. Si ha quindi:
lim
T →∞<| AT O | 2>
T= IA(ω0)∆ω (1.16)
Selezionando allora diversi valori di ω0 e facendo una media
tem-porale di |AT O|2 si ottiene lo spettro completo della fluttuazione A. In
questo senso IA(ω)dω ´e considerata la quantit´a di A nell’intervallo di
frequenze (ω,ω+∆ω).
In particolare se ET(t) ´e il campo elettrico dell’onda di luce diffusa,
il filtro ´e a banda stretta e se il rivelatore risponde quadraticamente all’impulso ricevuto si ha:
lim
T →∞<| ET O | 2>
T= IE(ω0)∆ω (1.17)
dove IE(ω0) ´e la densit´a spettrale della funzione di autocorrelazione
del campo elettrico e ω0 ´e definita dal filtro. Facendo filtrare frequenze
diverse si pu´o allora determinare IE come funzione di ω e ottenere
la funzione di correlazione del campo elettrico trasformando secondo Fourier: IE(ω) = 1 2π Z +∞ −∞ dτ < E ∗(t)E(t + τ ) > exp[iωτ ] (1.18)
Lo spettro della luce diffusa ´e determinato allora mediante la fun-zione di correlafun-zione del campo elettrico al rivelatore, da cui possono essere estratte importanti informazioni sulle propriet´a fisiche del mezzo diffusore.
Figura 1.3: Schema dell’apparato per la misura della densit´a spettrale
1.4
Realizzazioni di un esperimento
di scattering
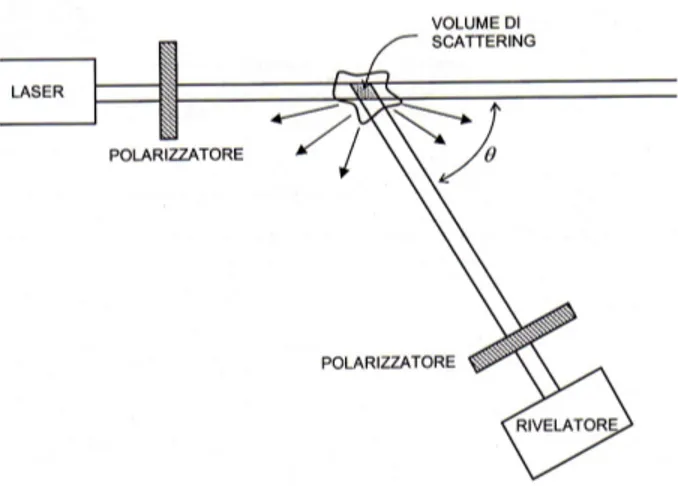
In un tipico esperimento di diffusione della luce, la radiazione polar-izzata linearmente prodotta da un laser incide sul mezzo diffusore. La luce diffusa passa allora in un filtro che seleziona una data polariz-zazione. A questo punto l’analisi della luce diffusa pu´o essere effettuata nel dominio delle frequenze, misurando la densit´a spettrale come de-scritto nella sezione 1.3, o nel dominio dei tempi, attraverso la tecnica della fotocorrelazione. Nel primo caso la luce diffusa viene filtrata, per esempio attraverso un monocromatore o un interferometro, e inviata ad un fotomoltiplicatore, nel secondo caso la luce incide direttamente sul rivelatore e il segnale passa ad un correlatore che calcola la funzione di correlazione dell’intensit´a diffusa. Le tecniche di fotocorrelazione co-munemente utilizzate sono quella omodina e quella eterodina, che il-lustreremo in dettaglio nel capitolo 3.
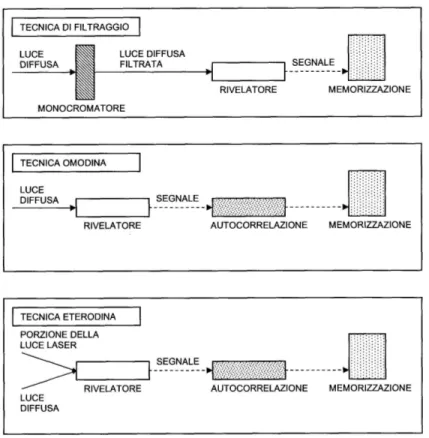
Riportiamo di seguito lo schema delle tre differenti tecniche utilizzate nella realizzazione di un esperimento di scattering:
1.4. REALIZZAZIONI DI UN ESPERIMENTO DI SCATTERING 13
Figura 1.4: Comuni realizzazioni di un esperimento di scattering:
In un esperimento di filtraggio la luce diffusa viene filtrata prima di incidere sul rivelatore.
In configurazione omodina la sola luce diffusa incide sul rivelatore e viene analizzata da un correlatore.
In configurazione eterodina parte della luce proveniente dal laser viene ag-giunta al facio di luce diffusa, i due fasci colpiscono il rivelatore e vengono analizzati da un correlatore.
Capitolo 2
Teoria base della diffusione
della luce
2.1
Risultati dalla teoria dell’elettromagnetismo
Immaginiamo di schematizzare il campo elettrico incidente su un mezzo non assorbente come un’onda piana monocromatica e polarizzata:
Ei(r, t) = niE0exp[ki· r] (2.1)
tale mezzo ha una costante dielettrica locale:
ε(r, t) = ε0I + δε(r, t) (2.2)
dove δε ´e il tensore di fluttuazione della costante dielettrica alla posizione r e al tempo t.
Si mostra 1
1Indicando con E,D,H i campi totali (campo elettrico, spostamento dielettrico e campo
magnetico), con Ei,Di,Hii campi incidenti e con Es,Ds,Hsquelli diffusi, poich´e sia il campo
totale che quello incidente verificano le equazioni di Maxwell, anche il campo diffuso le soddisfer´a per linearit´a:
∇ × Es = −1 c ∂Hs ∂t ∇ × Hs = 1 c ∂Ds ∂t ∇ · Ds = 0 ∇ · Bs = 0
(dove B ´e il vettore di induzione magnetica B=µH) se µ=1 allora Bs=Hs. Possiamo quindi
eliminare dalle prime due equazioni Hsda cui otteniamo:
∇ × (∇ × Es) = −1 c2 ∂2D s ∂t2 15
che il campo diffuso dal campione a grande distanza R dal volume di scattering, con polarizzazione nf, vettore d’onda kf, e frequenza ωf
´e: Es(R, t) = E0 4πRε0 exp[ikfR] Z V d 3r exp i(q· r−ω it)[nf·[kf×(kf×(δ ε(r, t)·ni))]] (2.3)
I vettori D ed E sono legati tra loro tramite la costante dielettrica:
D = (εI + δε)(Ei+ Es) = εEi+ εEs+ δεEi
dove abbiamo trascurato il termine δεEs' 0 poich´e Di=εEi, si ha che:
Ds= δεEi+ εEs
da cui, sostituendo Es si ha:
∇2D s− ε c2 ∂2D s ∂t2 = −∇ × ∇ × (δεEi)
Introducendo il vettore Π tale che:
Ds= ∇ × ∇ × Π si ha: ∇2Π − ε c2 ∂2Π ∂t2 = −δεEi
che pu´o essere risolta attraverso la funzione di Green: Π(R, t) = Z V d3rδε(r, tˆ 0)Ei(r, t 0) 4π | r − R |
dove t’=t-(n/c)| R-r| ´e il tempo ritardato che tiene conto del tempo che impiega il segnale a propagarsi nel mezzo e R ´e la posizione del rivelatore che consideriamo immerso in un mezzo di costante dielettrica ε.
Assumendo ora che | R |À| r| si ha: | R − r |=| R | −r · ˆR con ˆR= ˆkf versore nella direzione
di R. Il tempo ritardato diventer´a allora: t’'t-(n/c)(| R |-kf·r) e potremo scrivere:
Π(R, t) = E0 4π | R | Z V d3rδε(r, t0) ˆn iexp i(ki· r − ωit + ωi(n/c) | R | −ωi(n/c)r · ˆR)
Sviluppando ora δε in serie di Fourier le cui uniche componenti di frequenza Ωpche danno
contributo sono quelle tipiche del moto traslazionale e rotazionale del sistema, di molto inferiore alla frequenza della luce incidente, e definendo ωf ≡ ωi− Ωp, kp≡ (n/c)ωfkf,
qp≡ ki− kssi ottiene: Es(R, t) = ∇×∇×[ E0 4πε | R | X p Z V d3rδε
p(r)· ˆniexp iΩp(t−(n/c)(| R | −r· ˆkf)+iki·r−iωi(t−(n/c)(| R | −r· ˆkf)))]
da cui, poich´e Ωp¿ ωi, avremo:
Es(R, t) = E0
4πε | R |exp i(ks· R − ωit)ks× [ks×
Z
V
2.1. RISULTATI DALLA TEORIA DELL’ELETTROMAGNETISMO 17
dove V indica che l’integrale ´e fatto su tutto il volume di scattering. Il vettore q ´e definito come:
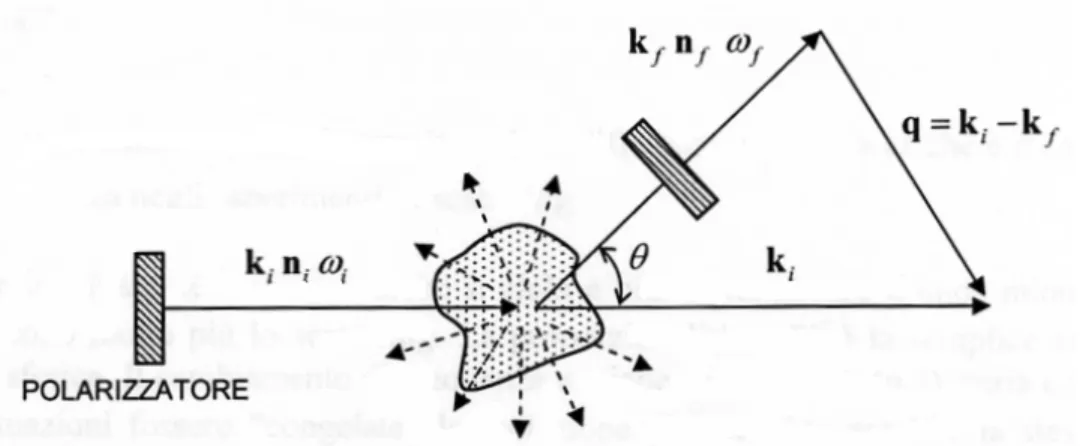
q = ki− kf
dove ki e kf sono i vettori d’onda della luce incidente e di quella
che raggiunge il rivelatore, rispettivamente; l’angolo tra di essi ´e detto angolo di scattering.
Figura 2.1: Vettori d’onda della luce polarizzata iniziale e finale
Assumendo che la lunghezza d’onda della luce vari poco nel processo di diffusione si ha
| ki |∼=| kf |
tale relazione porta alla condizione di Bragg: q = 2kisin
θ 2
Possiamo esprimere la (2.3) in termini della trasformata di Fourier di δε: δε(q, t) = Z V d 3rδε(r, t) exp iq · r come Es(R, t) = E0 4πRε0 exp[ikfR−ωit]{nf·[kf×(kf×(δ ε(q, t)·ni))]} (2.4)
e lavorando con i prodotti vettoriali2, otteniamo: 2
Es(R, t) = − k2 fE0 4πRε0 exp i(kfR − ωit)δεif(q, t) (2.5) dove: δεif(q, t) ≡ nf · δε(q, t) · ni (2.6)
´e la componente del tensore di fluttuazione della costante dielettrica tra le direzioni di polarizzazione iniziale e finale.
Si ottiene allora che la funzione di correlazione del campo elettrico diffuso dipende dalla funzione di correlazione delle fluttuazioni della costante dielettrica: < Es∗(R, 0)Es(R, t) >= k4 f | E0 |2 16π2R2ε2 0 < δεif(q, 0)δεif(q, t) > exp(−iωit) (2.7) trasformando secondo Fourier si ottiene la densit´a spettrale:
Iif(q, ωf, R) = I0kf4 16π2R2ε2 0 1 2π Z +∞ −∞ dt < δεif(q, 0)δεif(q, t) > exp(i(ωf−ωi)t) (2.8) dove I0≡|E0|2, si nota che:
1. Iif ∝λ−4 che spiega, ad esempio, perch´e la luce blu sia pi´u diffusa
di quella rossa, e anche perch´e gli esperimenti di diffusione effet-tuati con la luce visibile siano pi´u efficaci di quelli effeteffet-tuati con luce infrarossa
2. Iif ∝ R−2 che ´e l’attenuazione tipica di un’onda sferica
3. Iif dipende dalla frequenza ω=ωi-ωf che ´e non nulla solo se δε(q,t)
varia nel tempo, se infatti le fluttuazioni sono indipendenti dal tempo l’integrale della densit´a spettrale dar´a una δω, in tal ca-so la frequenza dell’onda diffusa coincider´a con quella dell’onda incidente
Quindi la densit´a spettrale della luce diffusa ´e proporzionale alla densit´a spettrale delle fluttuazioni della costante dielettrica:
Iifε(q, ω) = 1 2π Z +∞ −∞ dt exp(−iωt) < δε ∗ if(q, 0)δεif(q, t) > I(q, ωf, R) ∝ Iifε(q, ω) (2.9)
2.2. APPROCCIO MOLECOLARE ALLA DIFFUSIONE DELLA LUCE19
con costante di proporzionalit´a A = k4fI0
16π2R2ε2 0.
´
E chiaro quindi che sono proprio le fluttuazioni della costante dielet-trica di vettore d’onda q e frequenza ω a provocare l’evento di diffusione che produce un cambiamento di vettore d’onda q e di frequenza ω della luce incidente.
2.2
Approccio molecolare alla diffusione della luce
Quando la luce incide su una singola molecola, di polarizzabilit´a anisotropa definita dal tensore di polarizzabilit´a α, induce un momento di dipolo variabile nel tempo: µ(t) = α · E(t) che emette radiazione elettromag-netica.
Il campo elettrico della luce diffusa al rivelatore ´e proporzionale a αif(t)exp[iq·r(t)] dove:
αif(t) = nf·α(t)·ni (2.10)
´e la componente del tensore di polarizzabilit´a molecolare tra le di-rezioni ni e nf; r(t) ´e la posizione del centro di massa della molecola al
tempo t e q ´e il vettore di scattering. La variazione di αif nel tempo ´e
dovuto alle rotazioni e alle vibrazioni della molecola, mentre il termine exp(iq·r(t)) tiene conto della sua traslazione.
Se si considera l’accoppiamento elettronico tra le molecole debole, si conclude che la luce diffusa dall’insieme delle molecole contenute nel volume illuminato ´e data dalla sovrapposizione dei contributi proveni-enti da ogni singola molecola, ovvero il campo diffuso sar´a proporzionale a una somma di termini:
X j
0αj
if(t) exp(iq · r(t))
La densit´a spettrale del campo diffuso sar´a quindi proporzionale a: Iα if(q, ω) = 1 2π Z +∞ −∞ dt exp(−iωt)I α if(q, t) (2.11) dove: Iα if(q, t) =< δα∗if(q, 0)δαif(q, t) > e: δαif(q, t) = N X j=1 0αj if(t) exp(iq · r(t)) (2.12)
´e la componente spaziale di Fourier della densit´a di polarizzabilit´a: δαif(r, t) = N X j=1 0αj if(t)δ(r − rj(t)) (2.13)
Tale modello non tiene conto della distorsione della distribuzione di carica delle molecole conseguente agli urti subiti, che permane finch´e la molecola non attraversa il range effettivo dell’interazione intermoleco-lare (∼ 10−13s) dando luogo al cosiddetto scattering indotto da
colli-sioni.
2.3
Geometrie di scattering
´
E conveniente in molti casi pratici fare riferimento a geometrie di dif-fusione convenzionali. Riportiamo di seguito tali geometrie e le com-ponenti della fluttuazione della costante dielettrica che intervengono espresse in ciascun sistema di riferimento fissato.
Il piano individuato dai vettori d’onda iniziale e finale ´e detto piano di scattering ed in relazione ad esso si definisce una particolare geometria di scattering:
2.3. GEOMETRIE DI SCATTERING 21
Geometria I
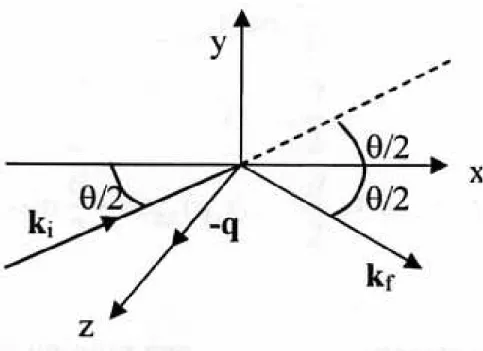
Figura 2.2: GEOMETRIA I:In questa geometria il piano XZ ´e il piano di scattering. L’angolo (ki,kf) ´e l’angolo di scattering, e il vettore di scattering q ´e antiparallelo all’asse Z
Geometria II
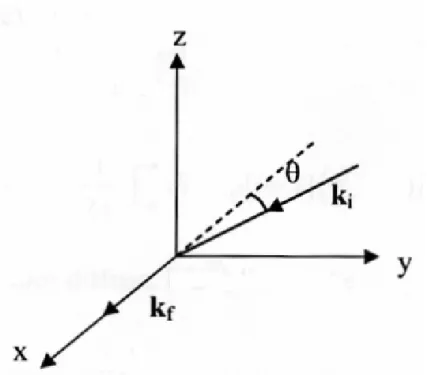
Figura 2.3: GEOMETRIA II:In questa geometria il piano XY ´e il piano di scattering. L’angolo (ki,kf) ´e l’angolo di scattering, e il vettore di scattering q non giace lungo nessun asse particolare
2.3. GEOMETRIE DI SCATTERING 23
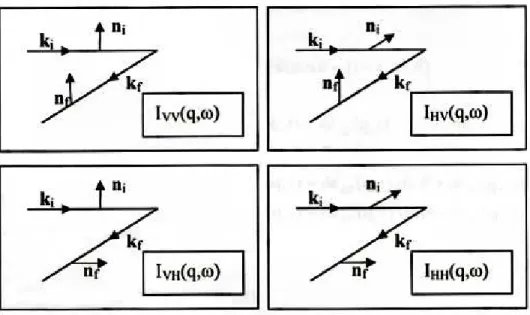
Possono essere definite quattro distinte polarizzazioni rispetto al pi-ano di scattering: al variare delle polarizzazioni dell’onda incidente e dell’onda diffusa si misureranno valori diversi dell’intensit´a della luce diffusa:
Figura 2.4: Quattro differenti direzioni di polarizzazione sono comunemente usate negli esperimenti di diffusione della luce
Le componenti della fluttuazione della costante dielettrica (o del-la fluttuazione deldel-la podel-larizzabilit´a) responsabili per l’intensit´a diffusa nelle varie configurazioni possono essere scritte, partire dalla (2.6), in termini dei sistemi dei riferimento definiti dalle due geometrie in figure 2.2 e 2.3: riportiamo di seguito tali espressioni in funzione dei valori delle proiezioni della fluttuazione della costante dielettrica sugli assi cartesiani xyz: per la geometria I: q=-qˆz δεV V(q, t) = δεY Y(q, t) δεV H(q, t) = δεY X(q, t) sin θ 2 − δεY Z(q, t) cos θ 2 δεHV(q, t) = δεXY(q, t) sin θ 2 + δεZY(q, t) cos θ 2 δεHH(q, t) = δεXX(q, t) sin2 θ 2− δεZZ(q, t) cos 2 θ 2+ [δεZX(q, t) − δεXZ(q, t)] sin θ 2cos θ 2(2.14)
e per la geometria II: q=q[ˆx(cos θ-1)+ˆy sin θ]
δεV V(q, t) = δεZZ(q, t)
δεV H(q, t) = δεZY(q, t)
δεHV(q, t) = δεXY(q, t) sin θ + δεY Z(q, t) cos θ
δεHH(q, t) = δεXY(q, t) sin θ + δεY Y(q, t) cos θ (2.15)
I simboli V e H corrispondono alle direzioni orizzontale e verticale rispetto al piano di scattering. IV V ´e detta componente polarizzata,
mentre IV H e IHV sono le componenti depolarizzate.
Vedremo in seguito come lo studio di tali componenti riesce a dare informazioni su gradi di libert´a diversi delle molecole del campione diffusore.
Capitolo 3
Esperimenti di diffusione
della luce
In un esperimento di fotocorrelazione l’onda elettromagnetica emessa da un laser incide sul campione, la luce diffusa da questo viene invia-ta ad un fotomoltiplicatore attraverso un’ottica di raccolinvia-ta; il segnale uscente dal fototubo passa ad un correlatore che calcola la funzione di autocorrelazione dell’intensit´a diffusa. Il metodo di rivelazione del fotofubo segue una legge quadratica in quanto il segnale da esso us-cente ´e proporzionale al quadrato del campo incidente: i(t)∝| E(t)|2.
Il correlatore che analizza il segnale in uscita dal fototubo dar´a quin-di informazioni sulla funzione quin-di correlazione dell’intensit´a del campo incidente sulla superficie del rivelatore:
< i(t)i(0) >∝<| E(t) |2| E(0) |2>
I processi indagati con la tecnica sopra descritta sono quelli carat-terizzati da una dinamica molecolare lenta, nel senso che si tratta della diffusione di macromolecole in soluzioni molto diluite che avviene su scale di tempo maggiori a 10−6 sec; dinamiche pi´u veloci, per cui
risul-ta insufficiente la velocit´a del rivelatore, vengono indagate tramite la tecnica del filtraggio ottico che si basa sulla misura della densit´a spet-trale descritta nella sezione (1.3).
In generale, in un esperimento di fotocorrelazione, si utilizzano due di-versi schemi di rivelazione: il metodo omodino, in cui il fascio diffuso dal campione colpisce direttamente il fototubo; e quello eterodino, in cui una parte del fascio incidente ´e aggiunta a quello diffuso.
3.1
Metodo omodino
Si definisce funzione di correlazione eterodina quella del campo diffuso: I1 ≡< Es∗(0)Es(t) > (3.1)
mentre la funzione di correlazione dell’intensit´a diffusa, ovvero la quantit´a direttamente misurabile, ´e detta funzione di correlazione omo-dina:
I2(t) ≡<| Es(0) |2| Es(t) |2> (3.2)
Nel metodo omodino solo il campo diffuso Es colpisce il fotocatodo,
quindi < i(t)i(0) >∝ I2(t); poich´e il campo diffuso ´e proporzionale alle
fluttuazioni della costante dielettrica, I2 dipender´a dalla funzione di
correlazione delle fluttuazioni della costante dielettrica.
Nel caso di approssimazione gaussiana, ovvero quando ´e possibile di-videre il volume illuminato in sottoregioni statisticamente indipendenti, in modo che il campo diffuso totale divenga la somma dei campi dif-fusi singolarmente da ogni sottoregione, si pu´o esprimere la funzione di correlazione omodina in funzione di quella eterodina:
I2(t) =| I1(0) |2 + | I1(t) |2 (3.3)
Nel caso di soluzioni diluite di macromolecole tale approssimazione ´e applicabile se nel volume illuminato c’´e un gran numero di molecole, ognuna delle quali diffonde un campo che contribuisce al campo totale diffuso. Nel caso in cui il decadimento della funzione di correlazione si pu´o assumere esponenziale, come appunto nel caso di macromolecole in soluzioni diluite, si avr´a:
I1(t) = A exp[− t τete ] da cui: I2(t) = A2(1 + exp[− 2t τete ])
ovvero la funzione di correlazione omodina segue sempre un decadi-mento esponenziale ma con un tempo di correlazione dimezzato rispet-to al tempo caratteristico di decadimenrispet-to della funzione di correlazione eterodina.
3.2. METODO ETERODINO 27
Se invece, nell’espressione della funzione di correlazione eterodina, ´e presente pi´u di un tempo di decadimento, ad esempio:
I1(t) = X
i
aiexp −t/τi
nella funzione di correlazione omodina compariranno nuove scale di tempo: I2 = X i X j aiaj[1 + exp −(1/τi + 1/τj)t].
3.2
Metodo eterodino
Nel metodo eterodino parte del fascio di luce proveniente dal laser, detto campo locale ELO, ´e aggiunto al fascio diffuso e raggiunge il
fotocatodo: dunque il correlatore analizzer´a il segnale i(t) proveniente dal fotomoltiplicatore, la cui funzione di correlazione sar´a:
< i(t0)i(t0+ t) >∝<| ELO(t0) + Es(t0) |2| ELO(t0+ t) + Es(t0+ t) |2>
(3.4) Rendendo l’ampiezza dell’oscillatore locale molto pi´u grande del-l’ampiezza del campo diffuso | ELO(t) |À| Es(t) |, considerando
trascur-abili le fluttuazioni del campo oscillante locale e statisticamente in-dipendenti i due campi, la (3.4) diviene:
< i(0)i(t) >∼= B[ILO2 + 2ILOReI1(t)] (3.5)
dove
ILO =<| ELO |2>
´e l’intensit´a del segnale oscillante locale, e ReI1(t) ´e la parte reale
di I1(t), detta funzione di correlazione eterodina.
Poich´e Es ´e proporzionale a δεif(q, t), o equivalentemente a δαif(q, t)
le funzioni di correlazione eterodina e omodina saranno rispettivamente proporzionali a :
Iif1(q, t) =< δε∗if(q, 0)δεif(q, t) > (3.6)
Iif2(q, t) =<| δε∗if(q, 0) |2| δεif(q, t) |2> (3.7)
Come gi´a visto, nel caso in cui valga l’approssimazione gaussiana le due funzioni di correlazione sono legate dalla relazione:
I2
3.3
Area di coerenza
Il grado di coerenza di un’onda luminosa ´e una misura di quanto questa sia vicina a un’onda monocromatica pura, ovvero di estensione spaziale e temporale infinita (quindi caratterizzata da una sola frequenza e da un solo vettore d’onda); le onde non coerenti hanno pi´u componenti di Fourier e le loro fasi e ampiezze fluttuano in modo random nel tempo e nello spazio.
Quando la luce proveniente da una sorgente estesa, nel nostro caso il volume illuminato che diffonde, incide su uno schermo, ad esempio la superficie del fotocatodo, su questo ´e prodotto un profilo di diffrazione che dipende, tra le altre cose, dall’estensione della sorgente, ovvero dalle dimensioni del volume illuminato. In un punto A del fotocatodo il va-lore del campo elettrico sar´a dato dalla somma dei campi provenienti da ogni centro diffusore; tale segnale sar´a pi´u o meno identico a quello rilevato in un punto B vicino ad A: i due segnali si dicono coerenti. Aumentando la distanza tra A e B la coerenza tra i due segnali verr´a meno. La lunghezza di coerenza lc ´e formalmente definita come la
dis-tanza oltre cui la funzione di correlazione spaziale dei campi elettrici in A e B ´e decaduta in modo significativo.
Diamo allora una stima della lunghezza di coerenza nel caso unidi-mensionale: il campo in un punto A dello schermo sar´a dato dalla sovrapposizione dei campi provenienti da ogni punto della sorgente:
E(A) = X
i
E(i) analogamente il campo in B sar´a:
E(B) =X
i
E0(i)
i due segnali provenienti dal medesimo punto i e ricevuti in A e in B, differiscono per un fattore di fase che dipende dalla differenza di cammino compiuta per raggiungere i due punti differenti. Si dimostra che la lunghezza di coerenza tra A e B ´e:
lc≈
λ
α (3.9)
dove α ´e l’angolo sotteso tra lo schermo e la sorgente e λ ´e la lunghez-za d’onda media della radiazione.
Se la sorgente fosse tridimensionale e la superficie di osservazione piatta come quella di un fotocatodo, si potrebbe definire un’area di coerenza tale che:
3.4. CONSIDERAZIONI SPERIMENTALI: IL CONTRASTO DINAMICO29
Acoh ≈
λ2
Ω (3.10)
dove Ω ´e l’angolo solido sotteso tra la sorgente e il rivelatore.
3.4
Considerazioni sperimentali: il contrasto
di-namico
Come abbiamo visto in precedenza, l’obiettivo di un esperimento di dif-fusione dinamica della luce ´e quello di misurare la dipendenza dal tempo della funzione di correlazione di una variabile dinamica del sistema, ad esempio dell’intensit´a luminosa([2]): I2(q, t) ≡< i(q, 0)i(qt) > questa
´e legata alla funzione di correlazione del campo diffuso I1(q,t) dalla
relazione di Siegert: I2(q, t) = I2+ | I1(q, t) |2 dove I ´e l’intensit´a del
campo diffuso. Si possono normalizzare tali funzioni ottenendo: ˆ I2(q, t) ≡ I2(q, t)/I2 ˆ I1(q, t) ≡ I1(q, t)/I Per definizione: ˆ I1(q, 0) = 1, Iˆ2(q, 0) = 2
Il valore sperimentale di I2(q,0) viene detto contrasto dinamico e risulta
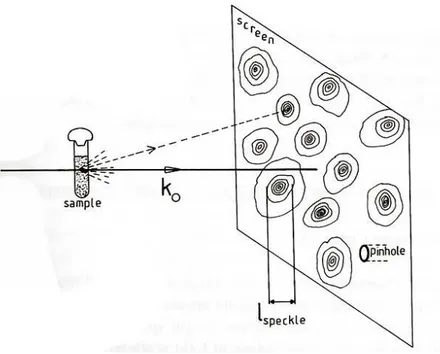
Figura 3.1: Macchie luminose proiettate sullo schermo di raccolta del seg-nale causate dall’interferenza tra i campi diffusi da diverse macromolecole in soluzione
Questo si spiega immaginando che il campione diffusore, ad esem-pio una soluzione colloidale come nel nostro caso, sia costituita da tanti centri diffusori, le macromolecole, i cui campi diffusi interferiscono tra loro. Ci´o produce sulla superficie di raccolta del segnale l’alternanza di macchie chiare e zone d’ombra, come risultato dell’interferenza costrut-tiva o distrutcostrut-tiva dei campi diffusi dalle varie molecole.
In generale la raccolta del segnale avviene sperimentalmente attraverso un pinhole, le cui dimensioni lineari non devono superare quelle delle macchie di luce, in quanto l’intensit´a rivelata attraverso molte macchie risulta inferiore a quella rivelata tramite una sola macchia. Vediamo come questo risulta essere legato alla geometria sperimentale.
3.4. CONSIDERAZIONI SPERIMENTALI: IL CONTRASTO DINAMICO31
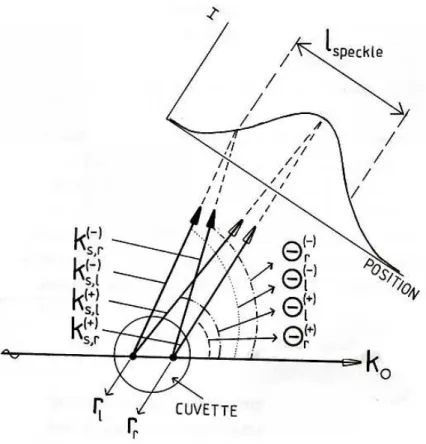
Definiamo rl e rr le posizioni di due punti alla sinistra e alla destra
del volume illuminato tali che |rl-rr|=12Vs1/3, dove V1/3s sono le
dimen-sioni lineari del volume di scattering. Supponiamo che il vettore d’onda della radiazione incidente k0 giaccia lungo la congiungente i due punti
considerati; e chiamiamo Θ+
l e Θ+r gli angoli di scattering per cui si ha
interferenza costruttiva dei campi provenienti dai punti rl e rr, mentre
Θ−
l e Θ−r saranno gli angoli di scattering per cui l’intensit´a della
mac-chia luminosa risulta minore. Le fasi, Φ±
l e Φ±r, della luce diffusa dai
punti rl e rr saranno: Φ± l,r = rl,r· (k0− k±s(l,r)) = rl,rk0[1 − cos Θ±l,r] Per definizione: Φ+ r − Φ+l = 2πn, Φ−r − Φ−l = 2π(n ± 1 2) con n intero. Quindi:
(Φ−
r − Φ−l ) − (Φ+r − Φ+l ) = ±π
Sia ora Θs l’angolo di scattering associato alla posizione del
pin-hole tra Θ+
l e Θ−r; per piccole differenze tra gli angoli di scattering
Θ±
l,r = Θs; sviluppando Φ±l,s in serie di Taylor, otteniamo che la
relazione precedente diviene:
| rrk0sin Θs(Θ+r − Θ−r) − rlk0sin Θs(Θ+l − Θ−l ) | = π
la taglia della macchia ´e:
lmacchia ≈ ld(Θ−r − Θr+) ≈ ld(Θ−l − Θ+l )
dove ld ´e la distanza tra il detector e il volume illuminato.
Ricor-dando che |rl-rr | = 12Vs1/3, troviamo che la taglia del pinhole per un
buon contrasto dinamico deve essere: lpinhole < lmacchia = 2π ld Vs1/3 1 k0 | 1 sin Θs |
Capitolo 4
Sistemi di molecole sferiche
Ci occupiamo ora descrivere, mediante la teoria della diffusione della luce, sistemi particolari contenenti molecole isotrope, a simmetria sferi-ca. In seguito estenderemo la nostra indagine anche a sistemi contenenti molecole anisotrope.4.1
Molecole sferiche
In generale il momento di dipolo indotto da un campo elettrico inci-dente sulla materia pu´o essere espresso da un vettore di componenti:
µx = αxxEx+ αxyEy + αxzEz
µy = αyxEx+ αyyEy+ αyzEz
µz = αzxEx+ αzyEy+ αzzEz (4.1)
ovvero, utilizzando una notazione vettoriale:
µ = α · E (4.2)
dove, in generale, α ´e un tensore. Nel caso di molecole sferiche ´e un tensore diagonale, e il momento di dipolo indotto nella materia risulta parallelo al campo incidente:
µ = αE (4.3)
In tale caso la (2.10) diviene:
αif = α ni · nf (4.4)
da cui la (2.12) risulta:
δαif(q, t) = (ni· nf)α N X j=1 0exp i[q · r j(t)] (4.5)
in cui l’apice indica che la somma ´e estesa solo alle particelle all’in-terno del volume illuminato. Sostituendo tale espressione nelle (3.1),(3.2) si ottiene che Iif2(t) e Iif1(t) sono rispettivamente proporzionali a:
F2(q, t) =<| ψ∗(q, t) |2| ψ(q, t) |2> (4.6) F1(q, t) =< ψ∗(q, t)ψ(q, t) > (4.7) dove: ψ(q, t) ≡ N X j=1 0exp iq · r j(t) (4.8)
Poich´e la somma ´e estesa alle sole particelle all’interno del volume illuminato, scrivendo la densit´a di particelle come ρ(r, t) = ρ0+δρ(r, t),
dove δρ ´e la fluttuazione della densit´a numerica, si pu´o scrivere: ψ(q, t) =
Z V d
3r δρ(r, t) exp iq · r = δρ(q, t) (4.9)
ovvero la ψ(q,t) ´e uguale alla trasformata di Fourier della flut-tuazione della densit´a numerica.
Al fine di considerare le sole particelle all’interno del volume illuminato introduciamo il parametro: bj(t) = ( 1 j∈V 0 j /∈V da cui: ψ(q, t) = N X j=1 bj(t) exp iq · rj(t) (4.10)
la somma ora corre su tutte le particelle, e non solo su quelle all’interno del volume illuminato.
4.2
Soluzioni diluite
Le macromolecole in soluzione hanno una polarizzabilit´a molto pi´u grande di quella delle molecole del solvente, e rispetto a queste si muovono molto pi´u lentamente. Questo comporta che le macromolecole
4.2. SOLUZIONI DILUITE 35
sono dei centri diffusori pi´u efficaci rispetto alle molecole del solvente, e che il loro moto domina l’andamento di F1 e di F2 a tempi lunghi.
Si pu´o quindi effettuare la somma della (4.10) solo sulle N macro-molecole in soluzione, considerando rj(t) la posizione del centro di
mas-sa della particella j al tempo t.
Nel caso di soluzioni molto diluite si considerano i moti delle particelle statisticamente indipendenti 1 gli uni dagli altri, si pu´o allora scrivere:
F1(q, t) =< N X j=1
bj(0)bj(t) exp iq · [rj(t) − rj(0)] > (4.11)
4.2.1 Funzione di correlazione eterodina
Facendo riferimento alla (4.11), cerchiamo di quantificare le scale di tempo che determinano il decadimento della funzione F1, prendendo in
esame i fattori in essa contenuti. Il termine bj(t)bj(0) passa dal
valo-re 1 al valovalo-re 0 come la particella j abbandona il volume illuminato, questo avviene in un tempo pari al tempo caratteristico del moto dif-fusivo τ = L2
D, dove D ´e il coefficiente di diffusione delle particelle. Il
termine esponenziale si allontana invece dal valore unitario quando lo spostamento rj(t) − rj(0) diventa confrontabile con la lunghezza q−1,
ci´o avviene in un tempo τete = (q2D)−1. Confrontando le due scale di
tempo si ottiene:
τ τete
= (qL)2 ∼ 106
dove sono stati considerati i valori tipici di L e q per un esperimento di diffusione di luce.
Poich´e il termine bj(t)bj(0) varia molto pi´u lentamente rispetto
all’e-sponenziale si possono fattorizzare i due termini e ottenere:
F1(q, t) =< N > Fs(q, t) (4.12)
dove la
Fs(q, t) ≡< exp iq · [rj(t) − rj(0)] > (4.13)
´e la trasformata di Fourier della distribuzione di probabilit´a Gs(R,t)
di una particella che compie uno spostamento di R nel tempo t: Gs(R, t) =< δ(R − (ri(t) − ri(0))) >
1Se due variabili sono statisticamente indipendenti: <xy>=<x><y>, poich´e < exp iq ·
infatti:
Z
d3R exp iq·R < δ(R−[rj(t)−rj(0)]) >=< exp iq·(rj(t)−rj(0)) >= Fs(q, t)
Utilizzando la teoria della diffusione cerchiamo un’espressione per Gs e per Fs. Al tempo zero si ha: Gs(R,0)=δ(R) e Fs(q,0)=1,
pos-siamo quindi considerare Gs(R,t) come la probabilit´a di trovare una
particella in un intorno d3(R) del punto R al tempo t, considerando
che inizialmente la particella era in un intorno dell’origine. Il moto di diffusione della particella sar´a un random walk, quindi ci si attende che la Gs sia soluzione dell’equazione di diffusione:
∂
∂tGs(R, t) = D∇
2G
s(R, t) (4.14)
trasformando secondo Fourier si ottiene: ∂
∂tFs(q, t) = −Dq
2F
s(q, t) (4.15)
considerando le condizioni iniziali si ha:
Fs(q, t) = exp −q2Dt = exp −t/τete (4.16)
da cui:
F1(q, t) =< N > exp −t/τete (4.17)
Dunque la funzione di correlazione eterodina ha un decadimento esponenziale con costante di tempo τete = (q2D)−1, che rappresenta la
scala di tempo su cui le particelle si spostano di una lunghezza pari a q−1. Secondo la teoria del moto Browniano il coefficiente di diffusione
per una soluzione diluita di macromolecole di forma sferica ´e:
D = kBT 6πηa
dove η ´e la viscosit´a del solvente e a il raggio delle macromolecole. Se vale l’approssimazione gaussiana allora la funzione di correlazione omodina ´e:
4.2. SOLUZIONI DILUITE 37
4.2.2 Funzione di correlazione omodina
La funzione di correlazione omodina per soluzioni diluite di macro-molecole, risulta essere dalle (4.10),(4.6):
F2(q, t) =< N X j,k,l,m=1 bj(0)bk(0)bl(t)bm(t) exp iq·[rk(0)−rj(0)+rl(t)−rm(t)] > (4.19) Per soluzioni diluite questa espressione ´e notevolmente semplificata:
F2(q, t) =< N X j,l=1 b2j(0)b2l(t) > + < N X j6=k=1 bj(0)bk(0) >| Fs(q, t) |2 (4.20) poich´e b2 l(t) = bl(t) e P lbl(t) = N(t), < N X j,l=1 b2 j(0)b2l(t) >=< N X j,l=1 bj(0)bl(t) >=< N(0)N(t) > analogamente N X j6=k=1 < bj(0)bk(0) >=< N (N − 1) > quindi si ottiene: F2(q, t) =< N (0)N(t) > + < N (N − 1) >| Fs(q, t) |2 (4.21) Esprimendo N(t) = N(0) + δN (t), si ha: < N (0)N(t) >=< N >2 + < δN(0)δN (t) >
Dall’ensemble grancanonico si trova che la probabilit´a PN di trovare
N particelle nel volume illuminato V non interagenti, ha una dis-tribuzione poissoniana:
PN =
< N >N
N! exp − < N > questa distribuzione gode delle seguenti propriet´a: 1. < N2 >=< N >2 + < N >
3. < N (N − 1) >=< N2 > − < N >=< N >2
che semplificano la (4.21):
F2(q, t) =< N >2 [1+ | Fs(q, t) |2]+ < δN(0)δN (t) > (4.22)
il primo termine di tale espressione ´e il risultato gi´a ottenuto in approssimazione gaussiana, vedi eq.(4.18), mentre il secondo rappre-senta la deviazione da tale approssimazione. Risulta che F2 decade
in due tempi: il primo tempo ´e dell’ordine di τomo = τete2 che
carat-terizza il tempo richiesto ad una particella per percorrere la distanza q−1; il secondo ´e il tempo necessario per l’attraversamento del volume
illuminato. si ha allora: F2(q, t) = 2 < N >2 + < N > t=0 < N >2 + < N > τ omo¿t¿τ < N >2 tÀτ
Il termine di correzione all’approssimazione gaussiana ´e dell’ordine di < N−1 > e pu´o quindi essere trascurato per soluzioni abbastanza
concentrate (ma non troppo da far cadere l’indipendenza statistica delle macromolecole).
In approssimazione diffusiva allora:
F2(q, t) =< N >2 [1 + exp −
t τomo
]+ < δN(0)δN (t) > (4.23) con τomo = (2qD)−1. Da un fit della funzione di correlazione ´e quindi
possibile ricavare il coefficiente di diffusione e da questo il raggio delle macromolecole in soluzione.
Capitolo 5
Sistemi contenenti molecole
anisotrope
La polarizzabilit´a di un sistema contenente molecole non sferiche ´e in generale un tensore non diagonale. La funzione di correlazione del campo diffuso da tale sistema dipender´a quindi, per quanto visto nella sezione (2.2), dalla proiezione del tensore di polarizzabilit´a αj della
molecola j, sulle direzioni iniziale e finale della polarizzazione della luce: αjif = ni· αj· nf = (ni)ααjαβ(nf)β
Quindi le componenti del momento di dipolo indotto non saranno in generale parallele al campo applicato:
µα = ααβEβ
Si pu´o tuttavia sempre trovare un sistema di assi in cui ci´o sia vero: tali assi vengono chiamati assi principali, e definiscono un ellissoide di polarizzabilit´a che ha la stessa simmetria della distribuzione di carica. Quando tutti e tre gli assi sono uguali si ha un sistema a simmetria sferica, e quindi con polarizzabilit´a isotropa; quando invece i tre assi sono differenti la polarizzabilit´a ´e anisotropa.
Restringendo il nostro campo d’indagine ai soli sistemi diluiti, e as-sumendo l’indipendenza statistica dei moti molecolari la
Iα if(q, t) = N X j=1 0 < αj if(0)αjif(t) exp iq · [rj(t) − rj(0)] > diviene: Iα if(q, t) = N X j=1 0 < αj if(0)αjif(t) > Fs(q, t) (5.1) 39
da cui, poich´e < αjif(0)αjif(t) > ´e uguale per ogni molecola equiva-lente del sistema, si ha:
Iα
if(q, t) =< N >< αif(0)αif(t) > Fs(q, t) (5.2)
Si nota che la funzione di correlazione < αjif(0)αjif(t) > varia perch´e la molecola cambia orientazione nello spazio, mentre la dipendenza dal moto traslazionale della molecola ´e contenuta nel termine Fs(q,t).
Analizzeremo di seguito il caso particolare di diffusione della luce da parte di molecole a simmetria cilindrica e di molecole lineari.
5.1
Scattering di molecole a simmetria cilindrica
Assumendo che nel sistema di riferimento della molecola il tensore di polarizzabilit´a abbia soli elementi diagonali αk lungo l’asse principale e
α⊥lungo gli assi ad esso perpendicolari, occorre trovare la relazione
geo-metrica che permette di esprimere tali componenti nel sistema di riferi-mento del laboratorio, al fine di calcolare la quantit´a < αifj (0)αjif(t) >. Scegliendo la geometria II introdotta nel paragrafo 2.3 fissiamo il vet-tore kf lungo l’asse x del sistema di riferimento fisso nel laboratorio,
mentre il vettore ki ´e contenuto nel piano zy; selezioniamo le
compo-nenti del campo diffuso tali che ni = nf = ˆz che danno IV Vα (q,t) e
ni = ˆz, nf = ˆy che danno IV Hα (q,t):
Iα
V V(q, t) =< N >< αzz(0)αzz(t) > Fs(q, t) (5.3)
Iα
V H(q, t) =< N >< αyz(0)αyz(t) > Fs(q, t) (5.4)
Possiamo interpretare la componente αzz(t) nel sistema del
labora-torio come la proiezione del momento di dipolo indotto da un campo unitario diretto lungo l’asse z sull’asse z stesso, infatti:
µz = ˆz · µ = ˆz · α · ˆz
analogamente per αyz(t).
Prendiamo l’asse principale x0 della molecola lungo la direzione θ
rispet-to all’asse z (nel piano zy) del sistema del laborarispet-torio, e gli assi y0 e z0
tali che il primo formi un angolo (π
2 − θ) rispetto all’asse z e il secondo
sia ortogonale ad esso.
Poich´e la molecola ´e simmetrica per rotazioni rispetto all’asse x0,
queste non influenzeranno lo spettro, pertanto le ignoriamo. Si ha che le proiezioni di ˆz e ˆy lungo x0y0z0 danno:
5.1. SCATTERING DI MOLECOLE A SIMMETRIA CILINDRICA 41
Figura 5.1: Gli assi XYZ individuano il sistema fisso nel laboratorio, mentre X’Y’Z’ il di riferimento ella molecola
ˆ z = cos θ sin θ 0 e ˆ y = sin θ sin φ − cos θ sin φ − cos φ da cui: αzz = (cos θ, sin θ , 0) αk 0 0 0 α⊥ 0 0 0 α⊥ cos θ sin θ 0 = αkcos2θ+α⊥sin2θ e
αyz = (sin θ sin φ, − cos θ sin φ, − cos φ) αk 0 0 0 α⊥ 0 0 0 α⊥ cos θ sin θ 0
= (αk−α⊥) sin θ cos θ sin φ
Si possono esprimere queste componenti in termini di armoniche sferiche del secondo ordine1 ottenendo:
1 Y2,0(θ, φ) = r 5 16π(3 cos 2 θ − 1) Y2,±1(θ, φ) = ∓ r 15
αzz = α + ( 16π 45 ) 1 2βY2,0(θ, φ) (5.5) αyz = i µ2π 15 ¶1 2 β[Y2,1(θ, φ) + Y2,−1(θ, φ)] (5.6)
dove sono stati introdotti i parametri α e β detti rispettivamente parte isotropa e parte anisotropa del tensore di polarizzabilit´a:
α ≡ 1
3(αk+ 2α⊥)
β ≡ (αk− α⊥) (5.7)
il parametro α ´e pari a un terzo della traccia del tensore di polar-izzabilit´a, e pertanto ha lo stesso valore in ogni sistema di riferimento, mentre il parametro β ´e una misura dell’anisotropia ottica del sistema, per molecole sferiche si ha αk = α⊥, quindi β=0.
Sostituendo quanto trovato nelle (5.3) si ottiene:
Iα V V = < N > [α2Fs(q, t) + 16π 45 β 2F(2) 2,0(t)(q, t)Fs(q, t)] (5.8) Iα V H(q, t) = < N > 2π 15β 2[F(2) 1,1(t) + F1,−1(2) (t) + F−1,1(2) (t) + F−1,−1(2) (t)]Fs(q, t)(5.9) dove Fm,m(l) 0 ≡< Ylm∗0(θ(0)φ(0))Ylm(θ(t)φ(t)) > (5.10)
´e una funzione di correlazione che riflette come gli angoli θ(t) e φ(t), che rappresentano l’orientazione dell’asse di simmetria della molecola, cambiano nel tempo.
5.2
Diffusione rotazionale di molecole lineari
Schematizzando la molecola lineare come una bacchetta rigida, ne speci-fichiamo la posizione attraverso un vettore unitario u. Il moto della bacchetta pu´o essere allora rappresentato da un punto sulla superficie della sfera di raggio u, che, in virt´u delle continue collisioni tra molecole, eseguir´a sulla superficie della sfera un random walk. Descriviamo tale diffusione rotazionale seguendo il modello di Debye, assumendo che le
5.2. DIFFUSIONE ROTAZIONALE DI MOLECOLE LINEARI 43
collisioni all’interno del liquido siano cos´ı frequenti, che la molecola pu´o ruotare di soli angoli piccoli prima di subire una nuova collisione che le dia un’orientazione diversa.
Considerando poi che l’insieme di molecole diffondano semplicemente sulla superficie della sfera, scriviamo l’equazione che governa tale moto:
∂c(r, t)
∂t = D∇
2c(r, t) (5.11)
dove c(r,t) ´e la densit´a di bacchette nel punto r=u sulla superficie della sfera unitaria al tempo t.
Esprimendo tale equazione in coordinate polari a | r |=1 fissato, si ottiene l’equazione di diffusione rotazionale:
∂c(r, t) ∂t = Θ 1 sin2θ[sin θ ∂ ∂θ(sin θ ∂ ∂θ) + ∂2 ∂φ2]c(r, t) (5.12)
dove Θ ´e chiamato coefficiente di diffusione rotazionale.
Nell’equazione (5.12) si riconosce l’espressione dell’operatore momento angolare orbitale adimensionale,( ˆI2Y
lm(u) = l(l+1)Ylm(u) e ˆIzYlm(u) =
mlYlm(u) con l=0,1,2,....∞ e ml=-l,...,0,...+l) pertanto si pu´o scrivere:
∂c(q, t)
∂t = −Θ ˆI
2c(u, t) (5.13)
la cui soluzione ´e:
c(u, t) = exp(−tΘ ˆI2)c(u, 0) (5.14)
dove ˆI2 agisce solo su u. Imponendo le condizioni iniziali e
uti-lizzando la relazione di chiusura valida per le armoniche sferiche si ha: c(u, 0) = δ(u − u0) = X lm Ylm(u0)Ylm∗ (u) da cui: c(u, t) = exp(−tΘ ˆI2)X lm Ylm(u0)Ylm∗ (u) (5.15)
che per le propriet´a delle armoniche sferiche diviene: c(u, t) =X
lm
exp −l(l + 1)Θt Y∗
lm(u)Ylm(u0) (5.16)
Si pu´o interpretare tale soluzione dell’equazione di diffusione come una probabilit´a di transizione e si definisce la densit´a di probabilit´a per
una bacchetta di avere un’orientazione u al tempo t, data l’orientazione iniziale u0 come: Ks(u, t|u0, 0) = X lm Ylm(u0)Ylm∗ (u) exp −l(l + 1)Θt (5.17)
La funzione di correlazione cercata nell’esperimento di diffusione di luce sar´a allora della forma:
< Y∗ l0m0(u(0))Ylm(u(t)) >= Z d2u 0 Z d2u Y
lm(u)Gs(u, t; u0, 0)Yl∗0m0(u0)
(5.18) dove Gs(u,t;u0,0)d2u0d2u rappresenta la probabilit´a di trovare una
bacchetta con orientazione u0 in d2u0 inizialmente, e con orientazione u
in d2u al tempo t. Si pu´o esprimere G
sin funzione di Kse della funzione
di distribuzione delle probabilit´a dell’orientazione iniziale p(u0):
Gs(u, t; u0, 0) = Ks(u, t; u0, 0)p(u0)
Per un insieme di molecole lineari all’equilibrio ci si attende un’ori-entazione uniforme delle molecole tale che p(u0)=4π1 .
Utilizzando allora la propriet´a di ortonormalit´a delle armoniche sferiche la (5.18) diviene: < Y∗ l0m0(u(0))Ylm(u(t)) >= Fl(t)δl,l0δm,m0 (5.19) dove: Fl(t) = 1 4πexp(−l(l + 1)Θt) (5.20)
La funzione di correlazione (5.10) vale allora: Fmm(2)0 = F2(t)δm,m0 = 1
4πexp −6Θt δm,m0 (5.21) Utilizzando l’equazione (4.16), otteniamo:
Iα V V = < N > {α2+ 4 45β 2exp −q2Θt} exp −q2Dt (5.22) Iα V H = 1 15 < N > β 2exp −6Θt exp −q2Dt (5.23) E si pu´o esprimere: Iα V V(q, t) = IISOα (q, t) + 4 3I α V H(q, t) (5.24)
dove il primo termine d´a informazioni sulla sola diffusione traslazionale mentre il secondo d´a anche informazione circa il moto rotazionale.
Capitolo 6
Risultati sperimentali
Il nostro studio si propone ora di analizzare la diffusione della luce da parte di un campione colloidale anisotropo.
Il campione utilizzato ´e una soluzione contenente il colloide Laponite: un colloide ´e un fluido contenente un gran numero di piccole particelle solide, le cui dimensioni variano tipicamente da 1 nm a 1000 nm, e sono costituite da aggregati di molecole oppure da macromolecole. Le gran-di gran-dimensioni delle particelle colloidali, rispetto agli atomi dei normali vetri, le rendono visibili al microscopio e fanno s´ı che il loro movimento sia molto pi´u lento.
Le sospensioni di Laponite sono costituite da dischi dalla superficie ca-rica negativamente, di spessore pari a 1nm e di diametro all’incirca pari a 30 nm.
Ci proponiamo di studiare la dinamica durante l’invecchiamento del colloide in soluzione investigando il rallentamento della diffusione ro-tazionale delle molecole nel campione tramite la funzione di corre-lazione g2
V H dipendente dalla IV H introdotta nel paragrafo (5.1).
6.1
L’apparato sperimentale
Per realizzare le misure di fotocorrelazione abbiamo utilizzato come sorgente un fascio di luce monocromatica e polarizzata verticalmente emessa da un laser a He-Ne, dalla potenza di 8-9 mW, e come campione una soluzione di acqua e laponite alla concentrazione del 2% . Il fascio diffuso ´e stato raccolto ad un angolo θ = π
2 e fatto passare attraverso un
polarizzatore in modo da selezionare la sola componente orizzontale, e quindi attraverso un pinhole, per aumentare il contrasto dinamico, per poi incidere sulla superficie del fotomoltiplicatore. Il segnale in uscita da quest’ultimo ´e stato elaborato da un correlatore digitale, interfacciato
ad un PC che permette di acquisire la funzione di correlazione del numero di fotoni raccolti dal rivelatore.
Nel preparare la soluzione di acqua deionizzata e laponite si ´e tenuto conto che questa ´e un’argilla, e come tale tende a trattenere acqua, circa il 16% in peso, si ´e pertanto aggiunta laponite in soluzione fino a raggiungere una percentuale del 2,378 in peso. Abbiamo lasciato in agitazione finch´e la soluzione non ´e diventata chiara, quindi abbiamo filtrato il campione mediante un filtro Millipore 0.45µm.
6.2
L’acquisizione dei dati
Come visto nel paragrafo (5.1) il campo elettrico diffuso da molecole anisotrope (quando ´e polarizzato linearmente nella direzione verticale) ha due componenti: quella polarizzata verticalmente EV V con ampiezza
proporzionale alla polarizzabilit´a media (vedi(5.8)), e quella depolar-izzata nella direzione orizzontale EV H la cui ampiezza ´e proporzionale
all’anisotropia intrinseca β delle particelle (vedi (5.7)); esperimenti di diffusione della luce polarizzata e depolarizzata misurano rispettiva-mente la funzione di correlazione temporale delle intensit´a VV e VH del campo diffuso:
g2 V V(q, t) =< IV V(q, t)IV V(q, 0) IV V(q)2 > g2 V H(q, t) =< IV H(q, t)IV H(q, 0) IV H(q)2 > .
Dall’equazione (5.24) ricaviamo che il contributo del moto rotazionale nella funzione di correlazione g2
V V ´e proporzionale a 43 IV H
IV V.
Sperimen-talmente abbiamo osservato che il numero di fotoni rivelati dal foto-moltiplicatore quando la luce diffusa ´e depolarizzata risulta inferiore rispetto al caso di luce polarizzata di circa un fattore cento, ´e quindi questo l’ordine di grandezza del rapporto IV H
IV V. Pertanto la dinamica
della funzione g(2)V V riflette principalmente la diffusione traslazionale, mentre la g(2)V H ´e determinata sia dal moto traslazionale che rotazionale delle molecole.
Abbiamo registrato la funzione di correlazione g(2)V H per diversi tempi di invecchiamento del campione: il fenomeno per cui alcune variabili del sistema dipendono dal tempo di attesa tw, pari al tempo trascorso
dalla preparazione del campione, ´e detto invecchiamento. L’invecchia-mento ´e legato al rallentaL’invecchia-mento della dinamica del sistema in esame, nel nostro caso dipendente dalla concentrazione delle molecole di laponite in soluzione: aumentando infatti il numero di molecole in soluzione il
6.2. L’ACQUISIZIONE DEI DATI 47
moto diffusivo di queste rallenta al diminuire del volume libero a loro disposizione. Uno studio recente [3] ha mostrato come il processo di invecchiamento evolve differentemente per alte e basse concentrazioni. Al fine di minimizzare il rumore presente nelle singole acquisizioni, dovuto al fatto che il numero di conteggi rilevati dal fotomoltiplica-tore risultava estremamente basso (circa 60 al secondo) a causa della scarsa intensit´a della luce diffusa con polarizzazione orizzontale, abbi-amo effettuato la somma dei dati registrati nell’arco delle 24 ore dal correlatore.
Le funzioni di correlazione cos´ı ottenute per diversi ”intervalli” d’in-vecchiamento sono riportate nelle figure seguenti.
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10
0,8 1,0 1,2 1,4 1,6 1,8 2,0 primo giorno g 2 V H t (s)
Figura 6.1: Funzione di correlazione del campo diffuso in polarizzazione VH, primo giorno di acquisizione dei dati
Per tempi di invecchiamento piccoli, i due tempi di rilassamento del sistema sono dello stesso ordine di grandezza e quindi difficilmente distinguibili. All’aumentare dell’invecchiamento, il secondo tempo di rilassamento cresce, come si nota dai seguenti fit:
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10
1,0 1,2 1,4 1,6 1,8 2,0 secondo giorno g 2 V H t (s)
Figura 6.2: Funzione di correlazione del campo diffuso in polarizzazione VH, secondo giorno di acquisizione dei dati
6.2. L’ACQUISIZIONE DEI DATI 49
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10
1,0 1,2 1,4 1,6 1,8 2,0 terzo giorno g 2 V H tempi (s)
Figura 6.3: Funzione di correlazione del campo diffuso in polarizzazione VH, terzo giorno di acquisizione dei dati
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10
1,0 1,2 1,4 1,6 1,8 2,0 quarto giorno g2 V H t (s)
Figura 6.4: Funzione di correlazione del campo diffuso in polarizzazione VH, quarto giorno di acquisizione dei dati
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10 0,8 1,0 1,2 1,4 1,6 1,8 2,0 quinto giorno g 2 V H t (s)
Figura 6.5: Funzione di correlazione del campo diffuso in polarizzazione VH, quinto giorno di acquisizione dei dati
La crescita del secondo tempo di rilassamento all’aumentare del-l’invecchiamento ´e tipica dei sistemi non stazionari: i moti cooperativi delle molecole che interessano le modificazioni della struttura micro-scopica (cui si faceva riferimento nella sezione (1)), coinvolgono tempi di rilassamento molto pi´u lunghi di quelli osservabili in laboratorio; pertanto tali sistemi sono per noi ”fuori dall’equilibrio” .
6.3
Conclusioni
Si riscontra dalle figure 6.1 - 6.5 un decadimento a due tempi della fun-zione di correlafun-zione che risulta via via pi´u evidente con l’aumentare dell’invecchiamento e che segue i due tempi di rilassamento dei gradi di libert´a traslazionali e rotazionali del sistema: mentre il primo tempo di rilassamento appare pressoch´e indipendente dall’invecchiamento, il secondo tempo ne risulta fortemente dipendente e cresce al crescere di tw.
Ipotizzando, come ´e stato fatto nello studio [3], che la funzione di corre-lazione decada in due tempi, di cui uno stretchato, abbiamo effettuato i fit dei dati sperimentali con la seguente funzione:
6.3. CONCLUSIONI 51 g2(q, t) = 1 + b[a exp −t τ1 + (1 − a) exp[−t τ2 ]β]2 (6.1)
dove τ1 ´e il tempo caratteristico associato alla dinamica veloce, τ2
quello associato alla dinamica lenta e β il parametro di stretching, il parametro a descrive infine il peso relativo dei due processi.
Riportiamo nella figura 6.6 i fit che descrivono l’andamento della fun-zione di correlafun-zione in funfun-zione del tempo all’aumentare del tempo di invecchiamento.
1E-6 1E-5 1E-4 1E-3 0,01 0,1 1 10
1,0 1,2 1,4 1,6 1,8 2,0 g 2 t (s) primo giorno secondo giorno terzo giorno quarto giorno quinto giorno
Figura 6.6: Fit della funzione di correlazione VH mediante la (6.1) a tem-pi di invecchiamento crescenti: all’aumentare dell’invecchiamento τ1 rimane costante, mentre τ2 cresce indefinitivamente, come ci si attende per un sistema non stazionario.
Si osserva che, con l’aumentare dell’invecchiamento, la dinamica lenta risulta rallentare, il secondo tempo di rilassamento τ2 ´e una
fun-zione crescente del tempo di invecchiamento; riportiamo l’andamento di τ2 in funzione di tw nella figura 6.7.