CARATTERIZZAZIONE DI MATERIALI POROSI
MEDIANTE ADSORBIMENTO DI GAS:
EVOLUZIONE DEL COMPOSITO NITRURO DI BORO - GRAFITE
OTTENUTO MEDIANTE HIGH ENERGY BALL MILLING
LAURA SILVESTRI, MARZIA PENTIMALLI, FRANCO PADELLA ENEA – Unità Tecnica Tecnologie dei Materiali
Laboratorio di Chimica e Tecnologie dei Materiali Centro Ricerche Casaccia, Roma
AGENZIA NAZIONALE PER LE NUOVE TECNOLOGIE, LʼENERGIA E LO SVILUPPO ECONOMICO SOSTENIBILE
CARATTERIZZAZIONE DI MATERIALI POROSI
MEDIANTE ADSORBIMENTO DI GAS:
EVOLUZIONE DEL COMPOSITO NITRURO DI BORO - GRAFITE
OTTENUTO MEDIANTE HIGH ENERGY BALL MILLING
LAURA SILVESTRI, MARZIA PENTIMALLI, FRANCO PADELLA ENEA – Unità Tecnica Tecnologie dei Materiali
Laboratorio di Chimica e Tecnologie dei Materiali Centro Ricerche Casaccia, Roma
I contenuti tecnico-scientifici dei rapporti tecnici dell'ENEA rispecchiano l'opinione degli autori e non necessariamente quella dell'Agenzia.
The technical and scientific contents of these reports express the opinion of the authors but not necessarily the opinion of ENEA.
I Rapporti tecnici sono scaricabili in formato pdf dal sito web ENEA alla pagina http://www.enea.it/it/produzione-scientifica/rapporti-tecnici
CARATTERIZZAZIONE DI MATERIALI POROSI MEDIANTE ADSORBIMENTO DI GAS: EVOLUZIONE DEL COMPOSITO NITRURO DI BORO - GRAFITE OTTENUTO MEDIANTE HIGH ENERGY BALL MILLING
LAURA SILVESTRI, MARZIA PENTIMALLI, FRANCO PADELLA
Riassunto
I materiali porosi sono noti per le loro proprietà dovute alla capacità di adsorbimento di specie liquide o gassose. Lo sviluppo di nuovi composti a elevatissima superficie specifica ha dato avvio a un rinnovato interesse scientifico e tecnologico dovuto alla possibilità di utilizzare solidi porosi quali substrati per l'accumulo di gas in fase condensata. In particolare, lo stoccaggio sicuro d’idrogeno in materiali adsorbenti in particolare, risulta cruciale per lo sviluppo delle tecnologie per la sostenibilità energetica. Il presente rapporto tecnico è costituito da due parti: nella parte A si danno alcuni cenni teorici di base sulle metodologie di caratterizzazione mediante adsorbimento di azoto; nella parte B è riportato lo sviluppo mediante High Energy Ball Milling e la caratterizzazione di un materiale composito con porosità di tipo a fenditura, costituito da nitruro di boro esagonale e grafite.
Parole chiave: materiali porosi; meccanochimica; nitruro di boro; grafite; fisisorbimento; accumulo di gas.
Abstract
Porous materials are well known for their capacity of adsorbing both liquid and gaseous species. The development of new classes of materials with very high specific surface promoted the interest of the scientific community in their use as solid substrates for gas storage. In particular, the safe hydrogen storage in adsorbing materials constitutes a crucial issue in enabling the use of new technologies for energy sustainability. This technical report is divided in two parts: in part A some theoretical basics are described on gas adsorption fundamentals for the characterization of porous materials; in B on the synthesis by High Energy Ball Milling and the characterization of a boron nitride-graphite composite with slit-like porosities are reported.
INDICE INTRODUZIONE 7 PARTE A. CENNI TEORICI 8 MATERIALI POROSI 9 Le isoterme di adsorbimento 12 Il meccanismo di adsorbimento 15
VALUTAZIONE DELL’AREA SUPERFICIALE MEDIANTE ADSORBIMENTO DI GAS 16
I modelli di adsorbimento 18 La determinazione della porosità 20
PARTE B.
UNA APPLICAZIONE SPERIMENTALE 25
EVOLUZIONE MORFOLOGICA DEL SISTEMA hBN/C SOTTOPOSTO AD HIGH ENERGY BALL MILLING 26
Materiali e metodi 26
Risultati e discussione 27
7
INTRODUZIONE
I materiali porosi sono noti da tempo per le loro proprietà dovute alla capacità di adsorbimento di specie liquide o gassose. Accanto alle più note e tradizionali applicazioni di purificazione, lo sviluppo di nuovi composti a elevatissima superficie specifica ha dato avvio a un rinnovato interesse scientifico e tecnologico dovuto alla possibilità di utilizzare solidi porosi quali substrati per l'accumulo di gas in fase condensata. Tale caratteristica risulta di particolare interesse in applicazioni energetiche. Infatti, un efficace stoccaggio in fase condensata dei gas combustibili ne permetterebbe una gestione non pericolosa, conseguenza dell'accumulo di elevate quantità in condizioni di bassa pressione. Lo stoccaggio sicuro d’idrogeno in materiali adsorbenti, in particolare, potrebbe dare un impulso rilevante allo sviluppo delle tecnologie per la sostenibilità energetica.
É stato calcolato che per ottenere fisisorbimento reversibile d’idrogeno in condizioni non estreme di temperatura e pressione l'energia d’interazione del gas con il substrato deve essere dell'ordine di 300-400 meV/molecola. Tali valori d’interazione, circa 3-4 volte più elevati di quelli sperimentalmente verificati nei normali substrati, sono potenzialmente ottenibili promuovendo l'interazione del gas con pareti doppie, a patto che tali pareti siano poste a circa 7 Å di distanza tra loro1.
Nel presente lavoro, dopo una breve rassegna delle metodologie di caratterizzazione mediante adsorbimento di azoto, è riportato lo sviluppo di un materiale con porosità di tipo a fenditura, caratterizzato da pareti statisticamente poste alla distanza ottimale per una doppia interazione con l'idrogeno molecolare. Due materiali a struttura esagonale simile (nitruro di boro esagonale e grafite) sono stati sottoposti a macinazione mediante High Energy Ball Milling. Il materiale sviluppato è stato studiato in termini di evoluzione microstrutturale, di fase e morfologica in funzione del tempo di macinazione. Le caratteristiche superficiali e microporose sono pure state studiate e riportate nel seguito.
8
9
MATERIALI POROSI
La porosità di un materiale è originata dalla presenza di pori: per essere definito poro, una cavità deve essere più profonda che larga. La presenza dei pori può essere intrinseca del materiale o dovuta all’aggregazione di più particelle. La porosità di un solido dipende da diversi fattori, tra cui: la densità, dimensioni e distribuzione delle particelle, la loro forma e il modo in cui si legano tra di loro2. Lo stato e la struttura dei pori dipendono principalmente dall’origine del poro (Figura 1). I pori comunicanti con la superficie esterna sono chiamati pori aperti (B, T, I in fig.1) e sono accessibili alle molecole o agli ioni,; quelli, invece non comunicanti con l’esterno sono detti pori chiusi (C in fig.1). I pori chiusi non con contribuiscono a determinare le proprietà di adsorbimento e permeabilità del materiale solido poroso, ma ne influenzano le proprietà meccaniche. Una ulteriore distinzione può essere fatta tra i blind pores (B in fig.1) e i pori interconnessi (I in fig.1) e quelli che si aprono da entrambi i lati di una particella, i through pores (T in fig.1).
Figura 1. Rappresentazione schematica della struttura dei pori
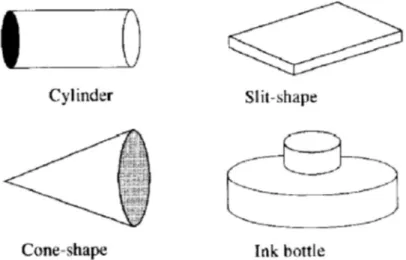
Per quanto riguarda la struttura dei pori, grande interesse riveste la caratterizzazione della geometria dei pori (figura 2). L’analisi dell’isteresi dell’isoterma di adsorbimento e l’osservazione al microscopio elettronico ha permesso di classificare la geometria dei pori in cilindrica (che può essere aperta da un solo lato o da entrambi), conica, a fenditura e ink bottle (a boccetta d’inchiostro)3.
10
Un’ulteriore classificazione dei pori è data dall’International Union of Pure and Applied Chemistry (IUPAC) che li classifica in base alle dimensioni e definisce: i micropori con larghezza al di sotto di 2 nm, i mesopori con larghezza compresa tra 2 e 50 nm e i macropori con larghezza maggiore di 50 nm. I micropori vengono, ulteriormente, classificati in super-micropori (tra 1 e 2 nm) e ultra-micropori (minore di 1 nm). Si noti che nel caso di pori cilindrici, la larghezza è data dal diametro del poro, mentre nel caso dei pori a fenditura, la larghezza è definita come distanza tra le pareti opposte. Esempi tipici di materiali porosi sono le zeoliti ed i carboni attivi, brevemente descritti nel seguito.
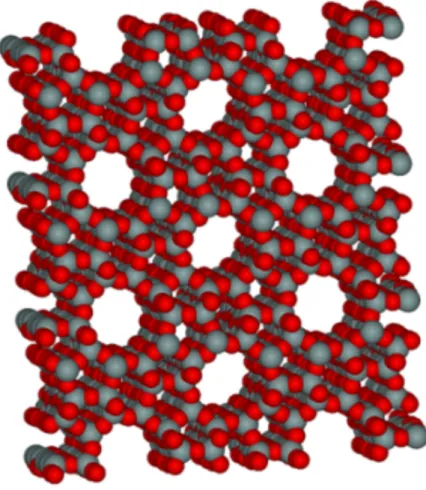
Le zeoliti4 sono una famiglia di minerali naturali o di sintesi con una struttura regolare e microporosa caratterizzati da un’enorme quantità di volumi vuoti interni ai cristalli. Nelle zeoliti i pori derivano dalla loro struttura cristallina intrinseca (figura 3). La struttura primaria delle unità delle zeoliti è costituta da tetraedri di silicio o di alluminio. Queste unità sono assemblate in una struttura secondaria poliedrica a cubo, prisma esagonale, ottaedro o ottaedro troncato. L’assemblaggio delle unità secondarie produce strutture cristalline regolari tridimensionali caratterizzate da elevata porosità.
Figura 3. Struttura molecolare microporosa della zeolite ZSM-5
Le zeoliti esibiscono importanti proprietà di adsorbimento in funzione della loro chimica superficiale. La superficie della struttura è costituita essenzialmente da atomi di ossigeno, mentre gli atomi di Si o Al si trovano posizionati nei piani interni della struttura. L’interazione della struttura zeolitica con un gas è essenzialmente dovuta alle interazioni di van der Waals tra le molecole di ossigeno e quelle del gas.
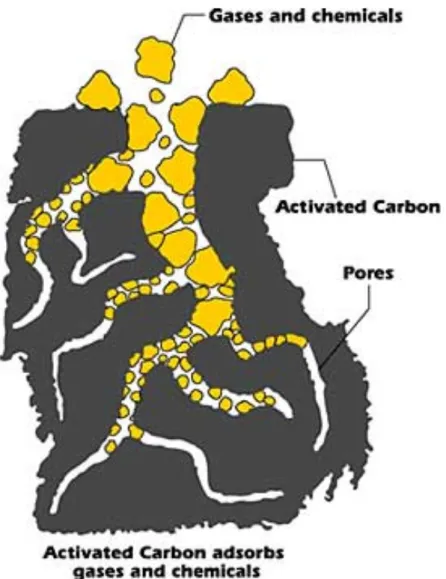
I carboni attivi4 sono materiali, contenenti principalmente carbonio sotto forma di micro-cristalli di grafite, trattati in modo da ottenere una struttura porosa con una vasta area superficiale interna. I
11
processi di produzione coinvolgono fondamentalmente i seguenti steps: la preparazione del materiale grezzo, la carbonizzazione a basse temperature e l’attivazione. I materiali di partenza per i carboni attivi sono costituiti da differenti tipi di materiali carbonacei. A partire dai pori inizialmente presenti nel materiale grezzo, attraverso diversi processi di attivazione, si fa in modo di creare ulteriore porosità secondo la particolare distribuzione desiderata. Due sono i metodi principali per l’attivazione, di tipo gas-assistita e di tipo chimico. L’attivazione tramite gas coinvolge la carbonizzazione a 400°-500° C per eliminare le sostanze volatili, e seguire una parziale gassificazione a 800-1000° C che sviluppa la porosità e aumenta l’area superficiale. Nel caso di attivazione chimica si utilizzano additivi inorganici in grado di degradare e deidratare i materiali cellulosici e, simultaneamente, prevenire il restringimento delle cavità durante la carbonizzazione. I carboni attivi sono caratterizzati da un’ampia area superficiale che può andare da valori dell’ordine di 300 fino a circa 4000 m2/g. Per i carboni attivi si trova di solito una distribuzione delle dimensioni dei pori di tipo polimodale (figura 4). I pori più larghi sono detti pori di trasporto o di caricamento, mentre, quelli più piccoli, senza sbocco, sono chiamati pori di adsorbimento.
Figura 4. Struttura dei pori in un tipico carbone attivato
La caratterizzazione delle proprietà in termini di area superficiale specifica e porosità può essere convenientemente realizzata mediante le misure di adsorbimento di gas. Questa metodologia è specificatamente applicata al calcolo dell’area superficiale, del volume poroso, e della distribuzione della dimensione dei pori (PSD) di materiali porosi.
12
Generalmente, viene utilizzato il processo di fisisorbimento in quanto è un processo reversibile con bassa specificità. Infatti, le molecole del gas adsorbente possono essere considerate delle vere e proprie sonde. Si tratta, comunque, di un metodo indiretto in quanto, per l’estrazione delle informazioni riguardanti le caratteristiche delle superfici del campione analizzato, vengono applicati alcuni modelli teorici ed empirici. Inoltre, la complessità fisica di molte superfici solide può rendere difficile ottenere una valutazione generale del significato fisico delle quantità derivate5.
Con il termine adsorbimento, s’intende il meccanismo chimico-fisico mediante il quale avviene l’arricchimento di una o più specie all’interfase tra due fasi distinte. Nel caso dell’adsorbimento di gas, le fasi sono chiamate: adsorbente (la fase solida su cui avviene l’adsorbimento di gas, il nostro materiale poroso) e adsorbato (il gas adsorbito sull’adsorbente). Secondo i tipi d’interazione che s’instaurano tra le molecole di adsorbato e adsorbente, si parla di: fisisorbimento o chemisorbimento.
Nel caso del fisisorbimento o adsorbimento fisico, a cui ci riferiremo d’ora in avanti, le forze d’interazione tra le molecole di gas e l’adsorbente sono dispersive, tipo van der Waals, e forze repulsive a corto raggio. Il fisisorbimento ha un basso grado di specificità. Inoltre, è un fenomeno di tipo esotermico e le energie in gioco sono dello stesso ordine di grandezza delle energie di condensazione del gas adsorbente. Il gas può adsorbirsi sull’adsorbente anche in forma di multi-strato, soprattutto ad alte pressioni relative, le molecole adsorbite non perdono la loro identità e desorbendo tornano alla loro forma originale.
In particolare, dato un materiale poroso adsorbente e fissato un gas sonda, è possibile determinato sperimentalmente l’isoterma di adsorbimento caratteristica di quella coppia solido-gas.
Le isoterme di adsorbimento sono delle curve che esprimono la quantità di gas adsorbita in funzione della pressione, a temperatura costante. Nel seguente paragrafo si mostrano le grandezze chimico-fisiche in gioco per la determinazione delle isoterme di adsorbimento e si definiscono i differenti tipi di isoterme in base alla classificazione IUPAC.
Le isoterme di adsorbimento
La quantità di gas adsorbito, na , da una massa, ms , di un solido dipende dalla pressione di equilibrio, p, dalla temperatura, T, e dalla natura del sistema gas-solido6:
na/ms= f(p,T,sistema) (1)
Per un dato gas adsorbito su un particolare solido a temperatura costante, si ha:
na/ms= f(p)T (2)
e se il gas si trova al di sotto della sua temperatura critica, è possibile scrivere:
na/ms= f(p/p0)T (3)
dove la pressione standard p0è uguale alla pressione di saturazione dell’adsorbato alla temperatura
T. Le equazioni 2 e 3 rappresentano l’isoterma di adsorbimento che è la relazione tra la quantità
adsorbita dalla massa unitaria di solido e la pressione di equilibrio (o pressione relativa), ad una certa temperatura.
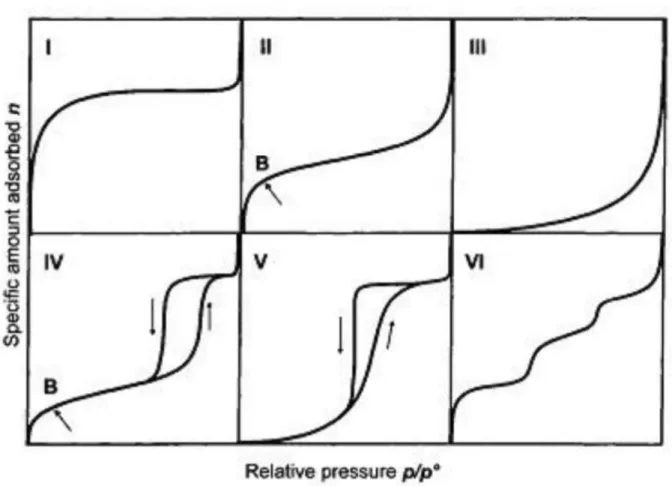
Le isoterme poste in grafico, misurate per un’ampia varietà di sistemi gas-solido, sono caratterizzate da una grande diversità di forme. Comunque, la maggior parte di queste isoterme risultanti da processi di adsorbimento fisico può essere raggruppata all’interno di sei classi in base alla classificazione IUPAC (figura 5)7.
13
Figura 5. Classificazione delle isoterme di adsorbimento secondo la nomenclatura IUPAC - L’isoterma di tipo I è caratteristica dei materiali totalmente microporosi. A pressioni relative
molto basse si osserva un aumento rapido, quasi verticale, delle moli adsorbite; a pressioni più elevate presenta un “ginocchio” oltre il quale si ha un plateau indicante che il materiale non sta adsorbendo più nonostante l’aumento di pressione; infine, a pressioni prossime a quelle di saturazione, l’isoterma tende a divergere. Raggiunta la pressione di saturazione, comincia la liquefazione del gas e dunque l’aumento repentino della curva è dovuto ad un aumento delle moli sottratte al gas che non passano allo stato di adsorbite ma a quello di liquido.
- L’isoterma di tipo II è associata ad un adsorbimento che avviene su un solido macroporoso o
non poroso. Indica la formazione di uno strato di materiale adsorbito il cui spessore aumenta progressivamente con la pressione relativa fino a valori di p/p0tendenti a 1. Il ginocchio di questo tipo di isoterma (punto B in figura 5) indica la pressione a cui si forma il mono-strato adsorbito e l’inizio della formazione del multistrato molecolare. L’ordinata del punto B dá una stima della quantità di adsorbato richiesto per coprire una massa unitaria di superficie di solido con uno strato completo monomolecolare (monolayer capacity).
- L’isoterma di tipo III ha una forma convessa e non presenta un ginocchio. Questa
caratteristica è rappresentativa di deboli interazioni adsorbente-adsorbato.
- L’isoterma di tipo IV è caratterizzata da un andamento iniziale simile a quella di tipo II e
tende a stabilizzarsi ad alti valori di pressione relativa. Esibisce un’isteresi; cioè, facendo il ciclo inverso, partendo quindi da pressioni relative prossime all’unità e decrescendo fino alle pressioni più basse (ciclo di desorbimento), la curva non ricalca il tracciato di adsorbimento ma, fissato un certo valore di p/p0, il sistema raggiunge l’equilibrio avendo sottratto alla fase gassosa un numero di moli maggiore del corrispondente punto nel ciclo di adsorbimento. Una tale tipologia di curva è tipica dei materiali mesoporosi. Durante il ciclo di adsorbimento, il gas tende inizialmente a riempire il monostrato, superato il punto B,
14
iniziano a riempirsi gli strati sovrastanti e ad una certa pressione (minore della pressione di saturazione p0), il gas adsorbito all’interno dei mesopori non è più stabile in fase vapore e condensa, risulta sull’isoterma come un gradino.
- L’isoterma di tipo V, come la III, è indicativa di deboli interazioni adorbente-adsorbato.
Esibisce un’isteresi che è associata al meccanismo di riempimento e svuotamento dei pori. - L’isoterma di tipo VI si ritrova in sistemi in cui la superficie adsorbente possiede
un’uniformità morfologica ed energetica e non presenta porosità. I gradini indicano rispettivamente gli stadi di riempimento dei singoli strati, mentre i tratti di plateau più o meno netti indicano che, per intervalli di pressioni relative diverse da quelle a cui si osservano i gradini, niente o poco del gas resta adsorbito.
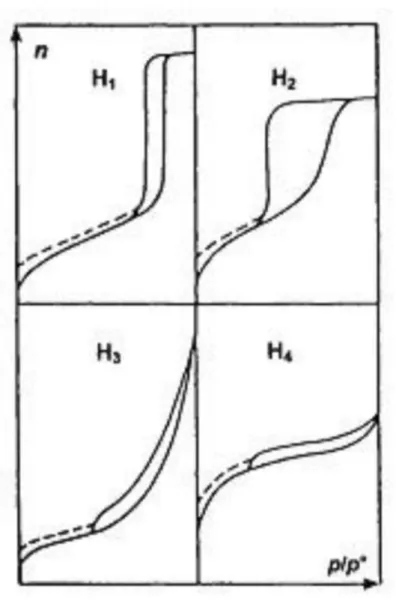
Oltre alla classificazione delle isoterme, esiste anche una classificazione IUPAC delle forme di isteresi (figura 6).
- H1 è attribuita ad una distribuzione di mesopori molto stretta;
- H2 è indice della presenza di mesopori interconnessi di diversa forma e grandezza;
- H3 viene spesso ricondotta alla presenza di pori interstiziali dovuti ad aggregati di particelle a foglietti; e
H4 viene attribuita alla condensazione capillare in pori interstiziali dovuti ad aggregazione di particelle.
15
Il meccanismo di adsorbimento
Da una prima analisi di tipo qualitativo di unaisoterma di adsorbimento ricavata sperimentalmente, si possono già ricavare alcune informazioni sulla natura porosa/non porosa del materiale analizzato. In particolare, confrontando la forma ottenuta con quelle classificate dalla IUPAC è possibile valutare se il materiale è poroso e di che tipo è la porosità (micro-, meso-, o macro-porosità). Ad esempio, le isoterme di tipo I sono tipiche di substrati microporosi, mentre i tipi IV, V e VI sono caratteristici di sistemi che presentano mesopori.
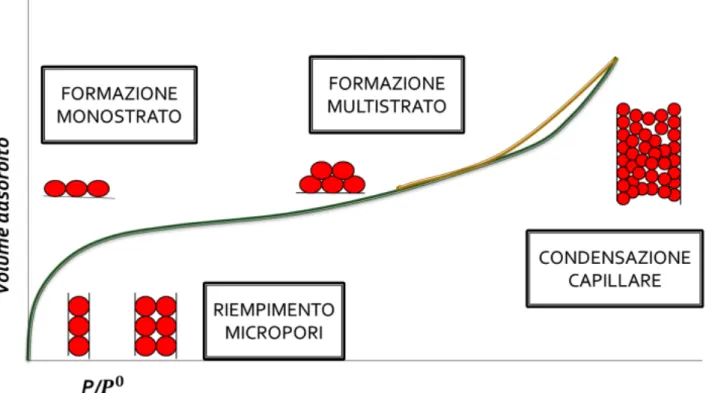
In seconda battuta, tenendo presente il processo di adsorbimento in funzione della pressione, è possibile ricavare ulteriori informazioni sul materiale studiato. Il processo di adsorbimento avviene approssimativamente come segue (figura 7): inizialmente, si ha il riempimento dei micropori che è dominato quasi interamente dalle interazioni tra adsorbato e pareti del poro; segue, a pressioni maggiori, la copertura della superficie esterna, consistente nella formazione del mono-strato o adsorbimento multistrato delle pareti dei mesopori e dei macropori, con condensazione capillare nel caso di mesopori.
Figura 7. Rappresentazione del processo di adsorbimento di un gas su una superficie porosa8.
Ciò vuol dire che, la zona a bassi valori di p/p0, corrispondente al riempimento dei micropori, è utile per ricavare il volume microporoso e il PSD; la zona corrispondente alla formazione del mono-strato, permette di calcolare l’area superficiale e la zona di condensazione capillare (presenza di isteresi sull’isoterma) viene utilizzata per caratterizzare i mesopori (figura 8).
16
Figura 8. Interpretazione isoterma di adsorbimento
VALUTAZIONE DELL’AREA SUPERFICIALE MEDIANTE ADSORBIMENTO DI GAS Il metodo volumetrico per la misura dell’adsorbimento
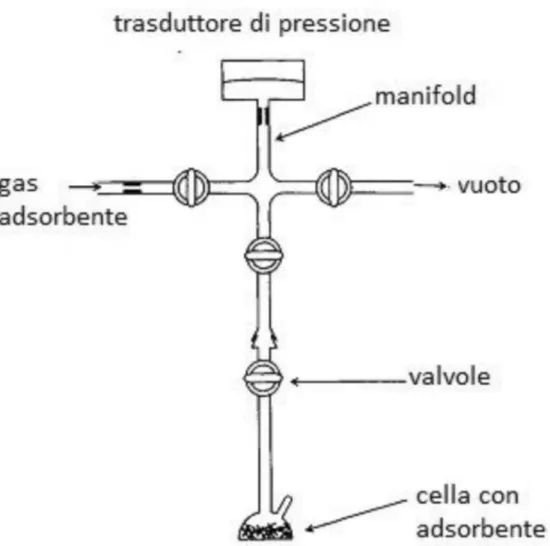
I parametri fisici che possono essere sfruttati per determinare un’isoterma di adsorbimento sono la pressione e la massa. Il metodo volumetrico è basato sulla misura della pressione.
L’apparato volumetrico è costituito da un volume tarato, manifold, a temperatura controllata, collegato ad un trasduttore di pressione. Il manifold è a sua volta connesso ad una pompa da vuoto, alla riserva di gas adsorbente (una bombola) e al campione tramite valvole da vuoto (figura 9). L’analisi viene effettuata tramite una procedura discontinua, o punto a punto:
• Aprendo la valvola di comunicazione tra riserva di gas ed il manifold si riempie lo stesso di una aliquota di gas adsorbente. Si chiude la valvola e si misura la pressione e la temperatura del gas nel manifold;
• Si mette in comunicazione il manifold con la cella contenente il campione, aprendo la valvola. Il gas si espande nel volume della cella ed entra in contatto con il campione. Una parte di esso viene adsorbita sulla superficie dell’analita. Al raggiungimento dell’equilibrio, si chiude la valvola di comunicazione tra il campione e il manifold e si misura la pressione e la temperatura del gas non adsorbito.
Iniziando la misura con la cella del campione a bassa pressione, l’isoterma di adsorbimento si ottiene ripetendo i due passi sopra descritti riempiendo il manifold a pressioni sempre più alte. La scelta delle quantità di gas aggiunte o rimosse è a cura dell’analizzatore e, dipende dalla quantità e dalla capacità di adsorbimento del campione. Oltre al numero di punti sperimentali desiderati, sono a discrezione dell’operatore i tempi di equilibrio e la determinazione delle fluttuazioni di pressione trascurabili per l’acquisizione della misura.
17
Figura 9. Schema di un apparato volumetrico per la determinazione dell’isoterma di adsorbimento
La forma delle isoterme
I parametri che caratterizzano un materiale poroso sono: area superficiale specifica, misurata in
m2/g; il volume microporoso, misurato in cm3/g; il volume dei pori, che rappresenta la somma dei
volumi dei micropori e dei mesopori dell’adsorbente e viene misurato in cm3/g; e la distribuzione
delle dimensioni dei pori (Pore Size Distribution, PSD). Il PSD è la rappresentazione grafica di
ΔVp/ΔDpcontro Dp, dove Vp rappresenta il volume cumulativo dei pori di dimensione Dp e misurato
in cc@STP/gÅ. L’unità cc@STP indica la quantità adsorbita misurata in centimetri cubici alle condizioni standard di pressione e temperatura (273.15 K e 760 Torr).
In genere, le isoterme di fisisorbimento sperimentali hanno una natura composita, sono caratterizzate, cioè, dalla sovrapposizione di due o più tipologie. In questi casi, è difficile trovare una corrispondenza con quelle classificate dalla IUPAC. In questi casi, risulta utile confrontare la curva sperimentale con alcune curve standard, in modo da determinare la deviazione dalla linearità6. I grafici costruiti con l’aiuto dei dati di uno standard vengono detti plot di confronto9. Quelli più rappresentativi sono: i t- e αs-plot. Ad esempio, come standard può essere utilizzata l’isoterma di
adsorbimento di solidi non porosi, presentante un chiaro punto B, struttura chimica simile e appartenenti alla stessa categoria (ad es. ossidi, metalli, grafiti) del campione da analizzare.
18
Nell’analisi t-plot10, la quantità adsorbita, W, di un’isoterma standard viene convertita in spessore dello strato di gas adsorbito, t, con l’equazione t=(W/WM)σt , dove σsrappresenta la spessore di un
singolo strato adsorbito che, nel caso di N2 è pari a 0.354 nm. Se ne ricava un grafico in cui
l’ascissa è rappresentata da t al posto di p/p0. Se l’isoterma in considerazione descrive un
adsorbimento multistrato, il t-plot sarà una linea retta passante per l’origine, la cui pendenza è proporzionale all’area superficiale. La deviazione dalla linearità del t-plot fornisce informazioni riguardo il tipo di pori, la dimensione media dei pori, l’area superficiale e il volume dei pori. Comunque, l’analisi con il t-plot ha una limitata applicabilità nei sistemi microporosi in quanto non viene fatta una considerazione esplicita sull’adsorbimento monostrato.
Sing e collaboratori hanno proposto, a tal proposito, un plot di confronto più generale, in cui si calcola il rapporto dell’adsorbimento a una determinata p/p0 con l’adsorbimento a p/p0=0.4,
designato con αs (=W/W0.4). L’isoterma, così, può essere ri-graficata con l’adsorbimento W contro
αs. Questo tipo di grafico viene detto αs-plot6. Per la sua costruzione non si ha bisogno di conoscere
la capacità del monostrato, così che è applicabile anche ai sistemi microporosi. Anche in questo caso se si ottiene una linea retta passante per l‘origine indica l’assenza di micropori o di mesopori; la deviazione dall’idealità, invece, da informazioni sulla struttura dei pori.
La misura dell’area superficiale si basa su una serie di teorie in grado di predire il numero di molecole di adsorbato richieste per coprire il solido con un singolo strato monomolecolare. Ugualmente importante, comunque, sarà il valore della cross-sectional area di ogni molecola o l’area effettiva coperta da ogni molecola adsorbita sulla superficie. L’area superficiale, poi, è il prodotto del numero di molecole presenti in un monostrato completo e la cross-sectional area effettiva di una molecola di adsorbato.
I modelli di adsorbimento
Il modello di Langmuir
L’approccio usato da Langmuir11 viene usato più precisamente nel caso del chemisorbimento in quanto ha come presupposto che i gas formino solo uno strato molecolare su un solido. La collisione di una molecola di gas con un solido viene considerata anelastica, cosicché la molecola di gas rimane in contatto con il solido per un tempo finito prima di ritornare nella fase gassosa. Questo tempo viene considerato responsabile per il fenomeno dell’adsorbimento. L’equazione 4 è ottenuta
(4)
mettendo in relazione la velocità alla quale le molecole urtano una superficie alla velocità alla quale esse lasciano la superficie. Va rappresenta la quantità di gas adsorbito alla pressione P, Vm è la
quantità di gas adsorbito quando l’intera superficie è coperta con uno strato monomolecolare e b è una costante empirica. L’equazione 4 può essere riarrangiata nella forma lineare:
(5)
Questa equazione rappresenta una linea retta su un grafico la cui ascissa è dato dai valori di P e l’ordinata da P/Va. I valori di b e Vm, invece, possono essere calcolati dalla pendenza e
dall’intercetta della retta ottenuta. La linea retta, però, viene ottenuta solo all’interno di un range limitato di valori. All’interno di questo, è possibile calcolare l’area superficiale conoscendo il valore di Vm, mediante l’equazione 5:
19
Dove σ è l’area della superficie occupata da una singola molecola di gas, NA è il numero di
Avogadro, m è la massa del campione adsorbito e V0 è il volume molare di gas. Nel caso in cui
viene utilizzato l’azoto come adsorbato si avrà:
(7)
Il modello BET (Brunauer, Emmett e Teller)
Brunauer, Emmett e Teller hanno esteso la teoria di Langmuir all’adsorbimento multi-strato. La teoria BET12 ipotizza che le molecole adsorbite che si trovano nello strato più alto sono in equilibrio dinamico con il vapore. Questo significa che dove la superficie è coperta con solo uno strato di adsorbato, questo sarà in equilibrio con il vapore; dove, invece, sono presenti più strati, solo lo strato esterno sarà in equilibrio. Dato che l’equilibrio è dinamico, la posizione dei siti superficiali coperti da uno o più strati può variare ma il numero di molecole in ogni strato rimarrà costante. Uguagliando la velocità di condensazione delle molecole di gas su uno strato adsorbito di gas alla velocità di evaporazione da quello strato e sommando per un infinito numero di strati, si ottiene la seguente espressione:
(8)
Dove C è una costante e P0è la pressione di saturazione del gas. Il valore di C nel più semplice dei
casi è pari a , dove ql è il calore di adsorbimento del primo strato, qL il calore di
liquefazione dell’adsorbato, R la costante dei gas e T la temperatura assoluta. L’equazione 8 può essere riscritta nella forma lineare:
(9)
Mettendo in grafico P/[Va(P0-P)] sull’asse delle ascisse e in ordinata P/P0 si ottiene una linea retta
con intercetta pari a 1/VmC e pendenza (C-1)/VmC. Il calcolo del volume del monostrato permette di
ottenere il valore dell’area superficiale del campione che viene ottenuta usando l’area occupata da una singola molecola di adsorbato.
Raramente, la teoria BET è in grado di riprodurre l’isoterma per tutto il range di pressioni relative. La BET e l’isoterma sperimentale risultano coincidenti nella regione di pressioni relative in cui il monostrato è completato (0.05< P/Po <0.35). In questa regione, risulta essere un utile strumento per il calcolo dell’area superficiale.
Se viene preso in considerazione l’esempio degli ultra-micropori, il riempimento dei pori avviene nelle zone a bassi valori di p/po dell’isoterma. Questo processo viene detto riempimento primario
dei pori ed è associato con un aumento delle interazioni tra adsorbato e adsorbente. Il processo di riempimento dei micropori più ampi, invece, come possono essere i super-micropori, è di tipo cooperativo che avviene a valori di p/po più alti e che può estendersi anche nella regione del
multistrato.
Comunque, numerosi fattori dovrebbero essere presi in considerazione tra cui: la struttura del solido adsorbente, la composizione della superficie, la dimensione e la forma dei pori, la polarizzabilità e
20
la polarità dei centri interagenti, le dimensioni molecolari e la forma dell’adsorbato, la temperatura a cui si opera. Inoltre, bisogna sempre considerare che la capacità di riempimento dei micropori dipende sia dal volume dei pori disponibile che dall’impacchettamento delle molecole adsorbite.
La determinazione della porosità
La teoria del riempimento dei micropori di Dubinin
L’adsorbimento di gas dai solidi microporosi corrisponde al processo di riempimento di un volume descritto dalla teoria di Dubinin13,14. Quest’ultima si basa sul potenziale termodinamico
e l’espressione fondamentale è l’equazione di Dubinin-Astakhov:
(10)
Dove Na (in mmol/g) è la quantità adsorbita a pressione relative p/p0 e temperatura T, Nao è la
quantità adsorbita al massimo volume microporoso W0=NaoVm, assumendo che Vm sia una valore
vicino al volume molare nello stato liquido, ed E è la cosiddetta energia caratteristica del sistema. Questa può essere riscritta come E=βE0, dove β15 è il coefficiente di affinità dell’adsorbato, usando
il benzene come riferimento (β(C6H6)=1). Per la maggior parte dei carboni attivati, l’esponente
dell’equazione 10 è pari a 2, e corrisponde all’equazione di Dubinin-Radushkevich (DR). L’equazione DR può essere riscritta nella forma lineare
(11)
E messa su un grafico la cui ascissa è data dal ln(Na/Nao) e l’ordinata da (A/β)2. Si dovrebbe ottenere
una linea retta dalla cui pendenza è possibile ricavare il valore di E0.
La forza della teoria di Dubinin sta nella semplicità dei parametri fisico-chimici e nel fatto che sia E che n non variano con la temperatura. Ciò significa che l’equilibrio di adsorbimento può essere predetto per un determinato intervallo di pressioni e temperatura.
Una correlazione empirica esiste tra la dimensione media dei micropori L0 (nm) ed E0 (kJ/mol),
secondo la seguente espressione:
(12) (0.4 nm < L0< 2 nm)
L’area superficiale dei micropori a fenditura, Smi (m2/g) corrisponde approssimativamente a:
(13)
La teoria di Dubinin non si limita al volume di riempimento dei micropori. Esiste, infatti, una forma modificata dell’equazione DR, chiamata equazione di Dubinin-Radushkevich-Kaganer (DRK), che può essere utilizzata per descrivere l’adsorbimento su superfici non porose,
(14)
La quantità Nam rappresenta la capacità di formazione del monostrato della superficie. Questa
21
relativa maggiori (0.05< p/po <0.35). Comunque, il suo uso è limitato a pochi adsorbati,
principalmente all’azoto a 77 K e il benzene a 293 K e, soprattutto, Nam(DRK) e Nam(BET) può
essere significativamente diverso.
Alcuni carboni microporosi danno un DR plot lineare in un ampio range di p/po, altri, invece, si
ottiene linearità solo per un intervallo molto ristretto. Usualmente, si ottiene un plot lineare a pressioni molto basse (p/po<10-2).
Metodo di Horvath-Kawazoe per la determinazione della distribuzione delle dimensioni dei pori
Horvath e Kawazoe (HK) hanno descritto un metodo analitico16, semi-empirico per il calcolo della dimensione effettiva dei pori nei materiali microporosi, partendo dall’isoterma di adsorbimento dell’azoto. L’approccio originale è basato sul lavoro di Everett e Powl e considera un fluido (l’azoto) confinato in un poro a fenditura.
Il metodo HK per la determinazione della distribuzione della dimensione dei pori (PSD) si basa sull’idea che la pressione relativa richiesta per il riempimento di micropori di una certa dimensione e forma è direttamente proporzionale all’energia d’interazione tra adsorbato e adsorbente. Ciò significa che i micropori si riempiono progressivamente con l’aumento della pressione dell’adsorbato. Praticamente, solo i pori con dimensioni più piccole di un determinato valore saranno riempiti ad un determinato valore di pressione relativa dell’adsorbato. Così, il metodo HK permette il calcolo del PSD dei micropori a basse pressioni.
Il metodo HK è basato sulla seguente equazione, includendo solo le interazioni di van der Waals, calcolate con l’aiuto del potenziale di Lennard-Jones:
(15)
Halsey e collaboratori hanno applicato il potenziale di Lennard-Jones al caso dell’interazione di una molecola di adsorbato con un piano infinito di molecole di adsorbente, ottenendo:
(16)
In seguito, Everett e Paul hanno esteso i risultati ottenuti da Halsey e collaboratori al caso di due piani infiniti di molecole separati da una distanza L:
(17)
Dove NAS è il numero di atomi/molecole per unità di area, e L è la distanza tra i due strati, σ =
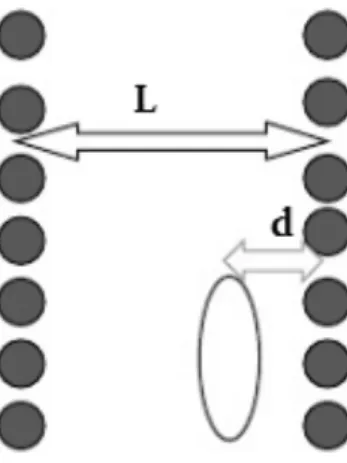
0.858d e con ds pari al diametro delle molecole di adsorbente e da il diametro delle
molecole di adsorbato. Inoltre, z rappresenta la distanza internucleare tra le molecole di adsorbato e di adsorbente, (L-ds) è la dimensione effettiva dei pori e AAS è la costante di dispersione, che tiene
22
Figura 10. Adsorbimento in un poro a fenditura Il termine AAS è calcolato con l’aiuto della formula di Kirkwood-Muller:
(18)
Dove m è la massa dell’elettrone, c la velocità della luce, αA e αs sono la polarizzabilità
dell’adsorbato e delle molecole di adsorbente, e χA e χs sono la suscettibilità magnetica
dell’adsorbato e dell’adsorbente.
Poi, Horvath e Kawazoe hanno proposto che al potenziale fosse aggiunto l’interazione adsorbato-adsorbato, suggerendo la seguente formula:
(19)
Dove NAA è il numero di molecole adsorbite per unità di area.
Anche AAA viene calcolato con l’aiuto della formula di Kirkwood-Muller, una costante caratteristica
dell’interazione adsorbato-adsorbato:
(20)
Il passaggio successivo è quello di ottenere l’energia d’interazione media. Ciò viene fatto :
(21)
dove ξ(L) è il potenziale medio in un dato poro e è il campo di adsorbimento all’interno di un poro a fenditura.
Infine, l’energia media può essere messa in relazione con la variazione di energia libera al momento dell’adsorbimento:
(22)
23
(23)
Dove NA è il numero di Avogadro.
Il metodo HK è uno strumento utile per la caratterizzazione di materiali microporosi che permette la stima delle dimensione dei pori dei materiali studiati.
Per calcolare la distribuzione delle dimensioni dei pori, viene prima calcolata la pressione relativa corrispondente a una determinata dimensione dei pori, L, usando l’equazione 23 poi, con l’aiuto dell’isoterma di adsorbimento sperimentale, la quantità adsorbita, na, corrispondente a quel
determinato valore di p/po viene ricavata. Da questo, differenziando la quantità adsorbita rispetto
alla dimensione dei pori, dna/dL, viene ottenuta la distribuzione delle dimensioni dei pori nel range
dei micropori.
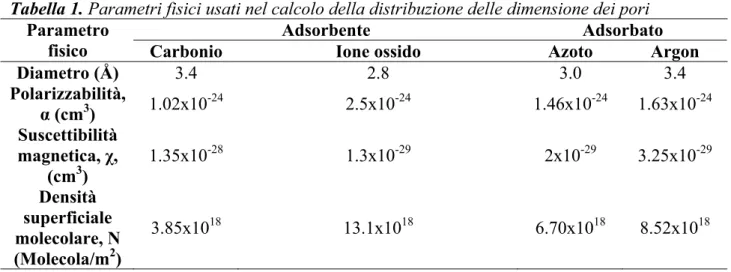
I parametri fisici da applicare all’equazione per cui alcuni sistemi adsorbato e adsorbente sono mostrati in tabella 1.
Tabella 1. Parametri fisici usati nel calcolo della distribuzione delle dimensione dei pori
Adsorbente Adsorbato
Parametro
fisico Carbonio Ione ossido Azoto Argon
Diametro (Å) 3.4 2.8 3.0 3.4 Polarizzabilità, α (cm3) 1.02x10 -24 2.5x10-24 1.46x10-24 1.63x10-24 Suscettibilità magnetica, χ, (cm3) 1.35x10-28 1.3x10-29 2x10-29 3.25x10-29 Densità superficiale molecolare, N (Molecola/m2) 3.85x1018 13.1x1018 6.70x1018 8.52x1018
Ad esempio, nel caso dell’adsorbimento di azoto a 77 K nei setacci molecolari carboniosi, l’equazione HK risulta in:
(24)
Estensione del modello HK ai pori cilindrici
Saito e Foley17 hanno esteso l’approccio HK ai pori cilindrici in modo che l’equazione HK possa essere utilizzata nello studio delle zeoliti.
Assunzioni addizionali rispetto al modello originale sono state fatte a tal proposito, tra cui: il poro viene considerato perfettamente cilindrico, di lunghezza infinita e la cui parete interna è costituita da un unico strato di atomi.
Anche in questo caso, però, l’energia potenziale media all’interno del poro cilindrico è rappresentativa dell’energia libera di adsorbimento.
24
Seguendo la logica della derivazione dell’equazione HK, Saito e Foley hanno derivato un’equazione simile che mette in relazione la pressione di riempimento dei micropori al diametro dei pori d=L-ds.
(25)
I parametri αke βk sono costanti definite come:
Con α0e β0 =1.
Estensione del modello HK ai pori sferici
Cheng e Yang18 (CY) hanno esteso l’equazione HK in modo che possa essere applicata ai pori sferici, in particolare le cavità delle zeoliti.
In tale modello, viene assunto che le interazioni avvengono solo tra adsorbato e atomi di ossigeno delle pareti delle cavità sferiche all’interno delle zeoliti e che queste cavità sono costituite da un singolo piano di atomi.
Così, il potenziale di Lennard-Jones viene integrato per lo spazio nelle cavità sferiche in modo da ottenere il potenziale adsorbato-adsorbente all’interno della cavità. Seguendo l’approccio di HK, il termine di interazione adsorbato-adsorbato viene incluso e si ottiene l’espressione per l’energia dal potenziale di Lennard-Jones. L’equazione del potenziale viene poi integrata per determinare l’energia potenziale media all’interno di una cavità sferica.
(26)
Dove è la variazione di energia libera, N1 è il numero di atomi di ossigeno sulla
superficie interna della cavità, N2 è il numero di molecole di adsorbato all’interno della cavità, è
il minimo dell’energia potenziale di interazione adsorbato-adsorbente, è il minimo dell’energia potenziale dell’interazione adsorbato-adsorbato, L il raggio del poro sferico e T1, T2, T3 e T4 sono
termini adimensionali la cui forma generale è
25
26
EVOLUZIONE MORFOLOGICA DEL SISTEMA hBN/C SOTTOPOSTO AD HIGH ENERGY BALL MILLING
Le polveri di nitruro di boro e grafite (hBN/C) sono state sottoposte a macinazione allo scopo di ottenere un composito lamellare a struttura porosa con pori a fenditura prodotti dall’intercalazione dei materiali di partenza. Di seguito è riportato lo studio del processo di intercalazione durante il trattamento di ball milling. É stata seguita l’evoluzione strutturale e di fase del sistema a differenti tempi di macinazione. L’analisi del comportamento all’adsorbimento ha permesso di ricavare le proprietà del composito in termini di porosità e superfice specifica, evidenziando come il prolungarsi del trattamento comporta un collasso della porosità dovuta a interazioni chimiche tra le lamelle dei differenti componenti.
Materiali e metodi
Le polveri di grafite (C) e nitruro di boro (BN) (Aldrich) sono state preventivamente trattate in stufa a 180° C in vuoto per 4 giorni, in modo da eliminare impurezze e umidità. I materiali (rapporto in peso 1/1), sono stati inseriti in una giara rivestita in teflon. Per la macinazione sono state utilizzate biglie di acciaio (diametro 3 mm), e un rapporto in peso polvere/biglie pari a 1/10. La macinazione ad alta energia è stata condotta in atmosfera di argon utilizzando un mulino SPEX 8000M. La miscela macinata si presenta sotto forma di una polvere scura molto fine e omogenea. Sono stati ottenuti differenti campioni hBN/C a diversi tempi di macinazione. In tabella 2 è riportato l’elenco dei campioni e la loro denominazione.
I campioni hBN/C e i materiali di partenza sono stati caratterizzati mediante diffrazione di raggi x utilizzando un difrattometro Seifert PAD VI con radiazione MoKα equipaggiato con un
monocromatore di tipo Johann posizionato nel fascio diffratto. L’identificazione delle fasi nei diffrattogrammi è stata eseguita per confronto utilizzando la banca dati PDF-2.
I campioni macinati sono stati analizzati mediante microscopia elettronica a scansione per studiare l’effetto della macinazione sulla morfologia del materiale. Le immagini SEM sono state acquisite mediante un microscopio SEM LEO 1530 equipaggiato con due rivelatori di ioni secondari ed un rivelatore dedicato agli elettroni retro diffusi e con ingrandimento 100kx.
Le misure di adsorbimento di azoto a 77 K sono state eseguite utilizzando l’apparecchiatura NOVA 2200e della Quantachrome. Prima di ogni misura, il composito è stato preliminarmente sottoposto a un trattamento termico a T=300°C in vuoto. I dati sono stati analizzati mediante l’applicazione dei modelli di BET per il calcolo dell’area superficiale, DR e HK per l’analisi della microporosità.
Tabella 2. Denominazione dei campioni di hBN/C ottenuti ai diversi tempi di macinazione.
Nome campione Tempo di macinazione (h) Rapporto in peso hBN:C
hBN/C-0.25 0.25 1:1 hBN/C-0.5 0.50 1:1 hBN/C-1 1 1:1 hBN/C-1.5 1.5 1:1 hBN/C-2 2 1:1 hBN/C-5 5 1:1 hBN/C-7 7 1:1 hBN/C-10 10 1:1 hBN/C-15 15 1:1 hBN/C-20 20 1:1
27
Risultati e discussione
In figura 11 sono riportati i diffrattogrammi delle polveri di nitruro di boro esagonale e grafite di partenza e confrontati con i diffrattogrammi dei campioni hBN/C macinati a differenti tempi di macinazione. All’aumentare del tempo di macinazione, si osserva una diminuzione di intensità e un allargamento dei picchi indicativi di una continua perdita dell’ordine strutturale del sistema. La direzione (002) sia per il BN sia per la grafite mostra la maggiore resistenza rispetto al trattamento
Figura 11. Diffrattogrammi XRD del BN, C e del composito hBN/C a diversi tempi di
28
di milling, attribuibile a una ri-condensazione dei piani. I picchi relativi alle altre direzioni cristallografiche tendono a scomparire per tempi di macinazione maggiori alle 2 ore. La spalla osservabile a circa 11° è attribuibile alla presenza di una struttura turbostratica del nitruro di boro indotta dalla macinazione e già osservata in precedenza19.
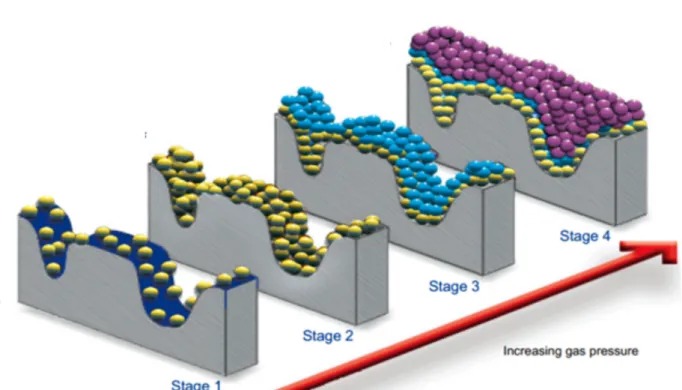
La figura 12 mostra le immagini SEM sull’evoluzione della microstruttura dei campioni hBN/C macinati in funzione del tempo di macinazione: a tempi brevi di macinazione, si nota la formazione di isole di BN (bianco in fig.12) su foglietti di grafite (grigio in fig.12); a tempi intermedi di macinazione, i foglietti tendono a intercalarsi e se ne riducono le dimensioni; a tempi di macinazione superiori alle 10 ore, invece, si perdono le informazioni morfologiche.
Figura 12. Immagini SEM del hBN/C macinato: a) 15 minuti, b) 2 ore e c)10 ore.
I campioni hBN/C macinati sono stati caratterizzati mediante misure di adsorbimento di azoto a 77K.
La figura 13 riporta il confronto tra le isoterme di 3 campioni rappresentativi. Si può notare come, aumentando il tempo di macinazione da 2 a 10 ore, il ginocchio della curva sia notevolmente più alto e la pendenza maggiore nel tratto di adsorbimento multistrato. A differenza di quanto osservato per il campione sottoposto a 2 ore di macinazione, la forma delle curve dei campioni hBN/C macinato 10 ore e 20 ore nel tratto iniziale, è riconducibile ad un’isoterma di tipo I secondo la classificazione IUPAC, caratteristica dei materiali microporosi. A valori di pressione relativa maggiori è presente un’isteresi (isoterma di tipo IV), indicativa della presenza di mesopori. L’isoterma del campione hBN/C-20 presenta un ginocchio più basso e un appiattimento nel tratto della curva oltre il ginocchio rispetto al campione macinato 10 ore, indicativo di un abbattimento della porosità a tempi lunghi di macinazione.
Alle isoterme misurate sperimentalmente, sono stati applicati i modelli BET, DR e H-K, per calcolarne l’area superficiale e le caratteristiche di porosità. L’area superficiale calcolata mediante l’equazione BET (tabella 3 e figura 14) aumenta in funzione del tempo di macinazione passando da valori di 20 m2/g per il campione macinato 0.25 h, raggiunge il valore di 300 m2/g dopo 10 ore di macinazione e poi decresce per tempi di macinazione più lunghi di 10 ore fino a 149 m2/g. Tale andamento è in accordo con le informazioni ricavate dall’analisi SEM e sembra indicare che tempi di macinazione prolungati oltre le 10 ore promuovono interazioni di tipo chimico tra i materiali macinati anche se non evidenziabili dall’analisi XRD.
29
Figura 13. Isoterme di adsorbimento-desorbimento di azoto a 77K per i campioni hBN/C a 2, 10 e 20
ore.
Tabella 3. Valori dell’area superficiale BET nei diversi campioni.
Nome campione Tempo di macinazione (h) Area BET (m2/g)
hBN/C-0.25 0.25 20 hBN/C-0.5 0.50 42 hBN/C-1 1 40 hBN/C-1.5 1.5 40 hBN/C-2 2 42 hBN/C-5 5 43 hBN/C-7 7 124 hBN/C-10 10 306 hBN/C-15 15 190 hBN/C-20 20 149
30
Figura 14. Variazione dell’area superficiale BET in funzione del tempo di macinazione.
La tabella 4 riassume i risultati dell’analisi ottenuti applicando i modelli HK e DR. I due metodi applicati hanno prodotto valori paragonabili. Il composito risulta microporoso già a bassi tempi di macinazione con il valore della grandezza media dei pori intorno a 7 Å. Il volume dei micropori, invece, aumenta con il tempo di macinazione, passando da 0.006 cm3/g (sia con HK che con DR) per il campione hBN/C-1 fino a un massimo di 0.100 cm3/g (DR) e 0.110 cm3/g (HK) del campione hBN/C-10. Il volume microporoso, per i campioni ottenuti a tempi di macinazione maggiori diminuisce.
Tabella 4. Analisi della microporosità dei campioni di hBN/C.
Campione Tempo di macinazione (h) HK Grandezza media dei pori (Å) HK Volume Massimo dei pori (p/p0=0.04851) (cm3/g) DR Volume dei micropori (cm3/g) hBN/C-0.25 0.25 hBN/C-0.5 0.50 hBN/C-1 1 7.2 0.006 0.006 hBN/C-1.5 1.5 hBN/C-2 2 hBN/C-5 5 6.7 0.014 0.010 hBN/C-7 7 hBN/C-10 10 7.1 0.110 0.100 hBN/C-15 15 6.9 0.078 0.099 hBN/C-20 20 7.0 0.052 0.060
31
Nelle figure 15, 16, 17 e 18 vengono riportati, a titolo d’esempio, i grafici ottenuti dall’applicazione dei modelli BET, DR e HK, descritti precedentemente, all’isoterma di adsorbimento di azoto a 77 K del campione con i migliori risultati in termini di area superficiale e microporosità, il hBN/C-10. Come si può notare dalla figura 16, per il calcolo dell’area superficiale, viene preso in considerazione il tratto corrispondente alla formazione del mono-strato. Questo corrisponde al
range di pressioni relative comprese tra 0.05 e 0.3. Infatti, solo in questo tratto si ha una
corrispondenza dell’isoterma sperimentale con quella calcolata dall’equazione BET. In questo caso, si è ottenuta una buona corrispondenza nel range: 0.052-0.2, ottenendo un coefficiente di correlazione pari a 0.9993.
Figura 15. Multi-point BET plot ricavato dall’applicazione dell’equazione BET all’isoterma
sperimentale di adsorbimento del campione hBN/C-10, nell’intervallo di p/p0pari a 0.052-0.2.
Il DR plot (Figura 16), invece, viene ottenuto dai valori di pressione relativa bassi (< 0.1). Con l’estrapolazione di un tratto rettilineo in questo range è possibile ricavare valori sulle dimensioni dei pori e sul volume poroso.
Infine, in figura 17 e 18, è mostrato il plot ottenuto dall’applicazione del modello HK. Come si nota dalle figure, la distribuzione delle dimensioni dei pori mostra un massimo intorno a valori di 3.4 (misura di metà poro), indicando che il materiale analizzato risulta essere microporoso.
32
Figura 16. Grafico ottenuto dall’applicazione del modello DR all’isoterma di adsorbimento del
campione hBN/C-10 nell’intervallo di p/p0 pari a 0.0003-0.0006
Figura 17. Grafico della distribuzione della dimensione dei pori (PSD) ottenuto applicando
33
Figura 18. Grafico del volume dei pori del campione hBN/C-10 ottenuto applicando all’isoterma
34
CONCLUSIONI
Nel presente rapporto è stata data una rassegna della metodologia di caratterizzazione delle superfici dei materiali mediante adsorbimento di gas. Sono stati brevemente descritti i principi della metodologia volumetrica e gli approcci teorici/sperimentali alla interpretazione dei dati di adsorbimento.
Utilizzando due materiali a struttura esagonale simile (hBN e grafite), è stato sviluppato un materiale composito con caratteristiche microporose indotte dal trattamento d’intercalazione via Ball Milling Il materiale sviluppato è stato studiato in termini di evoluzione microstrutturale e di fase, nonché morfologica, in funzione del tempo di macinazione. L’analisi ha evidenziato la possibilità di utilizzare con successo la via meccanochimica per ottenere strutture porose con pori a fenditura partendo da materiali differenti a simile struttura esagonale. Tuttavia trattamenti spinti inducono un collasso della porosità in precedenza sviluppata a causa d’interazioni chimiche non sufficientemente inibite nelle condizioni di trattamento. Sebbene sia necessario un affinamento delle condizioni energetiche applicate, il processo appare promettente nello sviluppo di una nuova classe di materiali porosi.
Nuove prove sono in corso per la valutazione delle proprietà di accumulo d’idrogeno allo stato solido del materiale sviluppato.
35
Bibliografia
1. Patchkovskii, S. et al. Graphene nanostructures as tunable storage media for molecular hydrogen. PNAS 102, 10439–10444 (2005).
2. Nimmo, J. R. Porosity and Pore Size Distribution. Encyclopedia of Soils in the Environment 295–303 (2004).
3. Kaneko, K. Determination of pore size and pore size distribution: 1. Adsorbents and catalysts.
Journal of Membrane Science 96, 59–89 (1994).
4. Yang, R. T. Frontmatter. Adsorbents: Fundamentals and Applications i–xii (2003).
5. Sing, K. S. W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure and Applied
Chemistry 57, 603–619 (1985).
6. Gregg, S. J. & Sing, K. S. W. Adsorption, surface area, and porosity. (Academic Press: 1991). 7. Russel, W. W. The Adsorption of Gases and Vapors. Volume I: Physical Adsorption (Brunauer,
Stephen). J. Chem. Educ. 21, 52 (1944).
8. Micromeritics Instrument Corporation Gas Adsorption Theory.
9. Condon, J. B. Chapter 1 - An Overview of Physisorption. Surface Area and Porosity
Determinations by Physisorption 1–27 (2006).
10. Lippens, B. ., Linsen, B. . & Boer, J. H. d. Studies on pore systems in catalysts I. The adsorption of nitrogen; apparatus and calculation. Journal of Catalysis 3, 32–37 (1964). 11. Langmuir, I. THE ADSORPTION OF GASES ON PLANE SURFACES OF GLASS, MICA
AND PLATINUM. J. Am. Chem. Soc. 40, 1361–1403 (1918).
12. Brunauer, S., Emmett, P. H. & Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am.
Chem. Soc. 60, 309–319 (1938).
13. Stoeckli, F. Dubinin"s theory and its contribution to adsorption science. Russian Chemical
Bulletin 50, 2265–2272 (2001).
14. Dubinin, M. M. Fundamentals of the theory of adsorption in micropores of carbon adsorbents: Characteristics of their adsorption properties and microporous structures. Carbon 457–
467doi:10.1016/0008-6223(89)90078-X
15. Odell Wood, G. Activated carbon adsorption capacities for vapors. Carbon 30, 593–599 (1992). 16. HorváTh, G. & Kawazoe, K. Method for the calculation of effective pore size distribution in
molecular sieve carbon. Journal of Chemical Engineering of Japan 16, 470–475 (1983). 17. Saito, A. & Foley, H. C. Curvature and parametric sensitivity in models for adsorption in
micropores. AIChE Journal 37, 429–436 (1991).
18. Cheng, L. S. & Ralph T, Y. Improved Horvath—Kawazoe equations including spherical pore models for calculating micropore size distribution. Chemical Engineering Science 49, 2599– 2609 (1994).
19. Xia, Z. P. & Li, Z. Q. Structural evolution of hexagonal BN and cubic BN during ball milling.
Edito dall’
Servizio Comunicazione
Lungotevere Thaon di Revel, 76 - 00196 Roma www.enea.it
Stampa: Tecnografico ENEA - CR Frascati Pervenuto il 15.2.2013