Introduction
1. A FRIENDLY GLOSSARY OF RECURRING TERMS
1.1 Nuclear factor-kappa B (NFkB)
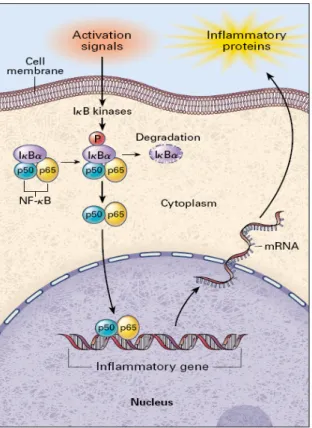
A cytoplasmatic transcription factor that, entering into the nucleus and binding to specific sequences in the promoter regions of target genes, transcribes products involved in inflammation and coagulation, including cytokines, chemokines, immune receptors, adhesion molecules and coagulation factors. Activation of NFkB is triggered by phosphorylation and subsequent degradation of an inhibitory protein, IKB, and is limited by NFkB-induced synthesis of IkBα, which transports activated NFkB back to the cytoplasm (Fig.1). Many stimuli activate NFkB, including cytokines, activators of protein kinase(PK) C, viruses. Antioxidants, such as pyrrolidine dithiocarbamate (PDTC) and acetylcysteine, may block at different levels NFkB activation suggesting that reactive oxygen species (ROS) might have an intermediary role in its function.
1.2 Cytokines
A group of proteins such as interleukins (ILs), tumor necrosis factors (TNFs), interferon (IFN), colony stimulating factors (CSFs), transforming growth factors (TGFs) by which cells communicate and influence the external environment acting in a autocrine, paracrine and juxtacrine manner. Cytokines are typically pleiotropic (a cytokine can trigger several different cellular responses depending on cell type, timing, and context), synergic (the association of two cytokines markedly amplifies their single activity) and capable of stimulating common receptor subunits.
1.3 Tumor necrosis factor alpha (TNF-α)
A cytokine produced by numerous cells, including macrophages, natural killer cells and T-lymphocytes that activates transcription factors such as NF-κB and mitogen-activated protein kinases (MAPK). TNF-α induces apoptosis and stimulates the expression of adhesion molecules, facilitates monocyte infiltration into the vascular wall by stimulating macrophage chemoattractive protein (MCP)-1 and their transformation to macrophages through CSF. Clinically, TNF-α contributes to cachexia in congestive heart failure (CHF) and impairs insulin sensitivity by downregulation of genes involved in insulin action, counteracting the effect of peroxisome proliferator activated receptor (PPAR)-γ and activation of lipolysis.
1.4 CD40 Ligand (L)
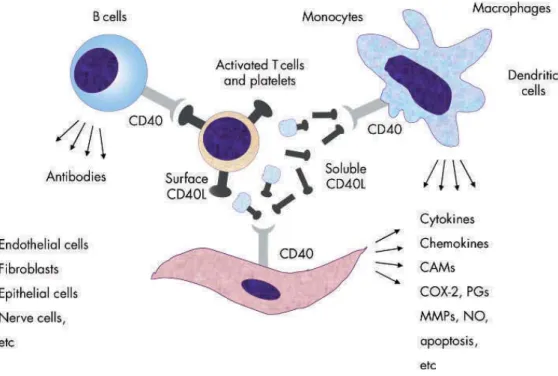
A component of the TNF family produced by T lymphocytes, platelets, vascular smooth muscle cell(VSMC)s, endothelial cell(EC)s and monocytes. CD40L binds to CD40, its receptor constitutively expressed by all cells involved in atherogenesis, and activates intracellular kinases, effector proteins and eventually NFκB thus stimulating the expression of inflammatory chemokines and cytokines, adhesion molecules and proteolytic enzymes. Once ligated to its receptor on macrophages, induces the expression of matrix metalloproteinase (MMP)s and the prothrombotic tissue factor (TF) (Fig. 2). CD40L released from activated platelets circulates in blood (sCD40L) and may predict cardiovascular events.
1.5 Transforming growth factor beta (TGF-β)1
It stimulates collagen deposition in extracellular matrix (ECM), inhibits cell proliferation, foam cell formation in cultured macrophages and adhesion molecules expression and promotes the expression of contractile proteins acting on ECs, VSMCs, macrophages, and T cells, a profile consistent with a role in the maintenance of normal vessel wall architecture and plaque stabilization.
1.6 Chemokines
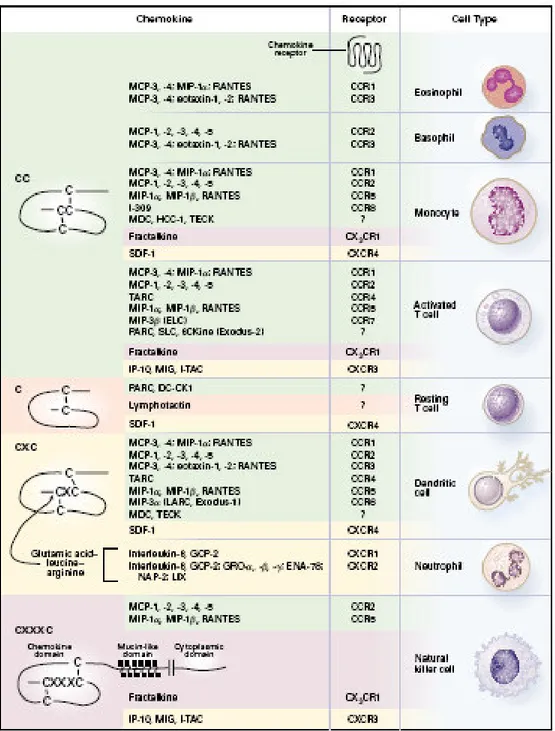
Small peptides produced by virtually all cell types, including the monocyte/macrophages of the atherosclerotic plaque, that orchestrate the migration of leukocytes into the intima and within atherosclerotic lesions, promote cell proliferation, enzyme secretion, induce ROS generation, interfere with VSMC migration and growth, and activate platelets (Fig. 3). Chemokines, most notably MCP-1 and RANTES (regulated on activation, normal T expressed and secreted), induce the conformational changes in integrins required for their adhesive functions, thus inducing firm adhesion of leukocytes to ECs which in turn results in a continued influx of monocytes into the plaque. As for several other inflammatory indices, MCP-1 have been proven to predict cardiovascular prognosis.
1.7 C Reactive Protein (CRP)
It is synthesized by hepatocytes under the influence of IL-6 within 24 and 72 h after infectious and non-infectious disorders, including myocardial infarction. Detection of both CRP mRNA and protein in VSMCs and macrophages within atherosclerotic plaques suggests its de novo synthesis in the vessel wall in which CRP may activate the complement system and/or interact with macrophages and other resident vascular cells. CRP, as an index of subclinical inflammation, predicts cardiovascular event risk in populations of otherwise healthy persons.
1.8 Peroxisome proliferator-activated receptor(PPAR)s
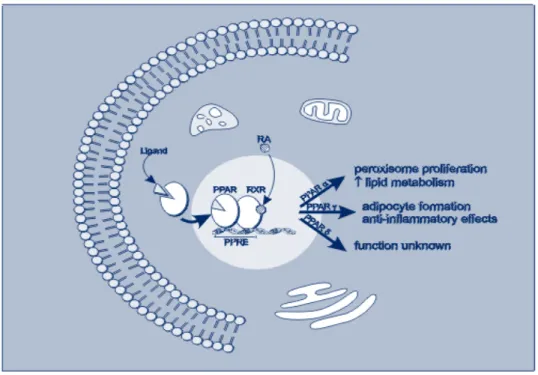
Ligand-activated transcription factors involved in lipid and glucose metabolism, overall energy homeostasis and inflammatory responses (Fig.4) including those mediated by angiotensin(A)II. Indeed PPAR-α and -γ agonists respectively prevented vascular inflammation and corrected structural abnormalities, normalized cell growth, and improved endothelial dysfunction in AII-infused rats.
1.9 Adhesion molecules
They allow adhesion of leukocytes to endothelium and their movement into vessel wall. Rolling of leukocytes is controlled by selectins and adhesion molecules expressed by both leukocytes and the ECs while their spreading and firm attachment to the endothelium is regulated by integrins expressed on leukocytes through the interaction with members of the immunoglobulin(Ig)-like family of adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) expressed by activated ECs.
1.10 Matrix metalloproteinase(MMP)s
These proteases are induced at transcriptional level by growth factors, cytokines, hormones, tumor promoters, and inhibited by heparin, TGF-β and corticosteroids, and are under control of tissue inhibitors of MMPs (TIMP). MMPs modulate the biological activity of ECM, cytokines, growth factors, cell surface receptors and adhesion molecules; their expression and activity increases in the atherosclerotic plaque by release from ECs, VSMC and macrophages. Ruptured plaques exhibit increased MMP-2 and -9 activities and proteolysis of collagens I and III by interstitial collagenases and gelatinases. MMPs can promote aneurysm formation by degrading elastic lamina in atherosclerosis-prone apolipoprotein E deficient (apoE-/-) mice.
1.11 Adiponectin
Produced by adipose tissue it improves hepatic insulin sensitivity, increases fuel oxidation and decreases vascular inflammation by binding to AdipoR1 and AdipoR2 receptors, the former highly expressed in skeletal muscle promoting lipid oxidation and the latter in liver, where it enhances insulin sensitivity and increases PPAR-α ligand activity. Adiponectin, which is an adipose-specific endocrine factor, links obesity with metabolic syndrome (MS), a cluster of cardiovascular risk factors including hypertension, insulin resistance or glucose intolerance, visceral obesity and atherogenic dyslipidemia, resulting in a prothrombotic, proinflammatory state.
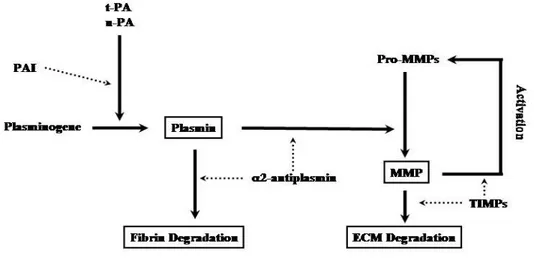
1.12 Plasminogen Activator Inhibitor(PAI)-1
A procoagulative agent and fibrinolysis inhibitor produced at various levels including adipose visceral tissue, hepatocytes and ECs (Fig. 5). IL-6 induction of PAI-1 synthesis within the adipose tissue may represent a possible link between obesity, inflammation and cardiovascular morbidity and mortality.
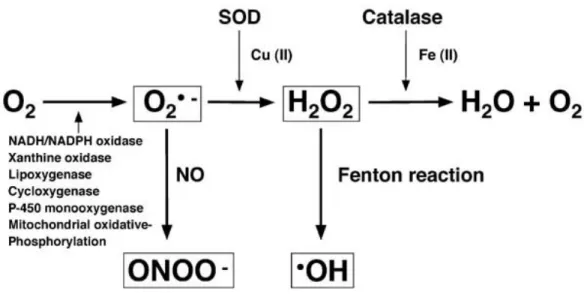
1.13 ROS as markers of oxidative stress and endothelial dysfunction
ROS are oxygen-containing molecules deriving by the mitochondrial electron transfer chain and other enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase and are represented by superoxide (O2-), hydroxyl radical (HO), hydrogen peroxide (H2O2), hypochlorous acid (HOCl) and also by reactive nitrogen species such as nitric oxide (NO) and peroxynitrite (ONOO-) (Fig. 6). ROS-producing enzymes are in balance with an antioxidant enzyme system such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX) and a disequilibrium between the oxidant and antioxidant system generates oxidant stress. In human, measures of oxidant stress are provided by 8-epi-prostaglandin(PG)F2, a biologically active isoprostane derived from nonenzymatic lipid peroxidation of arachidonic acid, or malondialdehyde (MDA), a measure of oxidized polyunsaturated fatty acids resulting in PG-like endoperoxides, or 8-hydroxy(OH)-deoxyguanosine (dG) serum levels, a biomarker of DNA oxidation, or forearm vasodilation to ischemia or muscarinic agonist at least to the extent that NO bioavailability is reduced by increased ROS generation.
2. AN OVERVIEW OF INFLAMMATION IN ATHEROSCLEROSIS
2.1 Atherosclerotic process
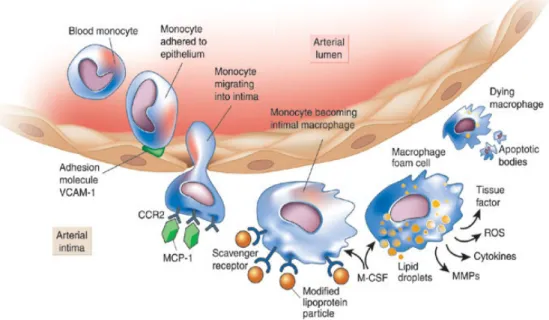
ECs, under the influence of a congeries of cytokines produced by vascular wall cells, express adhesion molecules such as ICAM-1 and VCAM-1 that attach circulating monocytes and lymphocytes to the endothelial lining (Fig. 7). Adhered monocytes/lymphocytes then migrate through diapedesis to the subendothelial spaces under the influence of chemokines such as MCP-1 produced by macrophages, VSMCs and other vascular wall cells 1. The macrophage/lymphocyte complex in interaction with ECs, also secrete MMP-9 which, by disrupting ECM, facilitate leukocyte infiltration through the endothelial layer and its basement membrane 2. Under the influence of CSF expressed by the inflamed intima 3, monocytes transform into macrophages and express scavenger receptors that incorporate oxidized lipoproteins 4, thus becoming foam cells, the hallmark of early-stage atherosclerosis. T lymphocytes, the effector arm of adaptive immunity, also enter atherosclerotic lesions in response to MCP-1 and are activated by cytokines such as TNF-α and IL-1β. T-helper 1 effector cells amplify the inflammatory response by producing IFN-γ and CD40L 5. VSMC migrated from the tunica media through ECM degraded by MMP-9 and other proteinases produce collagen in response to TGF-β and platelet derived growth factor (PDGF) released by macrophages and ECs thus facilitating the evolution of lipid-rich atherosclerotic lesions to a fibrotic and, ultimately, calcified stenotic plaque 6. IL-18 expressed by monocytes, ECs, VSMCs, and macrophages evokes VCAM-1, IL (-8/-6) and MCP-1, and MMPs (-1/-9/-13) synthesis and promote IFN-γ expression, a major proinflammatory cytokine, by T cells, macrophages and SMCs 6-8. Neovascularization arising from the artery’s vasa vasorum also contributes to atherogenesis providing another portal for leukocyte entry 9. Focal intra-plaque haemorrhages accelerate plaque growth by thrombin-activated production of CD40L, the chemokine RANTES and macrophage migration inhibitory factor by ECs, monocytes/macrophages, VSMCs and platelets 10. Platelets also contribute to atherosclerosis
by producing inflammatory mediators such as CD40L, myeloid-related protein-8/14, and PDGF as well as directing leukocyte incorporation into plaques through platelet mediated leukocyte adhesion 11. All cell types involved in atherosclerosis express CD40L that by binding to CD40 triggers expression of adhesion molecules and secretion of cytokines and MMPs involved in ECM degradation 12. ECs, macrophage and VSMCs under the influence of CD40L produce TF that, exposed to factor VII, induces thrombosis in macrophage-rich atheromata, the key histological feature of activated inflammation 13. Inflammation also impairs the integrity of the fibrous cap by destructing collagen fibers and blocking new collagen through IFN-γ produced by T-cells 14. Collagenases, including MMPs released by macrophages in response to T cell-derived CD40L and IL-1, contribute to plaque disruption and increase TF expression and activation of the coagulation cascade 15. Cap fracture and superficial endothelial erosion expose the lipid core or the subendothelial region of the intima 13
to blood and excess PAI-1, a serine protease inhibitor, further imbalances fibrinolysis and contributes to occluding thrombus 16.
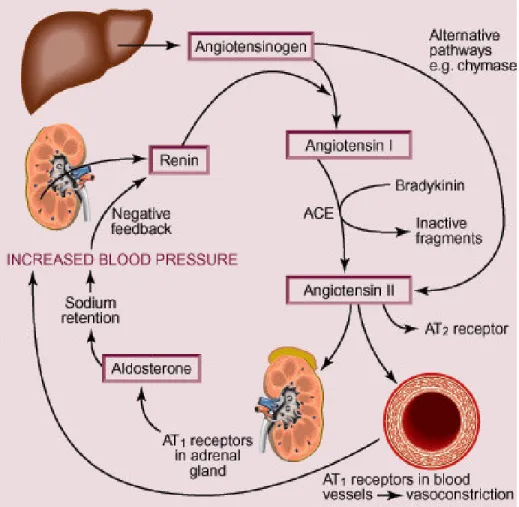
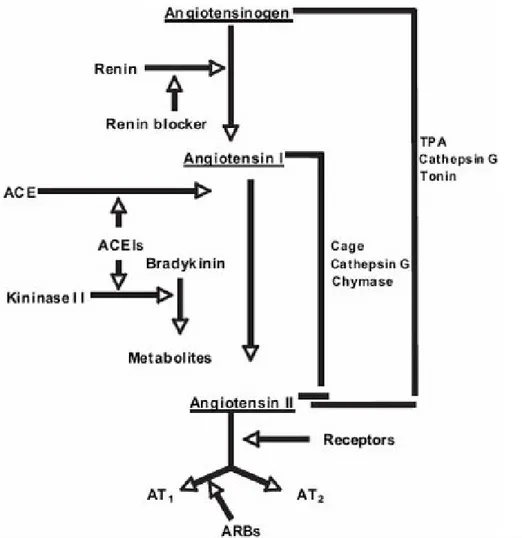
2.2 Renin Angiotensin System (RAS)
The RAS plays a vital role in the regulation of both cardiovascular and fluid homeostasis and thereafter of systemic blood pressure (BP) (Fig. 8). The final product of the system is represented by AII, which is cleaved from angiotensinogen by renin and angiotensin-converting enzyme (ACE). AII is a potent vasoconstrictor hormone that exerts its effects through 2 different receptors, AII type 1 receptor (AT1R) and type 2 (AT2R). The AT1R is the predominant receptor in the cardiovascular system and mediates most of the deleterious effects of AII such as vasoconstriction, stimulation of the release of catecholamines and antidiuretic hormone, endothelial dysfunction, and cell growth. The AT2R is the counter-regulator of the AT1R, exerting mostly beneficial actions like vasodilatation, anti-proliferation, and tissue regeneration 17.
2.3 Angiotensin II Receptor Blocker(ARB)s
Pharmacological RAS interference provides effective lowering BP, prevents target-organ disease and improves symptoms and prognosis in CHF, ischemic heart disease and renal insufficiency. Pharmacological RAS inhibition can be obtained at each significant step of AII biological function (Fig. 9) by direct renin inhibitors, ACE inhibitors and ARBs, the latter represented on the market by seven pharmacologically and pharmacokinetically heterogeneous compounds, all sharing the main property of blocking AT1R: candesartan (CAN), eprosartan (EPR), irbesartan (IRB), losartan (LOS), olmesartan (OLM), telmisartan (TEL) and valsartan (VAL).
ARBs selectively antagonize the interaction of AII at the AT1R, thereby relaxing smooth muscle, increasing salt and water excretion, reducing plasma volume, and decreasing cellular hypertrophy and so preventing the adverse AII-mediated effects in the cardiovascular system. Furthermore, selective inhibition of the AT1R not only inhibits these effects but also leaves the AT2R open to stimulation by AII, resulting in additional beneficial effects 17.
Clinically, ARBs represent a class of effective and well-tolerated orally active drugs that are widely used in the treatment of hypertension and hypertension-related end-organ damage by reducing peripheral vascular resistance usually without modifying heart rate or cardiac output. Most of the commercially available ARBs are able to control BP for 24 h after once daily dose without evidence of producing tolerance to the antihypertensive effect and with a low incidence of side effects (headache, upper respiratory infection, back pain, muscle cramps, fatigue and dizziness) even when used over the long term (3 years) and even in the elderly patients 18.
Despite their belonging to the same class, there are wide differences in pharmacologic properties among ARBs which are clearly due to their structural variety (Fig. 10). Most members of these non-peptide compounds such as LOS, VAL, CAN, OLM and IRB (but not TEL and EPR) include a biphenyltetrazole moiety, which is believed to aid in positioning the
molecule and presenting the active component to the AT1R. The biphenyltetrazole unit is attached to different substituents in each agent. The active part of the molecule differs among all ARBs; LOS carries a heterocycle imidazole, which is replaced by a nonplanar acylated amino acid in the case of VAL. Except for IRB, all active ARBs have a carboxylic acid group. Another difference is the fact that CAN cilexetil and LOS must be metabolized to become active, in contrast to other members of the group. The first is a prodrug and it is completely converted into the active metabolite CAN during gastrointestinal absorption and the second one has a metabolite (EXP3174) which is more active than the parent drug 18.
The variation in the molecular structure of the ARBs results in differences in the binding affinity to the AT1R, dissociation rate (“staying power”) and pharmacokinetic profiles. Possibly the most important difference between members of the class is their degree of selectivity for the AT1R.
The differences observed in lipid solubility, absorption/distribution, plasma protein binding, bioavailability, biotransformation, plasma half-life, and systemic elimination influence the time of onset, duration of action, and efficacy of the ARBs. On the basis of the daily mg dose, the antihypertensive potency of the ARBs follows the sequence: CAN cilexetil > OLM> TEL ~ LOS>IRB ~ VAL > EPR. After oral administration, the ARBs are rapidly absorbed (time for peak plasma levels = 0.5–4 h) but they have a wide range of bioavailability (from a low of 13% for EPR to a high of 60–80% for IRB). Most of these drugs have high plasma protein binding (95–100%); IRB has the lowest binding among the group (90%). The steady- state volumes of distribution vary from a low of 9 L (CAN) to a high of 500 L (TEL). Plasma elimination half-life is short for CAN cilexetil and LOS (1–4 h), intermediate for EPR and VAL (5–10 h), and longer for CAN, OLM, IRB and TEL (11–38 h); EXP3174 has a longer half-life than LOS itself. The drugs and their active metabolites do not accumulate to a significant extent after repeated dosing, except for TEL (100%) 18. The ARBs are an important treatment option for hypertension, being relatively safe and efficacious.
Fig.10 Structure of ARBs.
TEL
CAN cilexetil LOS potassium R1= CH2OH
R2=K
EPR OLM
3. THE PRO-INFLAMMATORY ROLE OF ANGIOTENSIN II
Besides the classical vasoconstrictor action, AII, exerts several other actions including a pro-inflammatory role documented by a wealth of evidence collected in the last two decades. AII amplifies vascular inflammation by acting at several levels including vascular permeability, leukocytes intima infiltration and tissue remodelling.3.1 Vascular permeability
Vascular permeability is altered mainly by pressure-mediated mechanical injury to the endothelium with the additional involvement of mediators such as eicosanoids, e.g. leukotriene C4, PGE2 and PGI2, and vascular permeability factor as well as increased tyrosine phosphorylation of protein at the cell-cell junction 19.
3.2 Leukocytes intima infiltration
Adhesion of monocytes and neutrophils to ECs is facilitated by AII 20 through upregulation of P- and E-selectin expression via AT1R and AT2R stimulation capturing free-flowing leukocytes from the blood and allowing rolling on ECs. The peptide also stimulates the expression of ICAM-1 and VCAM-1 on vascular ECs and VSMCs that allow leukocytes to accumulate at the sites of inflammation 19,21. AII facilitates leukocyte chemotaxis by stimulating MCP-1 in ECs, VSMCs, monocytes/macrophages, and cardiac myocytes and the inhibition of MCP-1 production limites macrophage infiltration into the arterial wall 22, an effect reproduced by AT1R blockade 23 and improving survival in mice with experimental myocardial infarction 24. AII increases expression of IL-8 25 RANTES 19 and osteopontin, a multifunctional molecule highly expressed in chronic inflammatory and autoimmune diseases recruiting monocytes-macrophages and regulating cytokine production in macrophages and T-cells in glomerular ECs 26, and IL-6 production, release and expression 19. Interestingly, IL-6 activates macrophages and adhesion molecules 19,27 and local angiotensinogen generation 28
and thereby local AII formation in the vascular wall, thus amplifying vascular inflammation. AT1R antagonism reduced atherosclerotic progression in DOCA-treated apoE-deficient mice, a low renin model of hypertension, suggesting the proatherogenic potential of locally generated AII even in the setting of suppressed circulating RAS 29. AII stimulates platelet binding to ECs and/or leukocytes thus releasing thrombin, the main effector of platelets, that augments the expression of P-selectin, E-selectin, VCAM-1 and ICAM-1 30.
3.3 Tissue remodelling
Stimulation of vascular growth, characterized by neointima formation and media thickening, inflammatory cells recruitment and endothelial dysfunction, is a third and additional contribution of AII to inflammation carried out by stimulation of vasoactive factors, such as endothelin and aldosterone, sympathetic nervous system activation and BP raise per se 31. Release of autocrine and paracrine growth factors such as PDGF, vascular endothelial growth factor (VEGF), TGF-β and its downstream mediator, connective tissue growth factor (CTGF), also contributes to cell hypertrophy/proliferation and tissue fibrosis in the heart and vasculature 31-35.
3.4 Pro-oxidant role of AII
It is now well-established that AII through AT1R-dependent and independent pathways increases ROS generation through stimulation of NADPH oxidases, an enzyme common to resident vascular wall cells such as VSMCs, monocytes, macrophages consisting of membrane (p22phox and gp91phox), cytoplasmic subunits (p47phox, p40phox, p67phox) and a small GTP-binding protein Rac 36-38. NFkB is sensitive to AII-induced ROS, aspirin-sensitive enzymes of the arachidonic acid metabolism, NF-kB inducing kinase, and IL1β 39. Several studies have shown the effect of AII-induced stimulation 40 on NFkB in SMCs, ECs, glomerular, tubular, and mononuclear cells and other investigations have identified its
overactivation in tissue of AII stimulated animals as a result of AT1R 41 and possibly AT2R activation 42. Recruitment of monocytes and macrophages may enhance the activity of the local vascular RAS, because monocytes and macrophages express angiotensinogen, renin, ACE, and AT1R, thus amplifying pro-inflammatory influences in an intracrine manner. The resulting ROS excess 36 also decreases NO bioavailability by both NO synthases (NOS), constitutive (eNOS) and inducible (iNOS) 40 thus impairing endothelial function and promoting atherosclerosis 42. In agreement with that view, antioxidants reduced aortic monocyte and macrophage infiltration and attenuated end-organ damage in experimental animals exposed to high AII levels 43. Actually, vascular inflammation may raise BP as suggested by animal experiments in which lack of T and B lymphocytes protected against the development of oxidative stress, endothelial dysfunction and hypertension induced by 2-week infusion of AII 44.
4. THE ANTI-INFLAMMATORY EFFECT OF ARBS
Interference with the pro-inflammatory action of AII by targeting all cell types involved in the vascular inflammatory process, including the macrophage/lymphocyte complex, ECs and VSMCs, may contribute to the ever-expanding indications of ARBs in cardiovascular and non cardiovascular diseases.
4.1 Candesartan (CAN)
In a randomized, double-blind, placebo-controlled, crossover study in uncomplicated hypertensive patients, CAN reduced plasma MDA, MCP-1, TNF-α, PAI-1 45, CRP, 8-epi-PGF2 and 8-OHdG 46. In diabetic and hypertensive patients, CAN reduced circulating MCP-1, ICAM-1, VCAM-1, PAI-1 and IL-6, urinary 8-epi-PGF2α, and improved albuminuria and lipid profile 47-49. In CHF patients, short-term therapy with CAN 50 reduced circulating sICAM-1 and sVCAM-1, TNFα and IL-6 although negative results were reported by other authors 51.
In in-vitro conditions, CAN attenuated macrophage accumulation and increased collagen deposition in the aortas of high-cholesterol fed rabbits undergoing aortic balloon injury 52, reduced the interaction between the pro-inflammatory cytokine TNF-α and the receptors for advanced glycation end products (RAGE) in human ECs and the expression of VCAM by inhibiting the binding of NF-κB to the RAGE gene promoter apparently independent of AT1R mechanisms 53. CAN coincubated with dipyridamole, a platelet inhibitor, downregulated Mac-1, the β2 integrin that binds ICAM-1 and promotes movement of neutrophils into the vascular wall, thus reducing the adhesion of neutrophils harvested from stroke patients to human vascular ECs 54. In brain microvessels and middle cerebral artery of spontaneously hypertensive rat (SHR), CAN for 1 month decreased macrophages adhesion and infiltration, endothelial AT1R, ICAM-1, TNF-α, IL-1β and NF-κB expression and normalized eNOS expression 55. CAN amplified brain antioxidant capacity reducing lipid peroxidation, restoring
glutathione 56 and improving ascorbic acid in chronic cerebral hypoperfused with bilateral occlusion of carotid arteries 57. In human aortic ECs, CAN decreased hypoxia-induced apoptosis by inhibiting caspase-3 activation and increasing bradykinin and eNOS expression perhaps by favouring AT2R stimulation by AII 58 suggesting a possible role of ARBs in neurodegenerative disorders characterized by high oxidative stress and inflammation. In diabetic rats and at sub-antihypertensive, CAN reduced oxidative stress markers, and normalized endothelium-dependent vasodilatation 59. Short (2 weeks) and long CAN (22 weeks) treatment of diabetic animals down-regulated renal expression of NADPH oxidase subunit p47phox and p22phox, iNOS, RAGE 60, and inhibited plasma lipid peroxidation products, renal H2O2 production and nitrotyrosine deposition, and reduced albuminuria 61. High-dosage CAN (75 mg/kg/day for 14 months) attenuated proteinuria, ameliorated renal fibrosis and suppressed MCP-1, RANTES, and NF-kB expression in unilaterally nephrectomized SHR 62. CAN decreased serum and epididymal leptin, a pro-inflammatory adipose-tissue derived adipokine, TNF-α and increased serum and epididymal adiponectin, fatty acid synthase, AT2R and PPARγ expression in rats 63. In rat pancreatic stellate cells, CAN attenuated the expression of CTGF and collagen type IV proteins therefore reducing proliferation and ECM protein synthesis aggravated by hyperglycemia 64.
Stress-induced gastric ulcerations and indomethacin-induced intestinal inflammation in rats was reduced by CAN through the inhibition of the interaction between AII and AT1R; TNF-α, CINC-1 (IL-8 family), ICAM-1, thiobarbituric acid-reactive substances, lipid peroxidation and neutrophil infiltration were reduced. In vitro flow system, adherence of neutrophils to human umbilical vein endothelial cell(HUVEC)s exposed to reoxygenation after anoxia was also reduced by CAN independently of antihypertensive effect 65,66. CAN inhibited in a dose dependent manner AII-, TNF-α- or EGF-induced MAPK phosphorylation and exerted anti-proliferative effect on human prostate cancer cells 67.
4.2 Eprosartan (EPR)
EPR reduced neutrophil O2- generating capacity, soluble MCP-1 and soluble VCAM-1 in newly diagnosed hypertensive patients with multiple risk factors for atherosclerosis and increased low-density lipoprotein oxidation lag time, suggesting an increased resistance to oxidation. Hydrochlorthiazide was neutral on those parameters 68, 69.
EPR decreased MCP-1 expression in the myocardium thus reducing macrophage recruitment. Stroke-prone SHRs fed a high-salt, high-fat diet developed heart failure characterized by left ventricular (LV) hypertrophy /pathology and hypocontractility. Suppression of MCP-1 expression might explain in part the reduction of the rate of morbidity/mortality and the preservation of LV systolic function and the reduction in cardiac remodeling/disease
progression 70.
4.3 Irbesartan (IRB)
IRB reduced plasma VCAM-1, TNF-α and O2- in patients with coronary artery disease (CAD) without affecting BP 71 and reduced the protein and activity of MMP-9 and the serum levels of IL-6, hsCRP 72,73.
IRB reduced in vitro generation of IL-6 after lipopolysaccharide(LPS)-stimulation 74 and attenuated cardiac IL1β, TGF-β and MMP activity in diabetic mice and normalized cardiac remodelling and improved LV function 75. The drug inhibited transmigration of monocyte/macrophage in the vessel wall and the expression and protein of MCP-1 and other angiogenic chemokines in aortic sinuses of ApoE knock out mice. IRB also inhibited MCP-1 production by human monocytes through an AII-independent mechanism 76. IRB effectively inhibited vasoconstriction and platelet aggregation induced by two different tromboxane(Tx)A2/PGH2 receptor agonist in canine coronary artery rings 77 and CAD patients 73.
4.4 Losartan (LOS)
In hypertensive, hypercholesterolemic patients, LOS treatment reduced circulating sCD40L 73 and decreased ICAM-1 and VCAM-1 plasma levels in normotensive atherosclerotic patients 78
. The drug improved endothelial function in hypertensive, hypercholesterolemic patients 79, 80
and decreased leukocyte L-selectin as a reflection of chronic inflammatory state 81 although plasma E-selectin and ICAM-1 leukocyte expression remained unchanged 82 in patients with CAD.
ICAM-1 and P-selectin expression and leukocyte rolling, adherence, and transmigration were reduced by LOS in SHR 83 although VCAM-1 expression, MCP-1 and IL-6 did not change in CRP-stimulated HUVEC 84. LOS inhibited basal and stimulated-MCP-1 production and circulating monocyte CD11b expression as well as fatty-streak formation likely through an AII-independent mechanism. CD11b expression was reduced in both peripheral blood and bone marrow monocytes in hypercholesterolemic monkeys 85. LOS ameliorated inflammation associated with low circulating levels of LPS in rat LV myocytes and prevented autoimmune myocarditis likely through AT1R inhibition, modulation of bradykinin degradation and/or decreases in AII levels and recruitment of inflammatory cells in mice 86,8). LOS decreased lipidic deposition, MMP-1 expression in the aorta of hypercholesterolemic rabbits by reducing ROS production and consequent NF-kB activation 88. In diabetic and stroke-prone SHR, LOS attenuated vascular remodelling by decreasing ROS production through inhibition of NADPH oxidase subunit p22phox and AII expression 89,90. LOS and its active metabolite EXP3174 stimulated NO release from platelets by the activation of platelet Ca2+-dependent eNOS and inhibited AII-independent platelet aggregation 91, 92. LOS reduced platelet aggregation and its active metabolite abolished AII dependent and independent generation of TxA2 and PGF2α 93. Both LOS and EXP3179 stimulated phosphorylation of Akt and eNOS through the activation of VEGFR2/PI3K/Akt in bovine and rat aortic endothelium. EXP3179 also reduced ECs apoptosis and suppressed TNFα-induced caspase-3 activation through
AII-independent since EXP3179 did not bind AT1R 94. In hypertensive type 1 diabetic patients with diabetic nephropathy, LOS decreased urinary CTGF 95. The drug also attenuated renal fibrosis and decreased CRP, osteopontin and TGF-β1 in experimental cyclosporine-induced nephropathy 96 and renovascular hypertensive animal model of hypertension 97. LOS slowed renal function decline in remnant kidney rat model in parallel with an interference with NADPH oxidase, NFkB, MCP-1, PAI-1, renal T cell, and macrophage infiltration 98.
In experimental acute pancreatitis, LOS reduced pancreatic and pulmonary myeloperoxidase (MPO) activities, serum IL-6 and IL-1, depletion of IkBβ, NF-kB p65, ICAM-1 and cyclooxygenase(COX)-2 protein expression as well as NF-kB binding activity through AT1R-dependent mechanisms 99. LOS exerted anti-inflammatory effects in mice with ulcerative interstitial cystitis 100. In rat fibrotic lungs LOS reduced the inflammation (MPO activity and protein content) in the bronchoalveolar lavage fluid and the collagen deposition (hydroxyproline content and less morphological changes) 101. LOS combined with captopril attenuated lung injury by suppressing cytokine expression and NF-kB activity in rats 102. In experimental models of acute arthritis, LOS reduced inflammation and suppressed TNF-α generation from inflamed human synovium 103.
4.5 Olmesartan (OLM)
Long-term OLM treatment reduced serum levels of inflammation markers as hs-CRP (15.1%), hs-TNFα (8.9%), IL-6 (14.0%) and MCP-1 (6.5%) in hypertensive patients characterized by increased hsCRP and decreased plasma levels of 8-iso-PGF2α, a biochemical marker of oxidative stress in patients with type 2 diabetes 104.
OLM decreased MCP-1 expression and macrophage infiltration in hyperlipidemic rabbits and in rats with hypertensive diastolic heart failure 105-107 and reduced TGFβ1 expression, NADPH oxidase activity, cardiomyocyte sizes and collagen area even at hemodynamically inactive doses 108. OLM treatment prevented the phosphorylation of ERK, angiotensinogen, IL-6 and iNOS expression induced by hypobaric hypoxia, in mice lung 108. In human ECs, OLM has been shown to downregolate TNFα-induced AGE and VCAM-1 expression at least partially through the inhibition of the binding of NF-kB to the RAGE gene promoter 53. In obese mouse, OLM improved TNF-α, PAI-1, MCP-1, and serum amyloid at least in part by reducing ROS production and NADPH oxidase subunits expression 109.
4.6 Telmisartan (TEL)
TEL suppressed retinal expression of ICAM-1 and VEGF in diabetic mice and of MCP-1 in glucose-stimulated murine brain-derived capillary ECs, an effect mediated by the inhibition of NF-kB pathway 110. In stroke-prone SHR, TEL enhanced eNOS and reduced NADPH oxidase subunit p22phox 90 and protected from experimental intracerebral hemorrhage while inducing eNOS and PPARγ expression and reducing TNF-α and COX-2 111. TEL also decreased vascular O2- production by reducing NADPH dependent oxidase activity and reduced urine and plasma 8-iso-PGF2α level in apoE-knock out mice 112.
In patients with CAD, TEL inhibited lymphocytes-expressed β2-integrin independently from AII stimulation suggesting an AT1R independent atheroprotective effect 113. TEL acted as a partial PPARγ agonist while increasing adiponectin and improved insulin sensitivity and lipid
profile in obese rats and patients with MS 114,115. Those effects were attributed to post-transcriptional PPARγ activation and, in support of that interpretation, TEL facilitated differentiation of preadipocytes, enhanced PPARγ-target gene and GLUT4 expression and 2-deoxy glucose uptake both in basal and insulin-stimulated in cultured and human adipocytes similar to pioglitazone 116. In hypertensive patients with MS, treatment with TEL increased adiponectin and reduced microalbuminuria and hs-CRP in ARB-treated patients supporting additional effects beyond AT1R blockade 117. In uncomplicated type 2 diabetic patients, TEL suppressed inflammatory and lipid peroxidation markers 118 and improved flow-mediated vasodilation and oxidative stress markers in type 1 diabetic patients 119. Saho et al. demonstrated amelioration of H2O2-induced PAI-1 expression in mesangial cells from wild-type and also AT1R knockout mice possibly owing to its lipophilic and antioxidant structure in absence of interference on PPAR-γ system 120. TEL inhibited cell proliferation and MAPK phosphorylation in prostate cancer cells and high concentrations of the drug exhibiting partial PPARγ agonism were less efficient in that respect 121. TEL attenuated hepatic steatosis and reduced indices of inflammation and fibrosis in experimental nonalcoholic steatohepatitis and also decreased both subcutaneous and visceral fat 122. The ARB protected the function of retinal neural in mice with LPS-induced uveitis, attenuated the expression glial fibrillary acidic protein and enhanced the expression of synaptophysin and rhodopsin essential for neural activities in Muller glial cells 123. Moreover, TEL, as well as VAL, suppressed laser-induced choroidal neovascularization and suppressed TNF- or LPS-laser-induced macrophage infiltration in the retinal pigment epithelium-choroid complex in human and murine tissues 124.
4.7 Valsartan (VAL)
In hypertensive patients, VAL reduced ROS-derived plasma MDA levels, enhanced plasma SOD and copper levels, an antioxidant trace element 125,126. The drug reduced vascular inflammation and fibrinolytic dysfunction induced by postprandial hypertriglyceridemia and attenuated the increase in P-selectin, PAI-1, and t-PA antigen in response to high-fat meal 127 and on longer term treatment reduced CRP levels and monocyte ROS formation and IL-6 production in both hypertensive and normal subjects 128 in whom VAL inhibited O2- anion generation, CRP levels and NF-kB activation 129. VAL treatment lowered plasma MCP-1 and IL-6 in parallel with urinary 8-epi-PGF2α, 8-OHdG, albumin, and type IV collagen excretion in diabetic patients with nephropathy 48. In healthy volunteers VAL attenuated hyperglycemia-induced endothelial dysfunction and reduced the release of IL-6 and TNF-α from leukocytes in response to hyperinsulinemia 130.
In hypercholesterolemic ApoE knock out mice, VAL blunted inflammation and oxidative stress independent of BP reducing the expressions of NADPH subunits p22phox and p47phox, MCP-1 and ICAM-1 and the production of O2-131. Similar to TEL, VAL suppressed retinal adhesion of leukocytes and the expression of ICAM-1 and VEGF in diabetic mice and of ICAM-1 and MCP-1 in glucose-stimulated murine brain-derived capillary through inhibition of AT1R/NF-kB pathway 110. Combined with fluvastatin, VAL inhibited neointimal formation induced by femoral artery stenosis in mice and the proliferation of rat VSMCs 132 and stimulated NO release through the activation of platelet Ca2+-dependent eNOS 91. MCP-1, TGF-β1 and IL-1β expression were lowered in stroke prone SHR 133.
VAL improved the renal interstitial fluid NO production 89 and reduced intrarenal level of TNF-α 134 in diabetic rats via direct action on TNF-α at the level of the kidney. As other ARBs, VAL suppressed laser-induced choroidal neovascularization 124 suggesting a favourable effect of ARB in improving microvascular complications in diabetes.
5. SPECIFIC BACKGROUND
Increased expression of VCAM 135 and increased ROS generation 136 are important contributing factors to the atherosclerotic process. The RAS through AII, its main effector, promotes both processes at the endothelial level in synergy with other pro-atherogenic biological systems 137. Therefore, vascular endothelium represents an ideal target for anti-atherosclerotic actions of ARBs 138, possibly through mechanisms unrelated to AT1R blockade 139. In this context, TEL, a lipophilic, highly selective ARB 140, is receiving attention because of an array of pharmacological properties unrelated to AT1R antagonism per se including partial agonism for PPAR-γ nuclear receptor system and other pleiotropic actions including modulation of oxidative stress and inhibition of pro-inflammatory stimuli 141. This study intended to analyze in greater detail the pleiotropic effects of TEL with particular reference to its AII independent anti-inflammatory and anti-oxidant potential.