CAPITOLO II
2.1
CAMPIONAMENTO
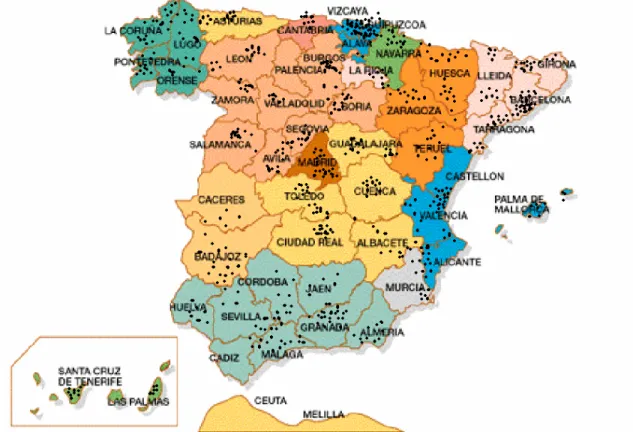
Il materiale oggetto di questo studio è DNA estratto da cellule dell’epitelio della mucosa boccale raccolto fra 300 spagnoli di ambo i sessi, non imparentati fra loro, residenti in tutte le 17 Comunità Autonome spagnole. La raccolta è avvenuta nei luoghi dove risiedono i donatori o nella Università Complutense nella quale tali persone studiano o lavorano. I donatori hanno dato il loro consenso al trattamento dei dati genetici in forma anonima per scopi scientifici. Sono stati scelti soggetti appartenenti alla stessa regione da almeno tre generazioni, più altri con i nonni di varia provenienza come rappresentanti del campione complessivo della nazione spagnola.
Secondo quanto spiegato nell’introduzione, i campioni sono stati raggruppati in base a tre distinti criteri di associazione:
1. un criterio storico-politico che raggruppa fra loro individui provenienti da Comunità Autonome con processi storici condivisi
2. un criterio geografico che raggruppa fra loro vaste zone con confini stabiliti in base all’orografia del terreno o alle caratteristiche climatiche tipiche di ciascuna area considerata
3. un criterio linguistico che raggruppa fra loro individui che parlano la stessa lingua, considerata tale secondo l’ordinamento linguistico spagnolo.
In un secondo livello di analisi l’intero campione tipizzato è stato confrontato con altre popolazioni del bacino del Mediterraneo al fine di stabilire la sua collocazione in tale panorama genetico. I raggruppamenti effettuati e le dimensioni di ciascuna popolazione sono descritti in modo esauriente nella Sezione Risultati.
La figura 2.1 mostra la distribuzione dei campioni tipizzati nel territorio spagnolo.
2.2
METODI MOLECOLARI
2.2.1 ESTRAZIONE DI DNA DA CELLULE DELL’EPITELIO DI DESQUAMAZIONE
Il DNA viene estratto utilizzando il kit BuccalAmpT M DNA Extraction Kit della EPICENTRE dotato di cottonfiok per la raccolta e soluzione per l’estrazione. Il materiale raccolto nel bastonc ino viene rilasciato per agitazione nella soluzione di estrazione e sottoposto ai seguenti cicli di riscaldamento e vortexing: 30 min a 65°C, 10 sec di vortex, 8 min a 98°C, 15 sec di vortex, 8 min a 98°C ed ancora 15 sec di vortex. In breve, l’estrazione è ottenuta mediante lisi delle cellule, digestione di proteine e RNA. Il DNA estratto è conservato a -20ºC dopo averne raccolto un’aliquota di 25µl da congelare e scongelare per l’analisi molecolare in modo tale da evitare il deterioramento del campione completo.
2.2.2 ANALISI MOLECOLARE
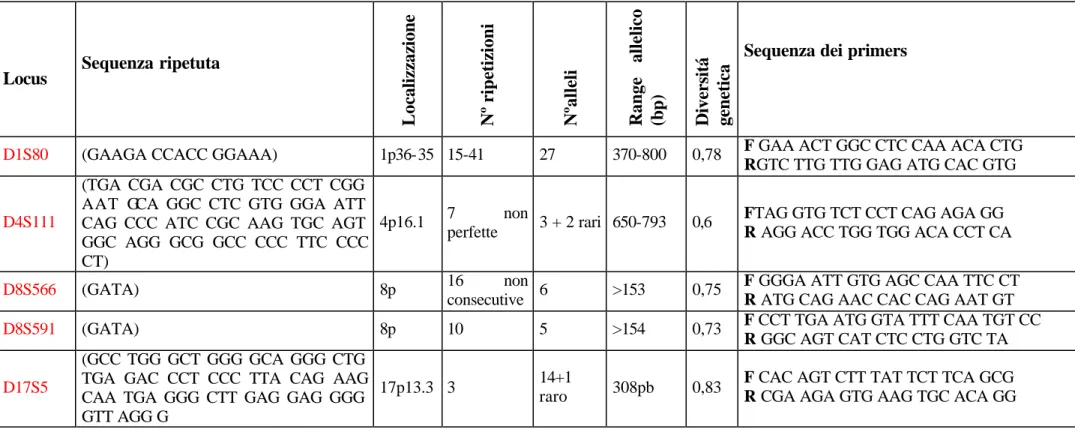
Il DNA genomico è stato tipizzato per tre sistemi VNTR e due sistemi STR per amplificazione con PCR (Horn, 1989). La descrizione dei marcatori è riportata in tabella 2.1.
- Amplificazione con PCR
La PCR è la reazione a catena della polimerasi (polimerase chain reaction) ed è oggigiorno il metodo più rapido ed efficace per l’amplificazione selettiva di un frammento di DNA ottenuta sfruttando la replicazione enzimatica effettuata da una polimerasi termoresistente estratta da microrganismi termofili (Taq polimerasi).
Per poter amplificare in modo unico la sequenza di interesse è necessario avere delle conoscenze a priori sulla stessa, in modo tale da poter costruire due sequenze oligonucleotidiche da utilizzare come primers (inneschi della reazione).
Tali inneschi, una volta aggiunti a DNA denaturato, si appaiano in senso opposto a sequenze di DNA complementari immediatamente fiancheggianti la regione da amplificare su ciascuno dei due filamenti. I primers vengono sintetizzati chimicamente uno in direzione 5’-3’ di un filamento (primer forward) e uno in direzione 3’-5’ del filamento opposto (primer reverse).
A seguire indichiamo in una tavola la descrizione dei marcatori con la sequenza ripetuta e la sequenza dei primers.
Si é utilizzata la “Taq PCR Master Mix” (Qiagen) che contiene nella stessa soluzione tutti i quattro nucleotidi trifosfato e il cofattore della polimerasi Mg++, sottoforma di MgCl nelle giuste proporzioni. Si conserva congelata a -20ºC.( La Taq deriva dalla polimerasi di un batterio che occupa la nicchia ecologica delle sorgenti d’acqua calda delle falde oceaniche: Thermus aquaticus).
La reazione avviene all’interno di un termociclatore che effettua ripetuti cicli sequenziali a distinte temperature, necessarie per le tre fasi del processo: denaturazione, appaiamento o annealing, estensione.
a) Denaturazione: consiste nella separazione della doppia elica in due filamenti di DNA che avviene solitamente intorno ai 95°C per il DNA genomico umano. b) Appaiamento o anneal ing: consiste nell’unione specifica fra i primers, presenti
temperatura di questo passaggio varia dai 50°C ai 70°C a seconda della temperatura di fusione
Locus Sequenza ripetuta
Lo
calizzazione
Nº ripetizioni Nºalleli Range allelico (bp) Diversitá genetica
Sequenza dei primers
D1S80 (GAAGA CCACC GGAAA) 1p36-35 15-41 27 370-800 0,78 F GAA ACT GGC CTC CAA ACA CTG
RGTC TTG TTG GAG ATG CAC GTG
D4S111
(TGA CGA CGC CTG TCC CCT CGG AAT GCA GGC CTC GTG GGA ATT CAG CCC ATC CGC AAG TGC AGT GGC AGG GCG GCC CCC TTC CCC CT)
4p16.1 7 non
perfette 3 + 2 rari 650-793 0,6
FTAG GTG TCT CCT CAG AGA GG R AGG ACC TGG TGG ACA CCT CA
D8S566 (GATA) 8p 16 non
consecutive 6 >153 0,75
F GGGA ATT GTG AGC CAA TTC CT R ATG CAG AAC CAC CAG AAT GT
D8S591 (GATA) 8p 10 5 >154 0,73 F CCT TGA ATG GTA TTT CAA TGT CC
R GGC AGT CAT CTC CTG GTC TA
D17S5
(GCC TGG GCT GGG GCA GGG CTG TGA GAC CCT CCC TTA CAG AAG CAA TGA GGG CTT GAG GAG GGG GTT AGG G
17p13.3 3 14+1
raro 308pb 0,83
F CAC AGT CTT TAT TCT TCA GCG R CGA AGA GTG AAG TGC ACA GG
c) (melting temperature Tm) della doppia elica in corrispondenza delle regioni di appaiamento, dipendente dal contenuto in G+C.
d) Estensione: consiste nella fase di polimerizzazione dei nuovi filamenti in direzione 5’-3’ nel tratto compreso fra i due primers, utilizzando la sequenza target come stampo. Il tutto avviene ad una temperatura di 70-75°C.
Al termine di tutti i cicli voluti, dopo un’ultima estensione prolungata, i prodotti amplificati vengono raffreddati per impedire la rinaturazione delle doppie eliche.
Il termociclatore usato per la reazione è il PTC-100, Programmable Termal Controller della MJ Research, Inc.
Poiché la quantità di DNA era spesso molto variabile, le reazioni di PCR sono state ottimizzate di volta in volta nell’intento di ottenere i migliori risultati. Si possono variare concentrazione dei primers e temperatura di annealing (tanto più specifica quanto più alta), ma, con la Taq Master Mix, non si può cambiare la concentrazione della polimerasi che deve sempre occupare la metà del volume della miscela di reazione.
-Visualizzazione dei frammenti amplificati
Al fine di valutarne resa, specificità e dimensioni, i prodotti amplificati sono stati visualizzati su gel di agarosio o poliacrilammide a seconda delle loro dimensioni.
-Visualizzazione su gel di agarosio
I minisatelliti D17S5 e D4S111 sono stati visualizzati su gel di agarosio al 2% in seguito a corsa elettroforetica orizzontale a 100V per una durata di 3 ore.
La tecnica prevede l’utilizzo di un gel con due file di pozzetti nei quali vengono caricati i campioni amplificati. Il gel è alloggiato in una vaschetta elettroforetica su un supporto rigido ed è completamente immerso in una soluzione tampone TAE 1X (Tris acetato 0.04 M; EDTA pH 8, 0.001 M). Due elettrodi posti ai lati del supporto, collegati ad una sorgente di corrente continua, pescano direttamente nella soluzione tampone che funge da ponte salino. Quando tra i due elettrodi viene applicata una differenza di potenziale si genera un gradiente di potenziale elettrico in presenza del quale qualsiasi sostanza dotata di carica migra attraverso il gel con una direzione ed una velocità dipendenti dalla tensione applicata, dal segno e dalla grandezza della carica, dalle dimensioni dello ione e dal pH del tampone utilizzato.
V= Eq/f
dove f rappresenta la forza d’attrito che si oppone al passaggio delle molecole, rallentandone il movimento, mentre Eq è la tensione applicata ai capi del dispositivo. Tale velocità è direttamente proporzionale alla carica elettrica ed inversamente proporzionale alla massa dello ione. Giocando sulla concentrazione dell’agarosio, si può migliorare il potere di risoluzione delle bande di diverso peso molecolare , realizzando un gel con una maglia più o meno stretta che trattiene in modo diverso le molecole nella loro corsa elettroforetica.
Il DNA amplificato viene appesantito con una soluzione di blu di bromofenolo che funge da tracciante del fronte di migrazione, consentendo di seguire la regolarità della corsa.
La soluzione del gel contiene 2µl di bromuro di etidio, un intercalante del DNA che assorbe nell’ultraviole tto ed emana nel visibile. I frammenti amplificati, catturati nel gel, vengono visualizzati mediante esposizione ad una fonte di luce ultravioletta (λ=312nm). La lunghezza dei frammenti di ogni banda viene dedotta dal confronto con le bande del marcatore di peso molecolare. Questo é un marcatore che va di 100 in 100 pb, con la banda di 500pb più intensa delle altre. È fornito dalla casa Estone Fermentas.
- Visualizzazione su gel di poliacrilammide
L’acrilammide è stato utilizzato come supporto per la corsa elettroforetica dei microsatelliti D8S591, D8S566 e per il minisatellite D1S80, dato il suo maggior potere di risoluzione rispetto all’agarosio. I principi fisici sui quali si basa la tecnica sono gli stessi indicati per la visualizzazione dei frammenti con l’uso di gel di agarosio.
La corsa elettroforetica su gel di acrilammide è verticale, si realizza con apparati dotati di due vaschette (una inferiore ed una superiore) che contengono il tampone, un tris di vetri nei quali sono alloggiati due gel ed un tappo dotato di due elettrodi di platino che vengono immersi nella soluzione di tampone, uno in ogni vaschetta, e collegati con la fonte di corrente; l’inferiore al polo positivo ed il superiore a quello negativo.
La soluzione di poliacrilammide utilizzata per la costruzione dei gel é al 30% con una proporzione 29:1 di acrilammide e N-Ndimetilbisacrilammide.
D1S80 é stato sottoposto a corsa elettroforetica in gel all’8%, a 90V per 17h. D8S591 e D8S566 sono stati visualizzati in gel al 12%, a 100V per 19h e 18h rispettivamente. Alla soluzione del gel si aggiunge tampone TBE (acido borico, tris, EDTA pH8), PSA
Anche in questo caso si carica in uno dei pozzetti di ogni gel un marcatore, standard, di peso molecolare. In questo caso, per i microsatelliti, si é utilizzato un marcatore che va di 20 in 20pb, da 20pb a 1000pb con la banda di 200pb e quella di 500pb piú intense. É il superladder-low 20pb prodotto da GenSura Laboratories, Inc. Per il minisatellite D1S80 si é utilizzato un marcatore allelico che marca in modo piú intenso le bande degli alleli piú frequenti.
I frammenti amplificati sono visualizzati mediante impregnazione argentica (Silver staining). Una volta terminata la corsa elettroforetica, il gel viene posto in una soluzione all’1% di acido acetico e al 45% di metanolo che serve a fissare la banda di DNA. Segue un bagno in una soluzione d’argento allo 0,2% durante il quale il metallo si intercala ai frammenti di DNA. Dopo averlo risciacquato per tre volte, il gel viene sommerso in una soluzione riducente di NaOH e formaldeide. L’Ag ridotto dà al gel un caratteristico colore bruno-arancione più scuro (nero) nelle bande. I gel vengono scannerizzati in uno scanner con luce sia dal basso che dall’alto. La lettura è facilitata dall’uso di programmi, quali Multi Analyst, per valutare la posizione relativa delle bande del campione rispetto a quelle dello standard.
- Marcatori molecolari
I marcatori molecolari scelti sono: D1S80, D4S111, D8S566, D8S591, D17S5 localizzati nei cromosomi 1, 4, 8 e 17 rispettivamente.
I programmi di amplificazione eseguiti per ogni marcatore sono indicati nelle seguenti tabelle: CICLI 1 2 3 4 5 6 7 TEMPERATURA 95ºC 95ºC 55ºC 72ºC ripetere 35 cicli 72ºC 4ºC DURATA 5 min 15 sec 15 sec 30 sec 10 min ∞
CICLI 1 2 3 4 5 6 7 TEMPERATURA 95ºC 95ºC 55ºC 72ºC ripetere 35 cicli 72ºC 4ºC DURATA 5 min 15 sec 15 sec 30 sec 10 min ∞
Tabella 2.3: Programma di amplificazione di D4S111e D17S5
CICLI 1 2 3 4 5 6 7 TEMPERATURA 95ºC 95ºC 65ºC 72ºC ripetere 40 cicli 72ºC 4ºC DURATA 5 min 1 min 1 min 1 min 10 min ∞
Tabella 2.4 : Programma di amplificazione per D1S80
Mini e microsatelliti
H2O
Taq PCR Master Mix Primer
DNA
Volume Totale
QUANTITÀ PER CAMPIONE (µl) 2,8
6
0,6 + 0,6 1
12
2.3
METODI STATISTICI
Lo studio della struttura genetica della popolazione spagnola ed il confronto di questa con altre popolazioni del bacino del Mediterraneo è stato effettuato su due livelli di analisi: un primo livello sull’analisi della diversità genetica (locus per locus) ed un secondo livello sullo studio degli aplotipi.
2.3.1 VERIFICA DELL’EQUILIBRIO DI HARDY-WEINBERG
Per la verifica dell’equilibrio di Hardy-Weinberg abbiamo scelto un test di probabilità che utilizza il principio del test esatto di Fisher: la probabilità di accettare l’ipotesi nulla Ho (unione casuale degli individui) corrisponde alla somma cumulata delle probabilità di tutte le tabelle aventi lo stesso numero di alleli che abbiano una differenza tra le medie delle distribuzioni minore o uguale a quella osservata.
E’ stato utilizzato il software Genepop 3.4 (Raymond e Rousset, 1995) che rappresenta un ottimo sistema di analisi per campioni di popolazioni costituite da un esiguo numero di individui. Questo software, inoltre, è in grado di minimizzare l’errore che può derivare dall’elaborazione dei dati dei sistemi ad elevato polimorfismo. Infatti, i sistemi utilizzati, essendo multiallelici, possono presentare frequenze teoriche di alcune classi genotipiche inferiori a 1, che portano per semplice effetto matematico ad elevati valori di χ2
.
Per il valore di χ2
cumulativo (per tutti i loci) è stato usato il metodo di Fisher (1954) che combina i valori di probabilità P secondo la formula:
∑
= − = k i P 1 2 ln 2 χ2.3.2 TEST DI DIFFERENZIAMENTO TRA LE DISTRIBUZIONI ALLELICHE E GENOTIPICHE (G TEST)
Il test di differenziamento tra le distribuzioni alleliche e genotipiche di due o più campioni ad un determinato locus valuta l’esistenza di uno scostamento statisticamente significativo rispetto all’ipotesi nulla di uguale distribuzione.
Formalmente la procedura è una estensione ad una tabella di contingenza r x k (r = numero di popolazioni, k = numero di alleli) del classico test esatto di Fisher sviluppato originariamente per una tabella 2x2.
In un test esatto, tutte le possibili tabelle di contingenza, aventi gli stessi totali marginali rispetto a quella data, vengono ordinate secondo i valori di una particolare statistica e viene calcolata la somma delle probabilità per le tabelle che presentano i valori estremi; tale somma è il valore di probabilità p del test.
Nel test esatto di Fisher tale statistica è la differenza tra le medie delle due distribuzioni di frequenza confrontate in due campioni; la probabilità della tabella data, sotto l’ipotesi di assortimento casuale, è calcolata come frequenza cumulata delle tabelle, fra tutte quelle ottenibili per permutazione, che abbiano una differenza fra le medie minore o uguale a quella osservata. Nell’ estensione ad una tabella r x k, dato il numero elevato di tutte le combinazioni possibili, è necessario procedere ad una approssimazione nella stima del parametro p. Lo spazio di tutte le possibili repliche della tabella e dei loro valori di probabilità viene esplorato con algoritmi che consentono una stima approssimata del valore di p condotta sulla base di un sottoinsieme casuale tra tutte le possibili soluzioni.
E’ stato utilizzato il software Genepop 3.4 (Raymond e Rousset, 1995), poiché rappresenta un ottimo sistema di analisi per campioni di popolazione costituiti da un esiguo numero di individui.
Per il valore di χ2 cumulativo (per i loci) è stato usato il metodo di Fisher (1954) che combina i valori di probabilità P secondo la formula:
∑
= − = k i P 1 2 ln 2 χ2.3.3 DIVERSITÁ GENETICA
Una delle misure più utilizzate per stimare il grado di polimorfismo di un locus entro una popolazione è l’indice di diversità genetica, o eterozigosi, che viene calcolata dalla somma dei quadrati delle frequenze di tutti i suoi alleli, sottratta all’unità:
H oss = 1 -2 1 i k i p
∑
=dove i=(1,…,k) è il numero di alleli e p la frequenza di ciascun allele.
Per un gene diploide in una popolazione in equilibrio, tale espressione corrisponde alla proporzione attesa di individui eterozigoti, per il locus considerato.
Il termine “Gene Diversity”, coniato da Nei e Roychoudhury nel 1974, non è che la definizione formale del valore di eterozigosità attesa in una popolazione. I termini eterozigosità attesa e Gene Diversity (Diversità Genica) sono spesso utilizzati in modo equivalente.poiché la nostra analisi ha riguardato un campione numericamente esiguo, la stima dell’indice di diversità genetica attesa è stato corretta per piccoli campioni, come suggerito da Nei (1987):
− − =
∑
= k i i p n n H 1 2 1 1dove n = numero di individui del campione in esame.
L’indice H assume valore zero in assenza di polimorfismo e si avvicina tanto più ad uno quanto maggiore è il grado di variabilità osservabile nel campione per quel determinato locus.
La stessa formula calcola la “diversità aplotipica” quando pi rappresenta la frequenza dell’i-esimo aplotipo in una popolazione.
La varianza per H associata al processo di campionamento è stata calcolata come indicato da Nei (Nei e Roychoudhury, 1974).
( )
(
) (
{
−)
[
∑
−(
∑
)
]
+∑
−(
∑
)
}
− = 3 2 2 2 2 2 2 2 2 1 2 2 2 i i i i x x x x n n n n h VQuando più loci sono tipizzati per lo stesso campione, il grado di polimorfismo medio esistente all’interno della popolazione può essere convenientemente espresso dalla Diversità Genetica media (“avereged Gene Diversity”) che è data dalla media aritmetica dei valori calcolati per i singoli loci (Nei, 1987) secondo la formula:
H =
∑
= k i i l h 1in cui l è il numero dei loci ed i indica un generico locus.
La varianza per H è stata calcolata secondo la formula di Nei e Roychoudhury (1974):
V(H)= 1
( )
ˆ ˆ /(
1)
2 1 − −∑
= k H h k k i 2.3.4 DISTANZE GENETICHEIn genetica di popolazioni esistono tanti metodi per calcolare la distanza genetica esistente tra le popolazioni e ciascuna si avvale di modelli mutazionali differenti (IAM, stepwise).
Il campione di questa tesi è stato studiato con il cosiddetto “metodo della corda” di Cavalli-Sforza e Edwards (1964).
Tofanelli et al.(2001) hanno osservato che la distanza genetica della corda –Dc- possiede valori di linearità e varianza tali da renderla ottimale per lo studio di problematiche evolutive che riguardano tempi di divergenza recenti. È per questo che si è fatto riferimento a tale tipo di distanza per descrivere la sottostruttura genetica del campione spagnolo composto da popolazioni molto vicine fra loro. Il metodo non segue nessun modello mutazionale (free model) e prevede la trasformazione dei dati di freque nza in una distanza angolare θ. La popolazioni sono concettualizzate in punti in uno spazio euclideo a m dimensioni (dove m è il numero totale di loci in entrambe le popolazioni). La distanza θ corrisponde all’angolo tra questi due punti, dove:
cos(?)= . . 1 1
∑
= i i y xe xi e yi sono le frequenze dell’i-esimo allele rispettivamente nelle popolazioni X e Y. Dc è una distanza geometrica che sarà misurata sulla linea diretta che unisce i due punti,
Dc = θ π 1 cos 2 2 −
La Distanza della Corda è sta calcolata con il programma Microsat 1.5d (Minch et al., 1997).
2.3.5 MULTIDIMENSIONAL SCALING (MDS)
Il multidimensional Scaling (Kruscal, 1964) è una procedura matematica che consente di rappresentare gli oggetti in esame in uno spazio euclideo, definito da un numero qualsivoglia ridotto di dimensioni, in modo tale che le distanze riprodotte riflettano con la maggior fedeltà possibile i valori osservati sperimentalmente. Il criterio è quello di collocare gli oggetti rispettando l’ordine reciproco tra tutte le misure a coppie date. Gli elementi della matrice analizzata possono essere costituiti da valutazioni che esprimono il grado di similarità o di differenziazione; nel nostro caso si tratta della distanze genetiche osservate tra coppie di popolazioni.
Sappiamo che dati n oggetti, definiti dalle distanze reciproche osservate sperimentalmente, essi, naturalmente, sono sempre perfettamente posizionabili in uno spazio euclideo a n-1 dimensioni. L’obiettivo del metodo MDS è di “costringere” questa rappresentazione, difficilmente visualizzabile ed interpretabile , all’interno di uno spazio caratterizzato da un numero inferiore di dimensioni, in ultima analisi, abitualmente, un sistema cartesiano (nel nostro caso bidimensionale), in modo però da introdurre la minor distorsione possibile rispetto ai rapporti originali.
La procedura di riduzione, a partire da una prima proiezione dei dati nel sistema spaziale definito, passa attraverso una serie di iterazioni (Guttman, 1968), nel corso delle quali gli oggeti vengono di volta in volta riarrangiati in nuove configurazioni. La fedeltà con cui ogni determinata configurazione riflette la matrice reale o, in altri termini il grado di adattamento tra le distanze a coppie riprodotte e le distanze osservate vengono valutati attraverso la misura di Stress (Phi) che è così definita:
In questa formula dij rappresenta la distanza osservata sperimentalmente tra due oggetti i e j (nel nostro caso le popolazioni) e δij è la distanza corrispondente alla rappresentazione spaziale ottenuta. Maggiore è l’adattamento tra le due matrici, quella osservata e quella derivata, minore è il valore di questo parametro. La sequenza di iterazione è governata da questo principio e viene interrotta nel momento in cui diventa relativamente “Improbabile” passare ad una distribuzione spaziale con un livello di Stress inferiore rispetto alla precedente. L’algoritmo opera confrontando continuamente la matrice relativa alle proiezioni ottenute con quella reale e misura la rapidità con cui diminuisce lo Stress. Quando tale velocità rimane costante al di sotto di una soglia minima prefissata la procedura ha termine (Guttman, 1968).
Per tale tipo di analisi è stato utilizzato il programma “Statistica” per Windows versione 5.1.