Capitolo 2
I primi passi del trattamento del dato e
le principali tecniche di normalizzazione
La facilità nel quantificare i dati provenienti da un microarray è fra le principali caratteristiche degli esperimenti che fanno uso di questi supporti e la rapidità con la quale questo processo viene eseguito consente di rendere minimo il tempo che intercorre fra la realizzazione dell’esperimento e l’ottenimento della risposta che si sta cercando.
Analisi tradizionale Analisi con microarray Dati Dati Esperimento Esperimento
Tuttavia queste caratteristiche non devono portare alla conclusione che le metodiche di estrazione e di trattamento del dato siano impostate
su basi matematiche semplici; in realtà, la teoria che è alla base della quantizzazione dei segnali acquisiti, del calcolo del rapporto delle loro intensità, della normalizzazione del dato e dell’estrazione del risultato è estremamente sofisticata e pone pesanti problemi soprattutto dal punto di vista dell’analisi statistica di dati generati con esperimenti simultanei su migliaia di geni.
Prima ancora di occuparsi di analisi statistica, è necessario comprendere quali siano i passi che portano ad ottenere un dato “ripulito” dagli errori generati dallo stesso esperimento e dal processo di estrazione. La mancata eliminazione di questi errori può rendere inconsistente l’analisi ed invalidare l’intero esperimento.
2.1 Diagramma del trattamento dei dati
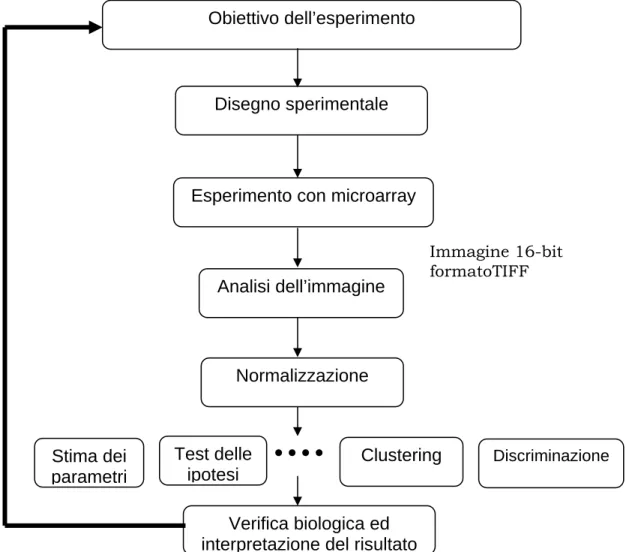
L’utilizzo sempre più diffuso dei microarray ha portato alla formulazione di un vero e proprio diagramma di flusso universalmente riconosciuto da chi lavora in questo settore, i cui passi consentono operativamente di eliminare gli errori sistematici e di preparare il dato per l’analisi conclusiva. Un esempio estensivo di tale diagramma è riportato in figura 2.1.
Obiettivo dell’esperimento
Test delle ipotesi
Verifica biologica ed interpretazione del risultato Esperimento con microarray
Disegno sperimentale
Figura 2.1: Diagramma di flusso operativo in un esperimento di microarray. Stima dei parametri Analisi dell’immagine Normalizzazione Immagine 16-bit formatoTIFF Clustering Discriminazione
2.2 Il processo di quantizzazione del dato
Gli scanner per microarray acquisiscono i dati di intensità di fluorescenza sottoforma di immagini in formato TIFF; l’immagine TIFF è una mappa d’intensità in due dimensioni della superficie del microarray e i segnali di fluorescenza sono immagazzinati nei suoi pixel. Ogni spot del microarray, che spesso individua un unico gene, è formato da diversi pixel ognuno dei quali contiene un’informazione quantitativa sullo spot.
Figura 2.2: Catena di generazione del segnale per eccitazione del fluorocromo con scanner laser.
L’analisi dell’immagine di un microarray può essere suddivisa in quattro fasi:
• posizionamento della griglia (gridding) dell’immagine; • segmentazione;
• estrazione delle intensità o quantizzazione; • correzione del background.
Fluorocromo Fotoni Elettro Segnale
Laser Fotomoltiplicatore conversione
2.2.1 “Gridding” dell’immagine
Dopo aver portato a termine il protocollo di ibridizzazione dei campioni sul microarray e aver acquisito l’immagine con lo scanner laser è necessario identificare la posizione di ogni spot sul supporto. Ciò avviene grazie all’allineamento sull’immagine di una griglia che viene generalmente fornita dal costruttore del microarray. Ogni cerchietto di questa griglia identifica l’ideale posizione dello spot, secondo quelle che sono le specifiche costruttive del microarray, e fornisce all’analista diverse informazioni sullo spot in esame grazie ad un file allegato che contiene, fra tante altre informazioni, anche il nome del gene corrispondente ad ogni spot e i suoi codici d’identificazione nelle banche dati genomiche.
Il corretto posizionamento della griglia permette di ricavare un dato consistente sugli spot; per questo motivo, spesse volte è necessario controllare l’allineamento spot a spot e intervenire manualmente su quegli spot che non vengono esattamente centrati o delimitati dalla griglia.
E’ fondamentale, come è facile intuire, che il processo di deposizione delle sonde sul supporto avvenga secondo un ben preciso schema di righe e colonne, in modo da agevolare l’identificazione degli spot.
2.2.2 Segmentazione
Una volta che gli spot sono stati identificati, è necessario separare il contributo del segnale da quello del background; per questo motivo deve essere riconosciuta la forma di ogni spot attraverso una “spot mask”.
Segnale Background
Figura 2.3: Separazione del background dal segnale attraverso una “spot mask”.
Generalmente si assume che gli spot abbiano forma circolare di diametro costante; coerentemente con questa ipotesi si identifica come segnale tutto ciò che cade all’interno del cerchio e come background tutto quello che è all’esterno, operando una segmentazione spaziale.
Figura 2.4: Segmentazione spaziale dello spot con griglia di forma prefissata.
Questa semplice assunzione viene raramente rispecchiata dagli spot sul vetrino e ciò è riconducibile solitamente ad errori nella fase di deposizione delle sonde. Per questo motivo molti software di analisi dell’immagine includono la possibilità di fare una segmentazione per intensità dei pixel: in questo procedimento si sfruttano i valori di intensità dei pixel per delimitare l’area da attribuire al segnale, utilizzando algoritmi di “Seeded Region Growing” (SRG) comuni a molti software di manipolazione di immagini.
Figura 2.5: Segmentazione per intensità con algoritmo SRG.
2.2.3 Estrazione delle intensità di segnale e di background Il processo che consente di passare dall’insieme delle informazioni relative ad uno spot al suo valore numerico, rappresentativo della concentrazione di mRNA di quel gene nel campione, prende il nome di
quantizzazione o quantificazione.
La quantizzazione assoluta porta ad ottenere un dato cumulativo dell’intensità dello spot su un canale (ad esempio il rosso) senza metterlo in relazione con quello ottenuto sull’altro canale (il verde); la quantizzazione relativa, invece, ricava il rapporto delle intensità assolute sui due canali, detto fold change, e serve ad avere informazioni sul livello di espressione in un canale rispetto all’altro.
I valori di segnale e di background possono essere calcolati in diversi modi, fra i quali il calcolo della media e della mediana sono fra i più comuni.
Il calcolo della media del segnale o “average intensity signal” consiste nel rapporto fra la somma delle intensità dei pixel identificati come segnale e il numero totale dei pixel che appartengono alla regione di demarcazione dello spot. Un calcolo analogo può essere fatto per la media del background prendendo in considerazione solo i pixel identificati come rumore di fondo dalla segmentazione. Per calcolare la mediana, invece, si ordinano per valore ascendente o discendente tutti i valori di intensità dei pixel della zona di demarcazione e si prende l’intensità del pixel che si posiziona a metà
dell’ordinamento come rappresentativa dell’intera zona. Il valore di mediana di uno spot è generalmente più robusto di quello di media e ciò è dovuto al fatto che il suo procedimento di calcolo scarta in maniera automatica quei pixel che vengono definiti contaminanti, cioè quelli che non sarebbero dovuti entrare a far parte della zona di demarcazione che si sta considerando.
Nel calcolo della media viene assegnato uno stesso peso sia a pixel buoni che a pixel che dovrebbero essere scartati attraverso la segmentazione; per questo motivo la media dei pixel si configura come un parametro poco affidabile per stabilire il valore di intensità rappresentativo dello spot. Una verifica sulla eventuale discrepanza fra i valori di media e di mediana è un buon metodo per stabilire se la fase di segmentazione è stata condotta correttamente o per valutare i limiti del programma che si sta utilizzando.
Dal punto di vista del formato del dato, ogni canale viene generalmente acquisito in immagini a 16 bit, cioè è possibile discriminare 65535 livelli d’intensità di segnale. Come regola generale i segnali che arrivano a livello 50000 vengono considerati come limite superiore per una rilevazione del dato affidabile; al di sopra di questo livello il segnale inizia ad andare in saturazione e perciò può essere meno attendibile.
In realtà sarebbe consigliabile mandare in saturazione il minor numero di spot e ciò può essere fatto modulando opportunamente il guadagno del tubo fotomoltiplicatore dello scanner in fase di acquisizione dell’immagine. E’ anche vero che mantenere un basso guadagno non permette di sfruttare a pieno la dinamica dei fluorocromi e impedisce la rilevazione di segnali deboli che spesso corrispondono a trascritti rari difficilmente identificabili.
2.2.4 Correzione del background
La presenza di un segnale di fondo sul microarray è dovuta a diversi motivi.
Può accadere, per esempio, che parte della soluzione contenente le sonde da depositare sul vetrino abbia contaminato aree esterne allo spot consentendo, in questo modo, l’ibridizzazione del campione marcato anche dove non dovrebbe avvenire
c) d) b) Background alto Segnale debole a)
Figura 2.6: a):Microarray con background alto dovuto ad ibridizzazione fuori dallo spot.
b) Microarray con “comete” dovute a spotting non preciso.
c) Microarray con depositi irregolari di soluzione buffer di spotting. d) Microarray con spot sovrapposti e di diametro irregolare.
Un esempio tipico è la rilevazione, in fase di scansione del microarray, delle cosiddette “comete”, che possono essere osservate in figura 2.6 a e b. Nella stessa figura si osservano altri esempi di problemi riconducibili alla fase di “spotting”.
Un altro fattore che contribuisce alla generazione di rumore è l’instaurarsi di legami aspecifici fra il supporto del microarray e il campione ibridizzato: in questo caso può essere utile sottoporre il vetrino ad un procedimento che prende il nome di pre-ibridizzazione, allo scopo di saturare questi legami e non renderli disponibili in fase di ibridizzazione del campione marcato.
Può ancora succedere che i reagenti utilizzati nella soluzione di “spotting” abbiano fluorescenza propria oppure siano riflettenti: anche in questo caso è possibile scambiare per segnale ciò che in realtà è esclusivamente background. Questi valori possono essere esclusi dall’insieme di dati che vengono in prima istanza considerati come segnale attraverso la correzione o sottrazione del background.
Nella sottrazione locale del background viene identificato un intorno sufficientemente ampio centrato sullo spot e viene considerata la media o la mediana del pixel esterni allo spot ma interni alla zona di demarcazione come valore locale del rumore; questo valore viene poi sottratto alla media o mediana dello spot canale a canale.
Figura 2.7: Intorno dello spot per il calcolo del background in diversi software di analisi.
Con questa operazione è possibile gestire la variabilità locale del rumore, tuttavia non è un procedimento privo di rischi; si pensi, per esempio, a spot con segnale debole: in questo caso la sottrazione di un livello locale di background che per qualche motivo risulti particolarmente alto porterebbe all’esclusione dello spot dall’insieme di quelli considerati accettabili. Inoltre è poco agevole fare un calcolo del genere quando il microarray è particolarmente denso, per evidenti difficoltà che si creano nell’identificare la zona sulla quale impostare il valore di correzione. Per ovviare a questi inconvenienti l’alternativa possibile è calcolare il valore di background su sotto-griglie del microarray; in questo modo si conduce il calcolo su un ambito meno locale e si può riuscire a ricavare una stima del rumore anche su array particolarmente densi di spot.
Figura 2.8: Sotto-griglia dell’array sulla quale calcolare il background.
Una via di mezzo fra i due procedimenti appena illustrati fa uso di un’area centrata sullo spot di diametro tale da includere un gruppo di spot. Lo scopo di questo procedimento è quello di mantenere il computo del background su un ambito abbastanza locale ma non troppo ristretto in modo da poter catturare anche la sua variabilità; grazie all’ampliamento dell’intorno è possibile applicare questo metodo anche su array densi.
Figura 2.9: Calcolo del background su un intorno ampio dello spot.
Esistono alcuni metodi di correzione che fanno uso di un fattore correttivo calcolato su aree nelle quali non sono presenti spot. Lo scopo è quello di stimare l’effetto dovuto all’interazione del substrato presente sulla superficie del vetrino con il campione marcato. Questo metodo, tuttavia, non è completamente affidabile in quando queste aree non sono rappresentative di ciò che realmente avviene dove sono presenti le sonde. Per questo motivo è più utile ricavare il valore di background su aree specifiche sulle quali vengono appositamente depositate sonde di controllo, che si sa non essere complementari alle sequenze del campione in esame.
E’ già stato illustrato l’effetto che produce la sottrazione del background su un segnale debole. Ciò deve invitare ad utilizzare cautela nell’applicazione di questo procedimento. Non fare la correzione, tuttavia, può corrompere i dati, dal momento che il contributo del rumore può modificare il dato relativo all’intensità dello spot generando falsi positivi o falsi negativi nella rilevazione dei geni differenzialmente espressi. L’approccio migliore consiste nel fare una correzione con un fattore che risulti da procedimenti globali, in modo da contenere al minimo gli errori introdotti da questa operazione.
Il parametro universalmente accettato per la misurazione degli effetti del rumore su un segnale è il rapporto segnale rumore (Signal to Noise Ratio – SNR), definito generalmente come:
SNR = Mediana del segnale / STD del rumore
dove al denominatore vi è la deviazione standard (STD) del rumore.
Molti software di analisi di microarray utilizzano il valore di SNR ricavato per ogni spot per escludere dal processo di normalizzazione quei dati che hanno un rumore troppo alto: in tal caso viene fissata una soglia per l’SNR, tipicamente pari a 3, in modo cha agli spot con un valore di SNR più alto venga applicata una “flag”, ossia un punteggio, che li esclude automaticamente dall’insieme degli spot utilizzati per la normalizzazione.
Sempre grazie all’applicazione di flag, che vengono assegnate in base alla rispondenza delle caratteristiche dello spot a quelle specificate dall’analista, è possibile escludere spot con forme irregolari, o con percentuale di pixel saturati superiore ad una soglia definita come accettabile. La selezione può escludere anche spot che vengono inseriti nel microarray esclusivamente per facilitare l’operazione di allineamento della griglia, oppure quegli spot che vengono lasciati appositamente vuoti. Si
effettua in questo modo un processo di controllo degli spot che prepara il dato alla successiva operazione di normalizzazione.
2.3 Normalizzazione dei dati
Molte variabili possono influire e distorcere i risultati di un esperimento di microarray:
disomogeneità del processo di deposizione delle sonde, quantità iniziali diverse di RNA,
diversa efficienza di incorporazione dei due fluorocromi durante il procedimento di marcatura dei campioni,
disomogeneità di ibridizzazione sul vetrino,
diversa efficienza di emissione dei due fluorocromi,
diversa efficienza dello scanner nel leggere i due canali di fluorescenza.
Tutti questi fattori possono influenzare pesantemente i dati causando spostamenti nelle distribuzioni dei rapporti delle intensità dei due fluorofori. E’, quindi, necessaria, prima di ogni tipo di analisi statistica, una normalizzazione dei dati atta ad eliminare distorsioni sistematiche. Un esempio di questo tipo di distorsioni si può vedere in figura 2.11 in cui è mostrato un grafico della dispersione dei dati, detto “scatterplot” dove sono messe a confronto le intensità dei segnali su due microarray ibridizzati con lo stesso RNA.
Figura 2.11: Scatterplot dello stesso mRNA ibridizzato su due microarray diversi.
Idealmente, le intensità relative ad ogni segnale dovrebbero coincidere sui due microarray e i punti che individuano i valori di tali intensità sullo scatterplot si dovrebbero posizionare sulla diagonale: quando ciò non accade significa che sono presenti errori sistematici, se la deviazione della diagonale è tutta dalla stessa parte, come in questo esempio, oppure errori casuali (random), quando i punti si allontanano dalla diagonale in entrambe le direzioni.
Questo errore diventa ancora più evidente quando si mettono a confronto le due intensità di segnale ottenute da un unico microarray su cui è stato ibridizzato lo stesso materiale marcato con entrambi i fluorocromi. Questo grafico viene denominato MA e presenta in ascissa il logaritmo della media geometrica delle due intensità (A), mentre in
A = ½ log (R*G) M = log (R/G)
Figura 2.12: Grafico MA di un array su cui è stato ibridizzato lo stesso RNA marcato con entrambi i fluorocromi
Il processo di normalizzazione è necessario anche per confrontare dati provenienti da repliche dello stesso materiale. Solitamente in un esperimento di microarray le repliche fra vetrini possono essere di due tipi:
repliche sperimentali: quando l’mRNA sui due vetrini proviene dalla stessa estrazione;
repliche biologiche: quando l’mRNA proviene da campioni biologici dello stesso tipo ma distinti (ad esempio individui diversi).
L’utilizzo di repliche biologiche consente di stimare l’errore random e maggiore è il numero delle repliche meglio si riesce a dare una stima della distribuzione di questo errore e del suo peso sui dati. Le repliche sperimentali servono, invece, ad ottenere una stima migliore dell’espressione
di un gene sulla base della corrispondenza dei dati relativi ai suoi spot su diversi microarray.
E’ necessario che la normalizzazione tenga conto del disegno dell’esperimento: la sua scorretta applicazione, infatti, invalida completamente il dato e, di conseguenza, i risultati.
Si parla di normalizzazione within-array quando la tecnica scelta viene applicata ad ogni vetrino singolarmente, nell’intento di correggere gli errori sistematici su ogni array preso come unità a sé e indipendentemente dal disegno sperimentale, mentre si fa una normalizzazione between-arrays quando si cerca di ottenere un dato uniforme considerando sia il disegno sperimentale applicato che il tipo di campione biologico.
In ciascuna di queste situazioni è necessario scegliere un gruppo di geni da utilizzare per la normalizzazione. Questi possono essere:
tutti i geni sull’array. Quasi tutti i geni sull’array possono essere
utilizzati per la normalizzazione quando è possibile prevedere che solo una porzione relativamente piccola di geni varierà significativamente in espressione fra i due campioni di mRNA.
geni espressi in maniera costante. Invece di utilizzare tutti i geni
per la normalizzazione si può scegliere di usare un piccolo sottoinsieme rappresentato dai geni housekeeping, cioè quei geni che mantengono lo stesso livello di espressione in condizioni sperimentali differenti. Non è facile identificare questo sottinsieme, ma spesso è possibile trovare un gruppo di geni che si comportano da housekeeping nelle condizioni sperimentali considerate. Una limitazione nell’utilizzo dei geni housekeeping è che essi tendono ad essere espressi molto e quindi potrebbero non essere rappresentativi di altri geni di interesse
controlli. Un’alternativa alla normalizzazione con geni housekeeping è l’utilizzo di controlli spiked o di una serie di sequenze di controllo a concentrazione scalare (titration). Nel metodo dei controlli spiked, sequenze sintetiche di DNA o sequenze selezionate da organismi differenti da quello studiato sono depositate sull’array e incluse nei due differenti campioni di mRNA in esame con identica concentrazione. Queste sequenze di controllo possono essere utilizzate per la normalizzazione perché daranno origine a segnali di uguale intensità nei due canali. Nell’approccio della serie titration, si utilizzano spot dello stesso gene a concentrazione scalare, con uguale intensità sui due canali nel range considerato, in modo da monitorare l’amplificazione lineare della risposta in intensità rispetto alla concentrazione.
2.4 Normalizzazione within-array
In questo caso la normalizzazione viene applicata separatamente su ogni array. Gli scopi principali sono la correzione del colore e dei problemi dovuti ad un’eventuale deposizione scorretta delle sonde.
2.4.1 Normalizzazione globale
I metodi globali di normalizzazione assumono che le intensità dei due fluorocromi siano proporzionali, cioè che valga la relazione R =K*G, dove con R si indica il canale rosso, con G il canale verde e K è la costante di
proporzionalità. In funzione di questa legge e nell’ipotesi che la maggior parte dei geni non si esprima differenzialmente, la normalizzazione globale sposta il logaritmo del rapporto dei due canali sullo zero, operando quello che viene tipicamente denominato “centraggio” della distribuzione dei dati:
log2 R/G - - - -> lognormalizzazione 2 R/G – c = log2 R/(KG)
dove c = log2 K.
Una particolare scelta per il parametro c è la mediana o, in alternativa, la media dei rapporti logaritmici delle intensità.
I metodi di normalizzazione globale non hanno un effetto intensità-dipendente sui dati e, quindi, non riescono a correggere le tendenze non lineari dei dati dovute al diverso comportamento che i fluorocromi presentano in emissione
Figura 2.13:
a) Distribuzione dei dati prima della normalizzazione (in rosso) e dopo lo
spostamento della media (in blu).
b) Scatterplot dei dati prima della normalizzazione (in rosso) e dopo la normalizzazione (in blu).
2.4.2 Normalizzazione intensità-dipendente
In molti casi gli errori sistematici riconducibili alla diversa efficienza di emissione dei fluorocromi sono dipendenti dall’intensità del segnale, come può essere evidenziato attraverso il grafico MA dei dati. In questi casi si possono correggere le distorsioni attraverso tre metodi:
interpolazione di curve e correzione, normalizzazione LOESS o LOWESS, normalizzazione a tratti.
2.4.2.1 Interpolazione di curve e correzione
La normalizzazione del colore può essere realizzata attraverso l’interpolazione di una curva sull’insieme dei dati dell’esperimento, da utilizzare successivamente per apporre le dovute correzioni. Visualizzando i dati su uno “scatterplot” che presenta in ascissa il log(R) e in ordinata il log(G/R), si può osservare che la distorsione dei dati introdotta dai fluorocromi ha generalmente un andamento esponenziale, come si può osservare in figura 2.14. Sulla base di questa osservazione si possono utilizzare i dati stessi per calcolare i parametri della funzione esponenziale che li interpola.
La procedura di normalizzazione comporta la suddivisione dell’asse delle ascisse in intervalli uniformi; questo corrisponde ad identificare sull’asse delle ordinate il sottoinsieme dei dati che ha al denominatore del rapporto logaritmico un valore di intensità compreso nell’intervallo fissato. Per ogni intervallo si calcola il corrispondente centroide dei dati in esso compresi, cioè un elemento rappresentativo di quel sottoinsieme di dati, e si determina la curva esponenziale interpolante l’insieme dei centroidi:
y = a + b * e
-c * xScopo di questa interpolazione è trovare la combinazione di valori a, b e c che genera la miglior curva esponenziale rappresentativa della dislocazione dei centroidi; una volta trovata, la relativa curva viene utilizzata per correggere i dati, così da posizionare i centroidi sull’asse orizzontale log (G/R)=0.
Figura 2.14: Normalizzazione per interpolazione di una curva esponenziale
2.4.2.2 Normalizzazione LOESS/LOWESS
La trasformazione LOWESS (LOcally WEighted polynomial regreSSion), così come la sua variante LOESS, divide i dati sull’asse delle ascisse in intervalli sovrapposti e interpola una funzione con una procedura simile a
quella usata nella normalizzazione con curva esponenziale, ma, in questo caso viene utilizzanta una funzione polinomiale:
y = a0 + a1x + a2x
2+ …..
I polinomi presentano la caratteristica di poter passare esattamente per tanti punti quanto è il loro grado. Tuttavia, questo approccio presenta il cosiddetto problema dell’“over-fitting”, ossia si ha un’approssimazione quasi perfetta della funzione bersaglio nei punti conosciuti, ma oscillazioni eccessive al di fuori di essi.
Per ovviare a ciò, l’approccio LOWESS utilizza polinomi di grado1, mentre il LOESS fa uso di parabole in modo da contenere l’over-fitting e l’eccessiva oscillazione fra i punti delle funzioni interpolanti.
Inoltre, poiché l’approssimazione polinomiale è precisa solo in piccoli intervalli intorno al punto scelto, può essere necessario dividere il dominio dei dati in finestre di dimensioni opportune con l’effetto collaterale di incrementare anche notevolmente il carico computazionale.
La divisione in piccoli intervalli ha inizio all’estremità sinistra dei dati con una finestra di larghezza data l e i dati che cadono in questi intervalli sono utilizzati per interpolare il polinomio applicando ad essi dei pesi diversi a seconda della loro posizione nell’intervallo: i dati prossimi al punto di stima hanno un peso maggiore di quelli lontani e ciò può essere realizzato utilizzando una funzione di peso w(x) della forma:
(
)
⎪⎩ ⎪ ⎨ ⎧ ≥ < − = 1 | | , 0 1 | | , | | 1 ) ( 3 3 x x x x wdove x è la distanza fra i punti stimati.
Il procedimento continua facendo scorrere la finestra verso destra e interpolando localmente di volta in volta un nuovo polinomio: il risultato è
una curva di “smoothing” attraverso cui correggere i dati. L’effetto dell’applicazione di questi metodi ad un insieme di dati può essere osservato in figura 2.15, dove si può notare come la normalizzazione agisca sui dati avvicinandoli all’asse delle ascisse ed eliminando o, comunque, riducendo drasticamente l’andamento non lineare.
LOWESS
Figura 2.15: Correzione dei dati con l’applicazione di una normalizzazione LOWESS.
Il vantaggio del metodo LO(W)ESS è che non ha bisogno di specificare una particolare funzione come modello: i soli parametri necessari sono il grado dei polinomi d e il fattore di smoothing q, che indica la larghezza della finestra.
Gli svantaggi del metodo LO(W)ESS includono il fatto che esso non produce una funzione di regressione o un modello che sia facilmente rappresentabile con una formula matematica. In particolare, il modello di correzione della distorsione del colore trovato su un particolare insieme di dati non può essere direttamente trasferito ad un altro: è necessario
riapplicare il metodo ogni volta che si ha un insieme di dati distinto e ciò produce sottili differenze ad ogni applicazione.
Un ulteriore svantaggio è legato al fatto che la procedura è computazionalmente molto pesante, anche se questo è un problema minore nel contesto di tutte le altre problematiche collegate all’analisi dei dati da microarray.
Il più importante svantaggio è la suscettibilità di questo metodo al rumore e agli “outlier”, cioè a quei dati che si discostano drasticamente dalla maggioranza dei dati acquisiti o, per essere più precisi, che si posizionano oltre 1.5 inter-quartile sopra il 75° percentile o sotto il 25° percentile della distribuzione dei dati.
Figura 2.16: Identificazione degli outlier sulla distribuzione dei dati
Malgrado siano state apportate notevoli modifiche al metodo il problema degli outlier resta tuttora irrisolto e, per questo motivo, è necessario eliminarli prima dell’applicazione del metodo.
2.4.2.3 Normalizzazione a tratti
La normalizzazione a tratti, o “piece-wise”,è molto simile alla LO(W)ESS, ma rispetto ad essa è computazionalmente più leggera, poiché elimina molti
calcoli che possono essere considerati in prima istanza ridondanti per i dati ricavati da microarray. Questo metodo sostituisce l’approccio a finestre mobili con un insieme fisso di finestre sovrapposte; in ognuno di questi intervalli i dati sono approssimati grazie ad una funzione lineare o quadratica. L’utente controlla le curve risultanti scegliendo il numero di tali intervalli e il loro grado di sovrapposizione.
Il vantaggio di questo tipo di normalizzazione è la generazione di una descrizione matematica compatta del modello dei dati che può essere memorizzata e utilizzata su differenti insiemi. Tale descrizione sarà una funzione lineare a tratti o quadratica con tanti tratti quanti sono gli intervalli specificati dall’utente.
Come la LO(W)ESS, anche la normalizzazione a tratti è sensibile agli outlier.
2.4.3 Normalizzazione “within-print-tip-group”
Ogni blocco in un array è depositato utilizzando le stesse punte o “print-tip”: modificando la configurazione secondo la quale viene realizzato l’array è possibile correggere il “layout” dell’array.
Possono esistere alcune differenze fra le punte, dovute per esempio a diversa larghezza dell’apertura o a deformazione dovuta ad usura. Solitamente i gruppi di punte generano effetti spaziali simili sul vetrino, per cui è necessario eseguire una normalizzazione fra gruppi in modo da eliminarli.
La normalizzazione “within-print-tip-group” dipende dal gruppo di punte e dal valore di A = ½ log (R*G) secondo un modello che può essere schematizzato con:
log2 R/G -> log2 R/G – ci(A) = log2 R/(Ki(A)*G)
dove la funzione di correzione ci(A) si trova facendo l’interpolazione LO(W)ESS di M e A sul blocco i-esimo.
2.4.4 Normalizzazione “within-slide”
Dopo aver realizzato la normalizzazione “within-print-tip-group”, tutte le distribuzioni relative a differenti gruppi di punte saranno centrate sulla media del gruppo, tuttavia è possibile che le medie relative a diversi gruppi di punte non siano uguali e si debba operare una riscalatura.
Un metodo per realizzarla può essere quello di assumere che tutti i rapporti logaritmici relativi all’i-esimo gruppo di print-tip si distribuiscano secondo una distribuzione normale con media nulla e varianza ai2σ2, dove σ2 è la
varianza dei rapporti logaritmici ed ai2 è il fattore di scala per l’i-esimo gruppo di print-tip.
Poiché è necessario ottenere una stima di questo fattore di scala, si procede massimizzando la funzione di verosimiglianza e si ottiene per ai la seguente stima: I I k n j kj n j ij i i i
M
M
a
∏∑
∑
= = ==
1 1 2 1 2ˆ
dove Mij denota il rapporto logaritmico j-esimo fra i due canali all’interno del gruppo di punte i-esimo, ni è il numero di griglie depositate con quella testina o gruppo di punte, e I è il numero di testine utilizzate per stampare l’array. Una robusta alternativa a questa stima è:
I I k i i i
MAD
MAD
a
∏
==
1ˆ
dove MAD (Median Absolute Deviation) è definita come:
MADi= medianj{|Mij – medianj(Mij)|}
Questa procedura assume che solo una piccola porzione di geni vari significativamente nel confronto fra due mRNA e inoltre, si ipotizza che la dispersione della distribuzione dei rapporti logaritmici sia pressoché la stessa per tutti i gruppi print-tip.
2.5 Correzione “paired-slide”
La correzione “paired-slide” si applica ad esperimenti nei quali due campioni diversi vengono ibridizzati su due microarray scambiando la marcatura, cioè il campione che sul primo microarray viene marcato in rosso, sul secondo sarà marcato in verde e viceversa per l’altro campione.
Si denoti con log2R/G-c il rapporto logaritmico normalizzato fra i due canali per il primo vetrino, con log2R’/G’-c’ quello per la seconda slide, mentre c e c’ siano le due funzioni di normalizzazione per i due vetrini. Nell’ipotesi che non vi siano comportamenti disuguali dei due fluorocromi sui due vetrini, deve valere che c ≅ c’ e che log2R/G= log2G’/R’,cioè:
(
)
[
]
[
]
(
')
2 1 /GR' RG' log 2 1 /R' G' log R/G log 2 1 /G'-c' R' log -c -R/G log 2 1 2 2 2 2 2 ≅ + = = M −MCon questo metodo è possibile scalare i livelli di espressione relativa per i due microarray senza esplicitare la normalizzazione: questo procedimento prende il nome di “self-normalization”.
La validità di questa assunzione può essere verificata utilizzando un insieme di geni con livelli di espressione costante sui due canali. Poiché l’assegnazione dei fluorocromi è invertita sui due microarray, ci si attende che su tali geni il rapporto logaritmico normalizzato sui due vetrini abbia uguale intensità ma segno opposto:
Quindi, riarrangiando l’equazione e assumendo ancora che c ≅ c’ è possibile stimare la funzione di normalizzazione c come:
c ≅
[
log R/G log R'/G']
2 1 2 2 + = ( ') 2 1 M M +Da un punto di vista operativo, la funzione c = c(A) di correzione su tutto il vetrino è stimata attraverso l’interpolazione LO(W)ESS di ( ')
2 1 M M + vs ) ' ( 2 1 A A+ .
2.6 Normalizzazione “multiple-slides” o “between arrays”
Dopo la normalizzazione “within array” le distribuzioni dei dati normalizzati di ogni microarray preso singolarmente saranno centrate sulla propria media. I metodi di normalizzazione “multiple-slides”, il cui scopo è quello di consentire confronti fra diversi array, servono per operare una scalatura fra i dati acquisiti con differenti vetrini quando i loro rapporti logaritmici normalizzati presentano dispersione. Non effettuare una normalizzazione tra array può indurre un peso non reale sui dati di un vetrino quando si effettua il confronto fra microarray.
a) b)
Figura 2.18: Box-plot degli array prima a) e dopo b) la normalizzazione between-arrays
E’ importante notare, tuttavia, che questo tipo di normalizzazione ha senso esclusivamente fra campioni identici (repliche sperimentali); per esempio, effettuare la normalizzazione tra array fra dati provenienti da un tessuto sano e uno malato è un errore poiché si sta involontariamente cercando di indurre un’omogeneizzazione delle dispersioni su campioni differenti, introducendo un artefatto.
Le tecniche utilizzate per la normalizzazione “multiple slides” sono uguali a quelle per i metodi “within array”.