Review
DNA
damage
response:
The
emerging
role
of
c-Abl
as
a
regulatory
switch?
Emiliano
Maiani
a,
Marc
Diederich
b,
Stefania
Gonfloni
a,*
aDepartmentofBiology,UniversityofRome‘‘TorVergata’’,ViadellaRicercaScientifica,I-00133Rome,Italy
bLaboratoiredeBiologieMole´culaireetCellulaireduCancer,FondationdeRechercheCanceretSang,HoˆpitalKirchberg,9rueEdwardSteichen,L-2540Luxembourg,Luxembourg
Contents
1. Theemergingcentralroleofc-AblinmodulatingthecellresponsetoDNAdamage... 1270
1.1. Surfingatthebreakpoint ... 1270
2. DNAdamageresponse:sensing,repairingorsignalingtodeath... 1271
2.1. DNAdamagenetwork... 1272
3. Ubiquitin-signalinginDDR... 1272
3.1. Makeubiquitinsignalsreversible–dynamicsthroughDUBs ... 1272
3.2. DefyingdeathafterDNAdamage:doesubiquitin-signalingsetthreshold? ... 1273
3.3. Workinghypothesis... 1273 4. Outlook... 1274 Acknowledgements... 1274 References... 1274 ARTICLE INFO Articlehistory: Received20May2011 Accepted1July2011 Availableonline7July2011 Keywords: DNAdamage c-Abl Histonemodifications DNArepair Germcells Chemotherapy ABSTRACT
A complex regulatorynetwork of signalingpathways safeguards genomeintegrity followingDNA damage.Whendoublestrandbreaksoccurseveralenzymesandmediatorsarerecruitedtothesitesof lesiontoreleaseanetworkofDNArepairprocessesreferredtoasDNAdamageresponse(DDR).c-Abl interactsinthenucleuswithseveralproteinsimplicatedindistinctaspectsofDNArepair.Thissuggests thatc-Ablmaybeinvolvedintheregulationofdoublestrandbreakrepair.Theinvolvementofc-Ablin DNArepairmechanismscameintothespotlightinfemalegermcellsundergenotoxicstress.Recent findingshaveimplicatedc-Ablinacisplatin-inducedsignalingpathwayelicitingdeathofimmature oocytes.Pharmacologicalinhibitionofc-AblbyImatinib(STI571)protectstheovarianreservefromthe toxiceffectofcisplatin.Thisimpliesthattheextentofc-Ablcatalyticoutcomesmaytipthebalance betweensurvival(likelythroughDNArepair)andactivationofadeathresponse.Manyobservations indicate thattimelyubiquitin-modifications andsignal decodingareimplicated inregulatingDNA repair.Here,wediscusssomeconnectionsbetweenphosphorylation-andubiquitin-mediatedsignaling atthedamagedsites.Wespeculateaboutmultipleinteractionsthatmayoccurbetweenc-Abl(and ‘sensor’ kinases) withubiquitin-related proteins involved in DDR. Additional workis required to understandthecomplexityofthephysiologicaloutcomesofc-AblinDDR.However,afine-tuningof nuclearoutcomes,throughpharmacologicalinhibitionofc-Abl,mayprovidenovelparadigmsforDDR and,potentially,therapeuticstrategiesforcancertreatment.
ß2011ElsevierInc.Allrightsreserved.
Abbreviations:53BP1,Tumorsuppressorp53-bindingprotein1;ATM,Ataxiatelangiectasiamutated;ATR,AtaxiatelangiectasiaandRad3-relatedprotein;BARD, BRCA1-associatedRINGdomainprotein1;BER,Baseexcisionrepair;BRCA1,Breastcancertype1susceptibilityprotein;BRCC36,BRCA1–BRCA2containingcomplexsubunit36;CtIP, CtBP-interactingprotein;DDB1,DNAdamage-bindingprotein1;DDB2,DNAdamage-bindingprotein2;DNAPK,DNA-dependentproteinkinase;DSBR,Double-strandbreak repair;ERCC6,Excisionrepaircross-complementing6;HERC2,HECTdomainandRCC1-likedomain-containingprotein2;MDM2,Doubleminute2protein;MMR,Mismatch repair;MRN,Mre11/Nbs1/Rad50complex;MSH5,DNAMismatchrepairprotein5;NER,Nucleotideexcisionrepair;PIAS,ProteininhibitorofactivatedSTAT;RAP80, Receptor-associatedprotein80;RNF8,RINGfingerprotein8;RNF20,RINGfingerprotein20;RNF40,RINGfingerprotein40;RNF168,RINGfingerprotein168;TopBP1,DNA topoisomerase2-bindingprotein1;UBC13,Ubiquitin-conjugatingenzymeE213;USP1,Ubiquitin-specific-processingprotease1;WRN,WernersyndromeATP-dependent helicase;YAP1,Yes-associatedprotein.
*Correspondingauthor.Tel.:+390672594319;fax:+39062023500. E-mailaddress:stefania.gonfl[email protected](S.Gonfloni).
ContentslistsavailableatScienceDirect
Biochemical
Pharmacology
j our na l ho me p a ge : w ww . e l se v i e r . com / l oc a te / b i och e mph a rm
0006-2952/$–seefrontmatterß2011ElsevierInc.Allrightsreserved. doi:10.1016/j.bcp.2011.07.001
1. Theemergingcentralroleofc-Ablinmodulatingthecell
responsetoDNAdamage
ThecellularresponsetoDNAdamage(DDR)reliesonanetwork ofmultipleinterconnectedsignalingpathwaysactinginconcertto
minimizethe dangerous effects of DNA double strands breaks
(DSBs).Thephosphatidylinositol3-kinases(PI3K)-relatedkinases ataxia-telangiectasiamutated(ATM),ATMandRad3-related(ATR) andDNA-activatedproteinkinase(DNAPK)areactivatedearlyby distinctDNAlesionsandstartacascadeofeventssignaledbythe rapidphosphorylationofseveralproteinsimplicatedinprocesses suchasDNArepair,cellcyclearrestandapoptosis[1–3].Although thePI3KrelatedenzymesareconsideredmajorplayersintheDNA damagecellresponse,afourthunrelatedkinase,c-Abl,hasmore recentlybeenassociatedtovariousaspectsoftheDDR[4].c-Ablis anon-receptortyrosinekinasethathasthepotentialtobindto several proteins [5]. It has been implicated in several cellular pathways,includingthoseoriginatingfromgrowthfactor stimu-lation,celladhesion,oxidativestressandDNAdamage[6–9];its activity is tightly regulated and it can be promptly activated followingionizingradiationandothertypesofgenotoxicinsults
[10,11]. c-Abl accumulation leads to cell cycle arrest and to programmedcell death in cultured cells. Several c-Abl targets
(YAP1,TP73, TP63,MDM2)areindeedimportantmodulatorsof
DNAdamage-inducedapoptosis.Atthesametime,manypartners
andsubstratesofc-Abl areknown mediatorsofDNArepair [5]
(amongthem,DDB1, DDB2, ERCC6,RAD9A, RAD51,RAD52 and
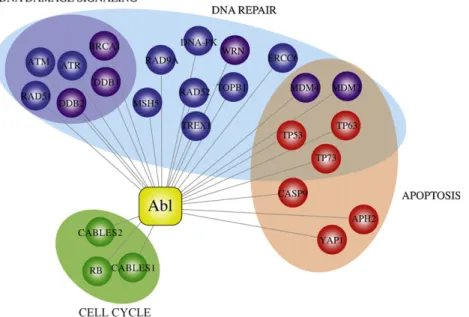
WRN,ATM,ATR,DNAPK,BRCA1,TopBP1,andMSH5,seeFig.1),
suggestingthatc-Ablmaybeimplicatedintheregulationand/or assemblyofDNArepaircomplexes.Inspiteofitsemergingcentral
role in DNA repair, the mechanistic details are still poorly
understood and the physiological functions, if any, of many of theinteractionsthathavebeenreportedremainselusive[12,13]. Wanget al.haverecentlyreportedthatc-Ablisinvolvedinthe
activation of ATM and ATR kinases following doxorubicin
treatment. c-Abl deficient primary MEFs, following genotoxic
stress,failedtoactivatebothATMandATRandtheirdownstream effectors[14].Theseobservationssuggestthatc-Ablmayhavea significantrole intheactivationofthekeyupstreammolecular
events governing the initiation and propagation of DDR [12].
Additional insights on the central role played by c-Abl in
modulating theinterplaybetweenDNA repair andinduction of
apoptosis came from the study of female germ cells under
genotoxic stress [15]. Intraperitoneal injection of cisplatin in newbornfemalemiceleadstodepletionofthefolliclereserveand tolong-terminfertility.Recentfindingshaveimplicatedc-Ablina cisplatin-inducedsignalingpathwayelicitingdeathofimmature oocytes [16]. A p53-related protein, TAp63, is a critical down-streameffectorofthis pathway.Inhibitionofc-Abl byImatinib (STI571) protects the ovarian reserve from the toxic effect of cisplatin.Thisimpliesthattheextentofc-Ablcatalyticoutcomes maytipthebalancebetweensurvival(likelythroughDNArepair) andactivationof adeath response.Ourcurrent modelsuggests thatc-Ablmayfunctionasahubassistingtheprogressionofrepair
but eventually promoting cell death when DNA breaks prove
irreparable[13].Althoughwehaveshownthatco-treatmentwith Imatinibhasa protectiveeffecton theovarianreserve[17],we
need toclarify themechanisms underlyingsuchan effect. The
kineticsofc-AblactivationfollowingDNAdamagerepresentsan
importantimmediate issuetobeaddressed.Additionalworkis
requiredtounderstandthecomplexityofthephysiologicalroleof c-AblinDDR,anditsinvolvementinthemodulationofthemany posttranslationalmechanisms,includingubiquitination, underly-ingtheDDR.
1.1. Surfingatthebreakpoint
ChromatinisacomplexscaffoldformedbychromosomalDNA
wrapped around the histone core. This scaffold is not static.
Chromatin modifications are essential for modulation of many
cellular processes including transcription, replication and DNA repair.Twoclasses ofenzymescanmodifychromatinstructure. Oneclassconsistsoflargemulti-proteincomplexesthatuseATP hydrolysistoalterthenucleosomepositionorcompositionwithin chromatin[18].Thesecondclassmediatescovalentmodifications of histone tails. Posttranslational modifications of histones are implicated in the DNA damage response [19,20]. In particular, histonemodificationinducedbymembersoftheubiquitinenzyme familyis oneofthemaindefensivestrategies adoptedby DNA-injured cells[21].Ubiquitin-conjugationseemstomodulatethe
Fig.1.Abl-interactingproteinsintheDNAdamageresponse.Inredellipseproteinsinvolvedinapoptosis,inbluethoseinvolvedinDNAdamagesignalingandDNArepair. Greenellipsecontainsproteinsinvolvedincellcyclearrest.Proteinsdirectlyinvolvedinubiquitin-signalingofDDRareinviolet.Alltheseproteinshavealsobeenreportedas AblsubstrateswiththeexceptionofTREX1,CABLE2,BRCA1,andDDB2.
assembly of themany components of thegenomesurveillance
system. Several ubiquitin-signaling paths influence various
aspectsofgenome-integritymaintenanceandboth
monoubiqui-tylationandpolyubiquitylationareemergingasversatile strate-giestomodulateprotein–proteininteractionnetworks[22–28].A modelofacomplex‘ubiquitinlandscape’atthedamagedsitesis
emerging, albeit incomplete and poorly understood [29,30].
Particularly noteworthy is the extensive crosstalk between
ubiquitin-modificationsandphosphorylation-mediatedpathways
in DDR. A complex web of molecular interactions determines
whetherandhowtorepairthedamageorratherlettheinjuredcell die [31–34]. Here, we discuss some connections between
phosphorylation- and ubiquitin-dependent signaling at the
damage sites. We speculate about multiple interactions that
mayoccurbetween c-Abl(and‘sensor’kinases)with ubiquitin-relatedproteinsinvolvedinDDR.
2. DNAdamageresponse:sensing,repairingorsignalingto
death
Intricatemechanismsaresetinmotionforcounteractingthe potentiallydangerouseffectsofDNAlesions.Thesemechanisms
arechallengedinchemotherapyregimensfor cancertreatment.
Crosslinkingagentsare amongthemostwidely usedand most
effectiveanticancerdrugs.Theyformcovalentadductsoncellular DNAeitheronthesamestrand(intrastrand)orbetweenthetwo complementarystrands(interstrand).Howaretheyrepaired?The mainplayersarenucleotideexcisionrepair(NER),baseexcision
repair (BER), mismatch repair (MMR) and double strandbreak
(DSB) repair. Interstrand crosslinks may induce double strand
breaksasan intermediatestepduring repair.So, cells mayuse severalDNArepairpathwaysinaconcertedway.Itisbeyondthe scopeofthisreviewtodiscusstheserepairmechanismsindetail. Interestedreadersaredirectedtoseveralreviewsonthissubject
[35–40].Here,wewillfocusonDSBssinceveryrecentstudieshave indicatedthattransientabrogationofc-AblactivitymodulatesDSB
repair pathway mediated by either homologous recombination
repair(HRR)ornonhomologous end-joining(NHEJ)mechanisms
[41,42].In addition,in germcells, DSBs occur normallyduring
meiosistopromotehomologousrecombinationandbydoingso
geneticdiversity[43]. Micedeficientinc-Abl exhibitdefectsin spermatogenesis[44].Thissuggeststhatc-Ablhasarolein the maintenanceofgenomeintegritybydealingwithDSBsinmeiotic cells.
Threedistinctproteincomplexesactassensors,transducersand effectorsofDDRinducedbyDSBs.Manycomponentsofthesethree
layers interact with each other and converge toward different
outcomesdependingontheseverityofthedamageandonthecell
type. The activation of checkpoints slows down cell cycle
progressionuntillesionsareresolved.IfunrepairedDSBspersist,
cells undergo either apoptosis or senescence to prevent the
accumulation of potentially tumorigenic mutations [45–47].
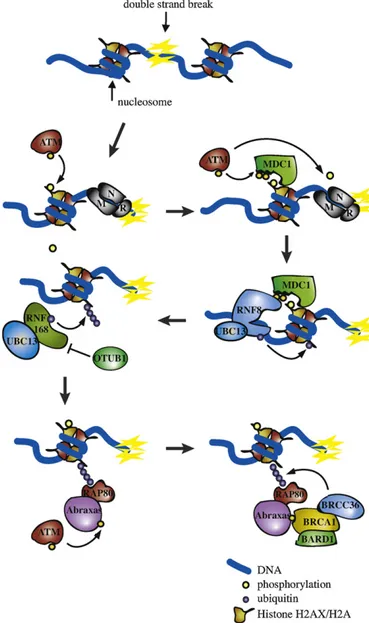
FemalegermcellsareextremelysensitivetoDNAinsultscompared with somatic cells [48]. In line with this, ovarian failure and infertilityareoftenoff-targetconsequencesofchemotherapeutic treatment.OocytesfromfolliclereservearearrestedinmeiosisI; DNAdamage iseitherquickly repairedortriggersa robustcell degeneration. Intriguingly, abrogation of c-Abl activity has a protectiveeffecton theovarian reserveundergenotoxic stress. Despitethediversityregardingthecelltype,theefficiencyofrepair andsignalingofthebreaksis enhancedbytheconcentrationof factorsintheproximityofthelesion.Atthedamagedsite,theDDR canbepresentedasasequentialassemblyofproteincomplexes (Fig.2).
DNA repair initiated by sensorsof breaks, – including MRN complex,ATM–reliesontheactivityofdifferentE3-ligasesnamely
RNF8,HERC2andRNF168.AmongthemanytargetsofATM,the
histone H2A variant H2AX is phosphorylated on Ser-139. This
modificationseemstobearecruitmentsignalforproteinswith dedicatedphospho-S/TrecognitiondomainssuchastheFHA[49]
orBRCTdomain[50].TheRING-typeubiquitinligaseRNF8[34,51– 54]ubiquitinatesH2AXandalsoseemstoshifttherecruitment modefrombeingphosphorylation-basedtobeingubiquitin-based. In spite of that, many reports indicate that phosphorylation of H2AXisnotessentialforDNArepair[55,56],suggestingthatother moleculescanorchestratetheassemblyofDNArepaircomplexes.
Noteworthy, DNA damaging complexes rely on protein
modularity associated to posttranslational modifications of
bindingpartners.Posttranslationalmodificationsarealso revers-ible,implyingasaconsequence,thedynamicnatureofanykindof
protein–protein interactions depending on such modifications.
Fig. 2. DSBs are recognized by Mre11–Rad50–Nbs1 (MRN) complex, which promotestheactivationofATM.H2AXphosphorylationbyATMprovidesadocking siteforMDC1.TheubiquitinligaseRNF8(recruitedthroughitsFHAdomain)in tandemwithUBC13ubiquitylatedH2AandgH2AX.Signalingofthebreaksisthen enhancedbytherecruitmentofE3ligaseenzymeRNF168(throughitsMIUdomain) thatactsbyextendingK-63ubiquitinchains.ThedeubiquitinatingenzymeOTUB1 suppressesRNF168-dependentubiquitinationbydirectinhibitionoftheE2ligase UBC13.RAP80associatestoubiquitinbyitsUIMdomainandrecruitstheBRCA1-A complex,throughtheinteractionwiththescaffold proteinAbraxas.TheBRCA complex containsthe ubiquitin protease BRCC36 that removes ubiquitin on histonesH2AandH2AX,antagonizingtheRNF8/RNF168-dependentubiquitination.
Largecomplexesaresobuiltthroughspecificrecognitionbetween
posttranslationalmodificationsanddecodingdomains.However,
following DDR progression, posttranslational modifications of
proteins,intimatelyinvolvedinDNArepair,canalsobeeditedby specificenzymesthusarrestingtherepairprocessandtriggering analternativepathwayleadingtocelldeath.Therefore, phospha-tases(PPI) and deubiquitylases (DUB)offer additionallevels of
complexity required for the fine-tuning of DDR pathways in
injuredcells.
2.1. DNAdamagenetwork
Inthebiologicalcontextmostproteinandgenenetworksdonot havethetopologicalpropertiesofrandomnetworksbutarerather
characterized by a high clustering coefficient and by a degree
distributionthatisscale-free[57].Ifwerestrictouranalysistothe DDRinteractions,mostoftheproteins(nodes)haveonlyfewedges (connections)whereasfewproteins(hubs),suchasATM,orp53
[58]haveavastnumberofconnections.However,theassemblyof large complexesin thevicinity of thelesionsfollows a strictly
hierarchical process [59] based on domain modularity and
localizedconcentrationoffactors.
Recently, the ‘phosphorylation landscape’ of DDR has been
expandedthroughtheidentificationofnovelputativesubstratesof ATMaswellasofsomeATMindependentsubstrates[60].These
observations underline the vast complexity of the cellular
responsesin theDDR pathwaysnecessarytomaintaingenomic
integrityandcellularhomeostasis.Rapidkineticsformostofthe phosphorylationeventssuggeststheexistenceofsimilartemporal patternsalsoforthedephosphorylationresponse[60].Shilohand colleagueshaverecentlyexploredsuchkineticsthroughanalysisof systemlevelnetworksofperturbedcells[60].Cellswereexamined afterradiomimetictreatmentatdistincttimepoints.Theanalysis ofisolatedphosphopeptides,throughlabel-freequantitativeLC– massspectrometry,wascarriedouttofollowdynamicsofdouble
strandbreaks(DSBs)-inducedphosphoproteome.Theyfoundthat
the dynamics of the DDR-induced changes are complex and
includeboth phosphorylationand dephosphorylationprocesses.
Theseevents, involvingmany interconnected proteins (or
com-plexes),indicatearobustandcomprehensivecellularresponseto
DNAdamage.Oneimportantobservationregardingthe
involve-mentofphosphatasesisthattheyareservingasshutoffsignalsof
DDR-signaling.Moreover,theauthorsfoundthat40% ofdouble
strand breaks (DSBs)-induced phosphorylation was not
ATM-dependentbutispotentiallyinducedbyseveralotherkinases.This suggeststhat,althoughATMsignalingisassociatedtoDSBs,onlya fractionofDSBsrepairisATM-dependent[61].Interestingly,the datafromShilohandcoworkersindicatethatthecontrolofDDR eventsisbasedonthesustainedactivityofATMoveranextended time.Thismechanismprobablyservestocounteracttheopposing effectsmediatedbyphosphatases.ProlongedATMactivitymaybe involvedinensuringitsretentionatthedamagedsitewhereATM actsasafuelforthesignalingcascade.
Ubiquitylation is alsoanimmediate modificationunderlying
theDDRprotein–proteinnetworks.Itsinterplaywith
phosphory-lation is crucial in damage repair and DNA signaling. Histone
decoration by ubiquitin-chains has been recently appreciated,
fuelled,inpart,bythediscoveryofenzymesresponsibleforthese modifications[28].Largecomplexesallowrecognitionandsetting
inmotion of mechanismsto mark(throughubiquitin-tags) the
sitesoflesionforanappropriateresponse[51–54].
3. Ubiquitin-signalinginDDR
Protein modification by a single ubiquitin moiety can have
severaldiverseoutcomes,rangingfromthecontrolofendocytosis
and intracellular trafficking to the regulation of chromatin
structure transcription and DNA damage processing [24,62].
However, the complexity of ubiquitin signaling is achieved
throughitsabilitytoformchains.Polymericchainscanbebuilt on all of ubiquitin’s seven Lys residues. Different linkages of ubiquitinmoietyorchainsadoptingdistinctgeometriesensurethe functionalcomplexityofsignaling(i.e.Lys-48chainsarelinkedto theproteasomedegradation,while,linearandLys-63chainsseem tomediatedifferentfunctions).Bothchainscanmodulateseveral
pathways related to genome stability [63]. Ubiquitin-chains
provide recognition sites for complexes assembly and are
necessary for signal propagation. Several types of
ubiquitin-bindingdomains(UBDs)havebeenrecentlycharacterized[64–66]. Notably,recognitioncanbedirectormodulatedthroughbinding withotherdomainsnecessarytogainspecificitytowardparticular
geometries of ubiquitin polymers. To date several
ubiquitin-modifications and signaldecoding are implicated in regulating
DNArepair[67].
3.1. Makeubiquitinsignalsreversible–dynamicsthroughDUBs
Ubiquitin-decoration is achieved through the sequential
cascade of activating (E1), conjugating (E2) and ligating (E3) enzymes;sucheventscanoccurthroughtheconjugationofsingle ubiquitinorpolyubiquitinchains(homotypicchains,or heterolo-gous,forked or mixed). The vast variety of ubiquitin-signals is recognizedanddecodedbydedicatedubiquitin-bindingdomains. Inaddition,tightcontrolismaintainedbytheactionofDUBsand bytheexistenceofcrosstalkbetweentheubiquitin-networkand other posttranslational modifications. In short, high levels of
specificity are achieved through (1) specific E2–E3 pairs, (2)
recognitionofcertainubiquitinbranchesmediatedbyindividual
UBD and eventually, (3) by a presumed relationship between
functional outcomes and distinct ubiquitin species [68]. Fine-tuningofubiquitin-pathwaysreliesonproteincomplexes,timely regulatedin space,mediatedbyscaffoldproteinsorchaperones
[69,70].TargetingofE2–E3pairsinresponsetospecificstressesis mediatedbyposttranslationalmodifications,recognitionthrough
surrounding domains and adaptors [68]. Ubiquitin-conjugation
canmediatenucleartranslocation;itcanalsoimpactonprotein activity,inducingconformationalchangeswithapositive[71]or
negative effect [72]. In some circumstances, phosphorylation
directlyregulatesE3ligaseactivity[73,74]orindirectly,controls
the timing of ubiquitin-attachment and removal by affecting
nucleartranslocationofdeubiquitylatingenzymes(DUB)[75]. Howtheversatilityofubiquitin-complexesatthesiteoflesion isaccomplished?SixclassesofUBDsareinvolvedintheresponse
toDNAdamage(UBA,UIM,MIU,UEV,UBM,andUBZ[67].Their
recognition occurs through binding of a hydrophobic motif on
ubiquitinandofspecificregionsonthesubstrate.Suchcomplexes
canbe modulatedby specificproteases(DUBs).DUB activityis
inducedthroughbindingwithsubstrate;a furtherregulationis
achieved throughposttranslational modifications
(phosphoryla-tion, ubiquitin or ubiquitin-like modifications) and/or specific
binding to accessory molecules that impinges on substrate
recognition and/or subcellular localization [25,68]. USP1
auto-deubiquitination isa remarkable example ofDUB regulationin
DNArepair[76].
DUBscanbedistinguishedintofivedistinctclassesdepending
on their domain structure [25]. Their importance in cellular
processesishighlightedbyrecentreports[77,78].DUBsoperate throughcleavageofubiquitinmoietyorubiquitin-linkedchains fromasubstrate.TheDUBsactivationimpingeson(1)recyclingof
free ubiquitin for cell homeostasis maintenance, (2) rescuing
proteinsfromdegradation,and(3) editingthelengthortype of
crucial for the fine-tuning of ubiquitin-conjugation directly affecting enzymatic activation or proteosomal targeting [79]. Largecomplexes,formedthroughubiquitinreceptors(UBDs)orby conjugationwithsmallubiquitin-likemodifier(SUMO),intandem withDUBsarebothrequiredforsignalingatdamagedsites.
MuchofthecurrentunderstandingofDDRisbasedonthestudy ofATMandATRkinases.Oneoftheearliesteventsisrecruitment andactivationoftheATMatthedamagedDNAsitesthroughthe
Mre11–Rad50–Nbs1 (MRN) sensor complex. This event clearly
illustrates the crosstalk between the ubiquitin-network and
posttranslational modifications of DDR. Within minutes after a
DSBgeneration,ATMphosphorylateshistoneH2AXtobecome
g
-H2AX.
g
-H2AXunleashesacascadeofchromatinmodulationandDNArepaireventsthroughtherecruitmentofMDC1(mediatorof DNAdamagecheckpoint1)[80].Thisisfollowedbyaccumulation
of two closely related RNF ubiquitin ligases, RNF8–RFN168
[26,52,54,81,82]intandemwiththeHECT-domainproteinHERC2
[83].FurtherrecruitmentofSUMO-ligasePIAS1andPIAS4[84,85]
thentriggers(andamplifies)bindingofubiquitinandSUMOonto
histones near the DNA lesions, allowing local recruitment of
importantrepairfactors,including53BP1andanotherubiquitin ligase,BRCA1[1].
Moyaletal.haverecentlyreporteda directpositiveeffectof ATMonmonoubiquitylationofH2Batdamagedsites.Theyobserve thattheE3ubiquitinligase,aheterodimericcomplexofthe
RING-finger–RFN20/RFN40 is phosphorylated by ATM. This event is
requiredforH2B monoubiquitylation,fortimelyrecruitmentof componentsinvolvedinthetwomajorDSBrepairpathways(NHEJ
and HR) so facilitating DNA repair via both mechanisms [74].
Interestingly RNF20 is also involved in the recruitment of
chromatin-remodeling factor SNF2h independently from H2AX
[86]. Depletion of RNF20 impairs resection of DNA ends and
recruitmentofRAD51andBCRA1.CellslackingRNF20orSNF2hor
expressing H2BK120R mutant exhibit pronounced defects in
homologous recombination repair (HRR) and an enhanced
sensitivity to radiation. Interestingly, thefunction of RNF20in HRRcanbepartiallybypassedthroughforcedchromatin
relaxa-tion.This suggests that RNF20-mediatedH2B ubiquitination at
DSBsplaysacriticalroleinHHRthroughchromatinremodeling
[86].
Chromatin modulationis a crucial event of the DNA repair
cascade.NonsensemutationsintheRNF168geneimpairretention
of 53BP1 and BRCA1 at sites of DSB repair [87]. This finding
supportstheroleoftheRNF8–RNF168–HERC2–BRCA1chromatin
ubiquitin-ligasecomplexes[26,85]forgenomeintegrity.Despite considerableefforts, theprecisefunction of BRCA1 in theDNA
damageresponseremains unclear.In addition,BRCA1seemsto
promote homologous recombination. BRCA1 has an
ubiquitin-ligase activity, it ubiquitylates CtIP a protein involved in DSB
resection [88].The 53BP1 protein promotesother pathwaysof
repairbyblockingresection,whereasthe53BP1sumoylationby
PIASproteins[83,84]maypromoteitsdisplacementfromDSBs,
releasingthebarriertoresection.
Inshort,non-degradativeubiquitylationplaysacentralrolein
theDNAdamageresponse.RNF8andRNF168,intandemwiththe
E2ubiquitinconjugatingenzymeUBC13catalyzetheformationof Lys-63linkedchainsattheDSBs sitestopromotetheirfaithful repair.Bycontrast,OTUB1,anovariantumorproteaseactingasa
DUB,counteractsRNF8/RNF168-dependentubiquitin-chains
for-mationatdamagedsites[89].Interestingly,OTUB1isnotinvolved inthecleavageofpolyubiquitinchainsbutdirectlytargetsUBC13
[77]. For this aspect, OTUB1 is an atypical DUB, that prevents ubiquitinligation,ratherthandetachingofboundubiquitin,andin thiswayinhibitsDNA repair.Inaddition,OTUB1istargetedby
phosphorylation, thus providing another level of control to
modulateitsaffinityforUBC13.Nakadaetal.foundthatinhibition
of OTUB1 expression restores the process of homologous
recombination in cells in which ATM kinase is inhibited [90].
Thus, OTUB1 depletion can in principle mitigate DNA-repair
defects.
Several DUBs have been reported to affect the ‘ubiquitin
landscape’ presentat DNAbreaks[68]. UCH37/UCHL1interacts
with chromatin-remodeling complex involved in nucleosome
sliding (INO80, inositol-requiring 80) [91]. Other DUB, such as
BRCC6(BRCA1–BRCA2containingcomplexsubunit36),mayacton
the RNF8–UBC13 ubiquitin ligase complex deubiquitylating
g
H2AX[92].Inaddition,DUBsinvolvedinDNAdamagesignaling areUSP1thattargetsPCNA(proliferatingcellnuclearantigen)[76],FANCD2 and FANCI (the Fanconi anemiaproteins) [93,94], and
USP3andUSP16thatdirectlydeubiquitylatehistoneH2A[95,96]. 3.2. DefyingdeathafterDNAdamage:doesubiquitin-signalingset threshold?
The experimental results compiled above suggest that the
interplaybetween pairactivitiesof phosphorylationor
dephos-phorylation (and also ubiquitination or deubiquitination) is
requiredforthefine-tuningofDDR.Itmaybepartofthereason bywhichtheDDRdecayinatimelymanner,afterdamagerepair, allowsasafetypathforthecells.Theimmediaterecruitmentof factorstoDSBs,andthelocalizedconcentrationofproteinsmight be particularlyimportantfor signalingamplification and toset thresholdlevelsofDNAdamage.
DDRdependsontherecruitmentofthesensors/transducersto thedamagedsite.Theiractivationleadscellstoadecisionpoint
betweensurvivalanddeath.Whicharethemechanisms
underly-ing such a decision? Survival of DNA-injured cells depends on
removal of the damage. A logical hypothesis is that the
amplificationofthesignalingcascadehasthefeasibilitytodrive cellstowarddeathasadefaultpathifnotattenuated.
Whyanattenuatedactivationofc-Ablendsinasurvivalpathin femalegermcells?c-Ablpresumablyaffectsdownstreamcascades
through phosphorylation of several proteins or substrates of
enzymesactivated/regulatedbyc-Abl.Pharmacologicalinhibition
of c-Abl could impact on distinct levels of such signaling. A
reasonable hypothesis is that c-Abl activation may impinge
directly or indirectly on ubiquitin-signaling of DDR. According tothis, arecent reportprovides evidencethatAbl regulatefoci
formation of protein like 53BP1, TopBP1, RAD51 and BRCA1
followingDNAdamage[14]. 3.3. Workinghypothesis
RecentfindingsfromWanget al.indicatethatc-Ablmaybe
necessary for the full activation of ATM and ATR and their
respectivedownstreamsignalingpathways.Accordingtothis,
c-Abl phosphorylates ATM, thus amplifying ATM activation and
signaling.PhosphorylationeventsmediatedbyATMare,inturn, necessary forrecruitmentofubiquitin-related enzymessuchas
RNF8, RNF20–RNF40 and BMI1 (polycomb group proteins) in
proximityofDNAbreaks.Inparticular,BMI1isinvolvedinDNA
damage-inducedmonoubiquitinationofH2A.BMl1interactswith
RING1B(RNF2)toformaheterodimerrequiredforPRC1mediated histoneubiquitination,thuscontributingtoefficientHRmediated DNArepair[97].LossofBMI1sensitizescellstoionizingradiation tothesameextentaslossofRNF8.IntheabsenceofBMI1,the
recruitment to damaged sites of 53BP1, RAP80 and BRCA1 is
stronglyimpaired[98].
Inaddition,c-Abldirectlymayimpinge(through phosphoryla-tionoritsbinding)onseveralproteinsand/orenzymesinvolvedin ubiquitin-signalingofDDR.Inlinewiththis,c-Ablinteractswith BRCA1a tumorsuppressor crucialforcell-cyclearrestand DNA
repair. BRCA1, in complex with another RING-domain BARD1 exhibitsubiquitin-ligaseactivity.Fewtargetsforthisactivityhave
beencharacterized invivo. The BRCA1/BARD1 can ubiquitylate
histones(H2AandH2B)inthecontextofnucleosome[99].This suggeststhatBRCA1mayalsoaffectdirectlynucleosomestructure anddynamicsthroughitsubiquitylationactivity.Inaddition,c-Abl directlyphosphorylatesubiquitin-relatedproteinssuchasDDB1
[100](involvedincomplexwithDDB2inDNArepairthroughNER
mechanism),WRNahelicasecontaininganUBDdomaininvolved
inDNArepair[101],andfinallytheE3RINGligaseMDM2[102]
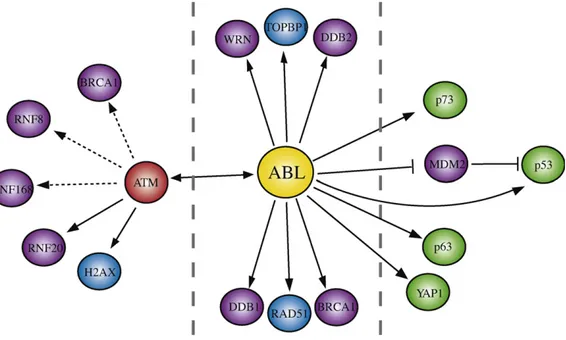
(Fig.3).
MDM2(alongwithMDMX)isapartofamulti-component
E3-complexthattargetsp53forproteasomaldegradation[103,104]. Recently,Mayoandcolleaguesfoundthatmulti-site
phosphoryla-tion of MDM2 by c-Abl is important for the MDM2–MDMX
complexformation[105].Oneofthetyrosineresiduesimportant
forcomplexformationisproximaltotheRINGdomainofMDM2.
Thissuggestsapossibleroleforthismodificationinmodulating RINGdomaininteractions.Interestingly,RINGdomain dimeriza-tionappearstobeageneralrequirementfortheassemblyofan activeligasecomplex[106].Thus,c-Ablphosphorylationprovides
a mechanism to regulate ubiquitination by modulating the
oligomerizationofE3MDM2–MDMXcomplexes.
4. Outlook
Severalcomplexcellularresponsescanbeunderstoodonlyby thinkingintermsofadensewebofinteractionsand feedbacks. Manyofthemostpressingissues,relatedtoDDRincells,cannot longerbesolvedsimplybybreakingsystemintoparts.Takingfew
major hubs out of the DNA damage network will simply
disassemble it in rather isolated protein–protein connections. Timelyseriesofubiquitin-modificationsandsignaldecodingare implicatedin regulatingDNA repair. Thecurrent model is that histoneubiquitylationservesasabeaconfortherecruitmentof effectorproteins. Futurestudies willlikely uncover newmotifs thatrecognizesingleorcombinatorialmodificationsonchromatin. SpecificE2–E3 pairs seemto berequired for distinct ubiquitin
chains,howeverresearchisneededtoclarifytheimportanceof ubiquitinbranchinginaphysiologicalcontextandtoidentifyand characterizemorepotentialDUBs.Weneedtoclarifyhowdifferent ubiquitin-marksaregeneratedanddecodedbyUBDsinthecells. Weneedtoknowhowmodifyingenzymesaretargetedtotheirsite ofactionandwhichenvironmentalormetabolicfactorsaffecttheir activity.
Here,wespeculateaboutsomeconnectionsoccurringbetween
phosphorylation- andubiquitin-mediated signalingatthe
dam-agedsites.Multipleinteractionsseemtooccurbetweenc-Abl(and ‘sensor’kinases)withubiquitin-relatedproteinsinvolvedinDDR.
The kinetics of c-Abl activation is certainly an important
immediateissuetobeaddressed.NovelparadigmsforDDRmay
arise from a better understanding of the crosstalk between
phosphorylationsignalsmediatedbyc-Ablandubiquitin-related changesonchromatin.
Acknowledgements
WethankGianniCesareniforcriticalreadingofthemanuscript.
We thankGiorgioMazzeo forhis support;CristinaFloreanand
Cindy Grandjenette for suggestions. We acknowledge support
fromAIRC(ItalianAssociationforCancerResearch)toS.G.Research inM.D.’slabissupportedbythe‘‘RechercheCanceretSang’’,the ‘‘RecherchesScientifiquesLuxembourgassociation,the‘‘EenHaerz firkriibskrankKanner’’association,theActionLions‘‘Vaincrele Cancer’’associationandbyTe´le´vieLuxembourg.
References
[1]JacksonSP,BartekJ.TheDNA-damageresponseinhumanbiologyanddisease. Nature2009;461:1071–8.
[2]LavinMF,KozlovS.ATMactivationandDNAdamageresponse.CellCycle 2007;6:931–42.
[3]HarperJW,ElledgeSJ.TheDNAdamageresponse:tenyearsafter.MolCell 2007;28:739–45.
[4]ShaulY,Ben-YehoyadaM.Roleofc-AblintheDNAdamagestressresponse. CellRes2005;15:33–5.
[5]ColicelliJ.ABLtyrosinekinases:evolutionoffunction,regulation,and speci-ficity.SciSignal2010;3:re6.
Fig.3.Modelforintegratedsignalingfunctionsofc-Abl.Ablmayregulatedoublestrandbreaksrepairand/orcelldeathtodamage.TheextentofAblcatalyticoutcomesseems toshiftthebalancebetweenlifeanddeath.Areasonablehypothesisisthatc-Ablpresumablyaffectsdownstreampathwaysthroughphosphorylationofseveralproteinsand/ orenzymesinvolvedinubiquitin-signalingofDDR.Solidline:directinteraction;dashedline:indirecteffect;ubiquitin-relatedproteinsarecoloredinviolet;modulatorsof DNAdamage-inducedapoptosisarecoloredingreen;DNArepairandDNAsignalingproteinsarecoloredinblue.
[6]GuJJ,RyuJR,PendergastAM.AbltyrosinekinasesinT-cellsignaling.Immunol Rev2009;228:170–83.
[7]SirventA,BenistantC,RocheS.Cytoplasmicsignalingbythec-Abltyrosine kinaseinnormalandcancercells.BiolCell2008;100:617–31.
[8]ZhuJ,WangJY.Deathby Abl:amatter oflocation.Curr TopDevBiol 2004;59:165–92.
[9]PendergastAM. TheAbl family kinases: mechanisms of regulationand signaling.AdvCancerRes2002;85:51–100.
[10]LiuZG,BaskaranR,Lea-ChouET,WoodLD,ChenY,KarinM,etal.Three distinctsignalingresponsesbymurinefibroblaststogenotoxicstress.Nature 1996;384:273–6.
[11]KharbandaS,RenR,PandeyP,ShafmanTD,FellerSM,WeichselbaumRR,etal. Activationofthe c-Abl tyrosinekinase in thestress responseto DNA-damagingagents.Nature1995;376:785–8.
[12]MeltserV,Ben-YehoyadaM,Shaul Y. c-Abltyrosine kinaseintheDNA damageresponse:celldeathandmore.CellDeathDiffer2011;18:2–4. [13]GonfloniS.DNAdamagestressresponseingermcells:roleofc-Abland
clinicalimplications.Oncogene2010;29:6193–202.
[14]WangX,ZengL,WangJ,ChauJF,LaiKP,JiaD,etal.Apositiveroleforc-Ablin ATM and ATR activation in DNA damage response. Cell Death Differ 2011;18:5–15.
[15]GonfloniS.Modulatingc-Ablnuclearactivityasastrategytopreservefemale fertility.CellCycle2010;9:217–8.
[16]GonfloniS,DiTellaL,CaldarolaS,CannataSM,KlingerFG,DiBartolomeoC, etal.Inhibitionofthec-Abl-TAp63pathwayprotectsmouseoocytesfrom chemotherapy-induceddeath.NatMed2009;15:1179–85.
[17]Woodruff TK. Preserving fertility during cancer treatment. Nat Med 2009;15:1124–5.
[18]LusserA,KadonagaJT.ChromatinremodelingbyATP-dependentmolecular machines.Bioessays2003;25:1192–200.
[19]Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693–705.
[20]MarmorsteinR.Proteinmodulesthatmanipulatehistonetailsforchromatin regulation.NatRevMolCellBiol2001;2:422–32.
[21]vanAttikumH,GasserSM.ThehistonecodeatDNAbreaks:aguidetorepair? NatRevMolCellBiol2005;6:757–65.
[22]PanierS,DurocherD.RegulatoryubiquitylationinresponsetoDNAdouble strandbreaks.DNARepair(Amst)2009;8:436–43.
[23]vanAttikumH,GasserSM.Crosstalkbetweenhistonemodificationsduring theDNAdamageresponse.TrendsCellBiol2009;19:207–17.
[24]UlrichHD,WaldenH.UbiquitinsignalinginDNAreplicationandrepair.Nat RevMolCellBiol2010;11:479–89.
[25]AtanassovBS,KoutelouE,DentSY.Theroleofdeubiquitinatingenzymesin chromatinregulation.FEBSLett2010.
[26]Al-HakimA,Escribano-DiazC,LandryMC,O’DonnellL,PanierS,SzilardRK, etal.TheubiquitousroleofubiquitinintheDNAdamageresponse.DNA Repair(Amst)2010;9:1229–40.
[27]DianovGL.RegulationofDNArepairbyubiquitination.Biochemistry (Mos-cow)2011;76:69–79.
[28]RamaekersCH,WoutersBG.RegulatoryfunctionsofubiquitinindiverseDNA damageresponses.CurrMolMed2011;11:152–69.
[29]LukasJ.TheinterfacebetweentheubiquitinfamilyandtheDNAdamage response.EMBORep2010;11:907–9.
[30]ShanbhagNM,Rafalska-MetcalfIU,Balane-BolivarC,JanickiSM,Greenberg RA.ATM-dependentchromatinchangessilencetranscriptionincistoDNA doublestrandbreaks.Cell2010;141:970–81.
[31]RichT, AllenRL, Wyllie AH. Defyingdeath after DNA damage. Nature 2000;407:777–83.
[32]LukasJ,BartekJ.DNArepair:newtalesofanoldtail.Nature2009;458:581–3. [33]BoultonSJ.DNArepair:decisionatthebreakpoint.Nature2010;465:301–2. [34]BennettEJ,HarperJW.DNAdamage:ubiquitinmarksthespot.NatStructMol
Biol2008;15:20–2.
[35]McHughPJ,SpanswickVJ,HartleyJA.RepairofDNAinterstrandcrosslinks: molecularmechanismsandclinicalrelevance.LancetOncol2001;2:483–90. [36]StojicL,BrunR,JiricnyJ.MismatchrepairandDNAdamagesignaling.DNA
Repair(Amst)2004;3:1091–101.
[37]RastogiRP,Richa,KumarA,TyagiMB,SinhaRP.Molecularmechanismsof ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010;592980.
[38]ReedSH.Nucleotideexcisionrepairinchromatin:damageremovalatthe dropofaHAT.DNARepair(Amst),doi:10.1016/j.dnarep.2011.04.029. [39]MladenovE,IliakisG.InductionandrepairofDNAdoublestrandbreaks:the
increasingspectrumofnon-homologousendjoiningpathways.MutatRes 2011;711:61–72.
[40]OzturkS,DemirN.DNArepairmechanismsinmammaliangermcells.Histol Histopathol2011;26:505–17.
[41]MeltserV,Ben-YehoyadaM,ReuvenN,ShaulY.c-Abldownregulatestheslow phaseofdouble-strandbreakrepair.CellDeathDis2010;1:e20.
[42]AmreinL,DavidsonD,ShawiM,PetruccelliLA,MillerJrWH,AloyzR,etal. Dualinhibitionofthehomologousrecombinationalrepairandthe nonho-mologousend-joiningrepairpathways inchroniclymphocyticleukemia therapy.LeukRes2011;35(8).doi:10.1016/j.leukres.2011.01.004. [43]SunH,TrecoD,SchultesNP,SzostakJW.Doublestrandbreaksataninitiation
siteformeioticgeneconversion.Nature1989;338:87–90.
[44]KharbandaS,PandeyP,MorrisPL,WhangY,XuY,SawantS,etal.Functional roleforthec-AbltyrosinekinaseinmeiosisI.Oncogene1998;16:1773–7.
[45]BartkovaJ,HorejsiZ,KoedK,KramerA,TortF,ZiegerK,etal.DNAdamage responseasacandidateanti-cancerbarrierinearlyhumantumorigenesis. Nature2005;434:864–70.
[46]GorgoulisVG,VassiliouLV,KarakaidosP,ZacharatosP,KotsinasA,LiloglouT, etal.ActivationoftheDNAdamagecheckpointandgenomicinstabilityin humanprecancerouslesions.Nature2005;434:907–13.
[47]d’AddadiFagagnaF.Livingonabreak:cellularsenescenceasaDNA-damage response.NatRevCancer2008;8:512–22.
[48]MoritaY,PerezGI,ParisF,MirandaSR,EhleiterD,Haimovitz-FriedmanA, etal.Oocyteapoptosisissuppressedbydisruptionoftheacid sphingomye-linasegeneorbysphingosine-1-phosphatetherapy.NatMed2000;6:1109– 14.
[49]HofmannK,BucherP.TheFHAdomain:aputativenuclearsignalingdomain found inprotein kinases andtranscription factors. Trends BiochemSci 1995;20:347–9.
[50]Bork P,Hofmann K,Bucher P, NeuwaldAF, Altschul SF, KooninEV.A superfamilyofconserveddomainsinDNAdamage-responsive cellcycle checkpointproteins.FASEBJ1997;11:68–76.
[51]WangB,ElledgeSJ.UBC13/RNF8ubiquitinligasescontrolfociformationof theRAP80/ABRAXAS/BRCA1/BRCC36complexinresponsetoDNAdamage. ProcNatlAcadSciUSA2007;104:20759–63.
[52]KolasNK,ChapmanJR,NakadaS,YlankoJ,ChahwanR,SweeneyFD,etal. OrchestrationoftheDNA-damageresponsebytheRNF8ubiquitinligase. Science2007;318:1637–40.
[53]HuenMS,GrantR,MankeI,MinnK,YuX,YaffeMB,etal.RNF8transducesthe DNA-damagesignalviahistoneubiquitylationandcheckpointprotein as-sembly.Cell2007;131:901–14.
[54]MailandN,Bekker-JensenS,FaustrupH,MelanderF,BartekJ,LukasC,etal. RNF8ubiquitylates histonesatDNAdouble-strandbreaksandpromotes assemblyofrepairproteins.Cell2007;131:887–900.
[55]CelesteA,Fernandez-CapetilloO,KruhlakMJ,PilchDR,StaudtDW,LeeA, etal.HistoneH2AXphosphorylationisdispensablefortheinitialrecognition ofDNAbreaks.NatCellBiol2003;5:675–9.
[56]YuanJ,AdamskiR,ChenJ.FocusonhistonevariantH2AX:tobeornottobe. FEBSLett2010;584:3717–24.
[57]BarabasiAL,BonabeauE.Scale-freenetworks.SciAm2003;288:60–9. [58]CollavinL,LunardiA,DelSalG.p53-Familyproteinsandtheirregulators:
hubsandspokesintumorsuppression.CellDeathDiffer2010;17:901–11. [59]SeebacherJ,GavinAC.SnapShot:protein–proteininteractionnetworks.Cell
2011;144(1000):e1.
[60]BensimonA,SchmidtA,ZivY,ElkonR,WangSY,ChenDJ,etal. ATM-dependentand-independentdynamicsofthenuclearphosphoproteome afterDNAdamage.SciSignal2010;3:rs3.
[61]RiballoE,KuhneM,RiefN,DohertyA,SmithGC,RecioMJ,etal.Apathwayof doublestrandbreakrejoiningdependentuponATM,Artemis,andproteins locatingtogamma-H2AXfoci.MolCell2004;16:715–24.
[62]MessickTE,GreenbergRA.TheubiquitinlandscapeatDNAdouble-strand breaks.JCellBiol2009;187:319–26.
[63]ChenZJ,SunLJ.Nonproteolyticfunctionsofubiquitinincellsignaling.Mol Cell2009;33:275–86.
[64]DikicI,WakatsukiS,WaltersKJ.Ubiquitin-bindingdomains–fromstructures tofunctions.NatRevMolCellBiol2009;10:659–71.
[65]HarperJW,SchulmanBA.Structuralcomplexityinubiquitinrecognition.Cell 2006;124:1133–6.
[66]HickeL,SchubertHL,HillCP.Ubiquitin-bindingdomains.NatRevMolCell Biol2005;6:610–21.
[67]HofmannK.Ubiquitin-bindingdomainsandtheirroleintheDNAdamage response.DNARepair(Amst)2009;8:544–56.
[68]GrabbeC, HusnjakK,Dikic I. Thespatialand temporalorganizationof ubiquitinnetworks.NatRevMolCellBiol2011;12:295–307.
[69]AvvakumovN,NouraniA,CoteJ.Histonechaperones:modulatorsof chro-matinmarks.MolCell2011;41:502–14.
[70]RansomM,DenneheyBK,TylerJK.ChaperoninghistonesduringDNA repli-cationandrepair.Cell2010;140:183–95.
[71]RabutG,PeterM.Functionandregulationofproteinneddylation.Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008;9:969–76.
[72]MeulmeesterE,MelchiorF.Cellbiology:SUMO.Nature2008;452:709–11. [73]GallagherE,GaoM,LiuYC,KarinM.ActivationoftheE3ubiquitinligaseitch
throughaphosphorylation-inducedconformationalchange.ProcNatlAcad SciUSA2006;103:1717–22.
[74]MoyalL,LerenthalY,Gana-WeiszM,MassG,SoS,WangSY,etal. Require-mentofATM-dependentmonoubiquitylationofhistoneH2Bfortimelyrepair ofDNAdoublestrandbreaks.MolCell2011;41:529–42.
[75]PastoriV,SangalliE,CoccettiP,PozziC,NonnisS,TedeschiG,etal.CK2and GSK3phosphorylationonS29controlswild-typeATXN3nuclearuptake. BiochimBiophysActa2010;1802:583–92.
[76]HuangTT,NijmanSM,MirchandaniKD,GalardyPJ,CohnMA,HaasW,etal. RegulationofmonoubiquitinatedPCNAbyDUBautocleavage.NatCellBiol 2006;8:339–47.
[77]BlackfordAN,StewartGS.Whencleavageisnotattractive:non-catalytic inhibitionofubiquitinchainsatDNAdoublestrandbreaksbyOTUB1.DNA Repair(Amst)2010;10:245–9.
[78]SowaME,BennettEJ,GygiSP,HarperJW.Definingthehuman deubiquitinat-ingenzymeinteractionlandscape.Cell2009;138:389–403.
[80]BartekJ,LukasJ.DNAdamagecheckpoints:frominitiationtorecoveryor adaptation.CurrOpinCellBiol2007;19:238–45.
[81]DoilC,MailandN,Bekker-JensenS,MenardP,LarsenDH,PepperkokR,etal. RNF168bindsandamplifiesubiquitinconjugatesondamagedchromosomes toallowaccumulationofrepairproteins.Cell2009;136:435–46. [82]RamachandranS,ChahwanR,NepalRM,FriederD,PanierS,RoaS,etal.The
RNF8/RNF168ubiquitinligasecascadefacilitatesclassswitchrecombination. ProcNatlAcadSciUSA2010;107:809–14.
[83]GalantyY, Belotserkovskaya R, Coates J,Polo S,MillerKM, JacksonSP. MammalianSUMOE3-ligasesPIAS1andPIAS4promoteresponsestoDNA doublestrandbreaks.Nature2009;462:935–9.
[84]MorrisJR,BoutellC,KepplerM,DenshamR,WeekesD,AlamshahA,etal.The SUMOmodificationpathwayisinvolvedintheBRCA1responsetogenotoxic stress.Nature2009;462:886–90.
[85]BartekJ,HodnyZ.SUMObooststheDNAdamageresponsebarrieragainst cancer.CancerCell2010;17:9–11.
[86]NakamuraK,KatoA,KobayashiJ,YanagiharaH,SakamotoS,OliveiraDV,etal. RegulationofhomologousrecombinationbyRNF20-dependentH2B ubiqui-tination.MolCell2011;41:515–28.
[87]DevganSS,SanalO,DoilC,NakamuraK,NahasSA,PettijohnK,etal. Homozy-gousdeficiencyofubiquitin-ligasering-fingerproteinRNF168mimicsthe radiosensitivitysyndromeofataxia-telangiectasia.CellDeathDiffer2011. [88]YuX,FuS,LaiM,BaerR,ChenJ.BRCA1ubiquitinatesits
phosphorylation-dependentbindingpartnerCtIP.GenesDev2006;20:1721–6.
[89]Rose A, Schlieker C. DNA repair: blocking ubiquitin transfer. Nature 2010;466:929–30.
[90]NakadaS,TaiI,PanierS,Al-HakimA,IemuraS,JuangYC,etal.Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010;466:941–6.
[91]YaoT,SongL,JinJ,CaiY,TakahashiH,SwansonSK,etal.Distinctmodesof regulationoftheUch37deubiquitinatingenzymeintheproteasomeandin theIno80chromatin-remodelingcomplex.MolCell2008;31:909–17. [92]ShaoG,LilliDR,Patterson-FortinJ,ColemanKA,MorrisseyDE,GreenbergRA.
TheRAP80–BRCC36de-ubiquitinatingenzymecomplexantagonizesRNF8– UBC13-dependentubiquitinationeventsatDNAdoublestrandbreaks.Proc NatlAcadSciUSA2009;106:3166–71.
[93]NijmanSM,HuangTT,DiracAM,BrummelkampTR,KerkhovenRM,D’Andrea AD,etal.ThedeubiquitinatingenzymeUSP1regulatestheFanconianemia pathway.MolCell2005;17:331–9.
[94]SimsAE,SpiteriE,Sims3rdRJ,AritaAG,LachFP,LandersT,etal.FANCIisa secondmonoubiquitinatedmemberoftheFanconianemiapathway.Nat StructMolBiol2007;14:564–7.
[95]NicassioF,CorradoN,VissersJH,ArecesLB,BerginkS,MarteijnJA,etal. HumanUSP3isachromatinmodifierrequiredforSphaseprogressionand genomestability.CurrBiol2007;17:1972–7.
[96]Joo HY,ZhaiL, YangC, NieS, Erdjument-Bromage H, TempstP, etal. RegulationofcellcycleprogressionandgeneexpressionbyH2A deubiqui-tination.Nature2007;449:1068–72.
[97]GinjalaV,NacerddineK,KulkarniA,OzaJ,HillSJ,YaoM,etal.BMI1is recruitedto DNAbreaks and contributesto DNA damage-inducedH2A ubiquitinationandrepair.MolCellBiol2011;31:1972–82.
[98]Ismail IH,Andrin C, McDonald D, Hendzel MJ.BMI1-mediated histone ubiquitylation promotes DNA double strand break repair. J Cell Biol 2010;191:45–60.
[99]ThakarA,ParvinJD,ZlatanovaJ.BRCA1/BARD1E3ubiquitinligasecanmodify histonesH2AandH2B inthenucleosomeparticle.JBiomolStruct Dyn 2010;27:399–406.
[100]WangH,ZhaiL, XuJ,JooHY,JacksonS,Erdjument-BromageH,etal. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligasefacilitates cellularresponseto DNA damage.Mol Cell2006;22: 383–94.
[101]ChengWH,vonKobbeC,OpreskoPL,FieldsKM,RenJ,KufeD,etal.Werner syndromeprotein phosphorylation by abl tyrosine kinase regulates its activityanddistribution.MolCellBiol2003;23:6385–95.
[102]GoldbergZ,VogtSionovR,BergerM,ZwangY,PeretsR,VanEttenRA,etal. TyrosinephosphorylationofMDM2byc-Abl:implicationsforp53 regula-tion.EMBOJ2002;21:3715–27.
[103]GuJ,KawaiH,NieL,KitaoH,WiederschainD,JochemsenAG,etal.Mutual dependenceofMDM2andMDMXintheirfunctionalinactivationofp53.J BiolChem2002;277:19251–4.
[104]WaningDL,LehmanJA,BatuelloCN,MayoLD. ControllingtheMDM2– MDMX-p53circuit.Pharmaceuticals(Basel)2010;3:1576–93.
[105]WaningDL,LehmanJA,BatuelloCN,MayoLD.c-Ablphosphorylationof MDM2 facilitates MDM2–MDMX complex formation. J Biol Chem 2011;286:216–22.
[106]KentsisA,GordonRE,BordenKL.Controlofbiochemicalreactionsthrough supramolecular RING domain self-assembly. Proc Natl Acad Sci USA 2002;99:15404–9.