59
4. Gli inosilicati idrati di calcio
Gli inosilicati idrati di calcio formano un gruppo comprendente dieci specie mineralogiche differenti, oltre ad alcune fasi sintetiche. Tutte queste specie sono caratterizzate da catene tipo wollastonite, sia singole che doppie (Tab. 4.1).
Tab. 4.1 – Inosilicati idrati di calcio
Inosilicati a catena singola Inosilicati a catena doppia Incertae sedis Foshagite Clinotobermorite Oyelite Hillebrandite Tobermorite Tacharanite
Jennite Xonotlite
Plombièrite Riversideite
Fra queste specie se ne possono individuare alcune caratterizzate da un comune assetto strutturale e che possono essere inserite nel gruppo informale della tobermorite. Queste fasi (clinotobermorite, oyelite, plombièrite, riversideite, tacharanite e tobermorite) saranno trattate in maniera più approfondita nei prossimi capitoli. Qui ci limiteremo allo studio dei rimanenti silicati ed in particolare all’esame di foshagite, hillebrandite, jennite e xonotlite.
I campioni studiati sono riportati in tab. 4.2. Oltre alla provenienza e ad una loro sommaria descrizione, vengono riportate le tecniche analitiche impiegate per la loro caratterizzazione. Occorre subito evidenziare come la scarsa qualità dei campioni disponibili non abbia consentito di raccogliere dati strutturali relativi a queste fasi che sono quindi state esclusivamente identificate sulla base di diffrattogrammi di polvere e di analisi chimiche semi-quantitative in modalità EDS. Unica eccezione è rappresentata dai cristalli di xonotlite della cava Grolla (Spagnago, Vicenza) che hanno consentito anche studi di cristallo singolo con tecnica Weissenberg.
Tabella 4.2 – Campioni di silicati idrati di calcio del gruppo della wollastonite studiati nel corso di questo studio
Minerale Provenienza Descrizione Tecniche analitiche
Foshagite Crestmore (California, USA) Cristalli fibrosi bianchi XRPD, SEM-EDS Foshagite Fuka mine (Okayama Prefecture,
Giappone)
Venette fibrose bianche XRPD, SEM-EDS
Foshagite Mihara (Okayama Prefecture, Giappone)
Venette fibrose bianche XRPD
Hillebrandite Fuka (Okayama Prefecture, Giappone) Vene fibrose bianche SEM-EDS Xonotlite Mihara (Okayama Prefecture, Cristalli fibrosi bianchi con XRPD
60
Fig. 4.1 - Struttura della wollastonite-2M, vista lungo c
Giappone) calcite
Xonotlite Groppo di Gorro (Berceto, Parma) Cristalli fibrosi bianchi XRPD, SEM-EDS, TG-DSC
Xonotlite Montecastelli (Pomarance, Pisa) Cristalli fibrosi bianchi XRPD, SEM-EDS, TG-DSC
Xonotlite Cava Grolla (Spagnago, Vicenza) Cristalli fibrosi bianchi XRPD, SC-XRD, SEM-EDS
Come già detto, le fasi trattate in questo capitolo mostrano la presenza di catene tipo wollastonite. Quindi, anche se quest’ultimo minerale non è un composto C-S-H ma bensì un silicato anidro di calcio, riteniamo necessario premettere una veloce descrizione della sua struttura. Sarà così possibile introdurre anche la terminologia della teoria OD, teoria sviluppata a partire dal lavoro di Dornberger-Schiff (1956) in seguito all’osservazione, da parte di Jeffery (1953), di alcune singolarità proprio nei diffrattogrammi della wollastonite.
La struttura di questo silicato è caratterizzata da catene singole con periodicità pari a tre tetraedri; utilizzando la terminologia proposta da Liebau (1985), possiamo definire queste catene come dreierketten. Esse possono essere descritte come formate dall’unione di gruppi disilicato (paired tetrahedra) [Si2O7] con tetraedri ponte (bridging tetrahedra) [SiO4
Le catene singole tipo wollastonite possono connettersi a formare doppie catene condividendo un numero variabile di tetraedri; in particolare potranno condividere uno, due o tre tetraedri. In quest’ultimo caso si formeranno catene completamente condensate (es. elpidite, Na
]. Le catene silicatiche sono legate a nastri di poliedri di Ca a coordinazione VI (fig. 4.1).
2Zr[Si6O15].2H2O, ed epididimite, Na2Be2[Si6O15].H2O), caratterizzate da un forte grado di rigidità (fig. 4.2a). Le catene meno condensate, come quelle formate dalla condivisione di due tetraedri su tre (fig. 4.2b) oppure di un solo tetraedro ogni tre (fig. 4.2c), potranno invece assumere differenti configurazioni. Merlino & Bonaccorsi (2008) hanno descritto catene contratte con
61
Fig. 4.2 – Differenti topologie delle catene tipo wollastonite nella epididimite (a), nell’okenite (b) e nella xonotlite (c) (da Merlino & Bonaccorsi, 2008).
simmetria [.2/m.], tipiche della xonotlite, catene estese con simmetria [.2/m.], presenti nella clinotobermorite, ed infine catene estese con simmetria [2mm], presenti nella struttura della tobermorite.
La periodicità delle catene silicatiche, pari a 7.3 Å, è esattamente il doppio della lunghezza dello spigolo dei poliedri Ca, 3.65 Å. Pertanto le catene silicatiche potranno connettersi ai moduli contenenti i cationi Ca2+ secondo due differenti modalità geometriche, traslate l’una rispetto all’altra di 3.65 Å nella direzione [010]. Questo aspetto cristallochimico è alla base della natura OD e del conseguente politipismo caratteristico della wollastonite e di un gran numero di composti C-S-H.
4.1 Cenni della teoria OD
La teoria OD fu proposta per la prima volta da Dornberger-Schiff (1956) per fornire una adeguata spiegazione delle proprietà anomale manifestate dai diffrattogrammi della wollastonite (fig. 4.3; Jeffery, 1953), proprietà anomale che si sono poi rivelate comuni a molti altri minerali.
Le caratteristiche riscontrabili dall’attento esame dei diffrattogrammi sono:
• compresenza di macchie nette e righe (streaks) diffuse oppure di macchie nette e riflessi più o meno diffusi;
• assenze sistematiche non di gruppo spaziale;
• simmetria della diffrazione maggiore di quella corrispondente al gruppo puntuale ed alla legge di Friedel (spesso questo fenomeno interessa solo una porzione del reticolo reciproco e viene indicato come partial enhancement of simmetry);
• esistenza di una serie di riflessi comuni nei diffrattogrammi ottenuti da cristalli diversi di una stessa famiglia (riflessi di famiglia); essi si presentano nelle medesime posizioni e con la stessa intensità nei vari membri della famiglia;
• evidenza di geminazioni, spesso polisintetiche; • evidenza di fenomeni di politipismo.
62
Tutte queste caratteristiche, che spesso si manifestano contemporaneamente, sono diverse manifestazioni della natura OD del cristallo studiato (Dornberger-Schiff, 1956; Dornberger-Schiff, 1964; Dornberger-Schiff, 1966; Merlino, 1997; Ferraris et al., 2004).
Ogni struttura normalmente ordinata può essere descritta in termini di strati strutturali bidimensionali. Allorché strati adiacenti possono susseguirsi secondo differenti modalità geometricamente (e quindi anche energeticamente) equivalenti non si avrà un’unica struttura bensì un numero infinito di possibili sequenze di strati, ordinate o disordinate, nelle quali coppie di strati adiacenti sono geometricamente equivalenti. L’intero insieme di tali sequenze costituisce una famiglia OD. Ogni possibile sequenza, ordinata o disordinata, rappresenta un individuo della stessa famiglia; gli individui corrispondenti alle infinite sequenze ordinate rappresentano i vari politipi. In ogni famiglia OD esiste poi un piccolo numero di politipi principali, detti anche “semplici”, “standard” o “regolari”, generalmente riscontrati più frequentemente; la teoria OD fornisce un criterio geometrico che permette di riconoscere tali politipi, chiamati “strutture MDO” (strutture a massimo grado di ordine, Maximum Degree of Order). Essi sono infatti gli individui nei quali non solo coppie ma anche triple (ed in generale ennuple) di strati consecutivi sono geometricamente equivalenti.
Ogni politipo presenta una sua simmetria; risulta tuttavia più interessante concentrare l’attenzione sulle proprietà di simmetria comuni all’intera famiglia, dalle quali seguono quelle del singolo
politipo. Le operazioni di simmetria che riportano uno strato Lp su se stesso sono indicate come λ-POs; la sigla POs sta per “operazioni parziali” (Partial Operations) in quanto le leggi di simmetria vigenti all’interno dello strato strutturale non necessariamente si estendono all’intera struttura; invece le operazioni di simmetria che riportano uno strato Lp su uno strato adiacente Lp+1 sono
Fig. 4.3 - Diffrattogramma di cristallo singolo della wollastonite. A sinistra la sovrapposizione degli strati h0l e h2l; a destra lo strato h1l (da Jeffery, 1953).
63
indicate come σ-POs. L’insieme degli operatori parziali λ- e σ- ci fornisce le proprietà di simmetria comuni all’intera famiglia di strutture OD, secondo un simbolo che si dispiega su due righe. Nella prima riga vengono definite le λ-POs con l’indicazione del tipo di reticolo e degli operatori di simmetria corrispondenti alle direzioni x, y e z; nel caso di famiglie caratterizzate da strati OD con reticoli quadrati o trigonali/esagonali sono necessarie più di tre posizioni per indicare le operazioni relative alle varie direzioni, e precisamente cinque nel caso di reticoli quadrati e sette nel caso di reticoli trigonali/esagonali. La direzione lungo la quale si ha la successione degli strati viene esplicitata ponendo il corrispondente operatore di simmetria fra parentesi tonde. Nella seconda riga vengono invece riportate le σ-POs. Per esse si usa un’ovvia estensione delle notazioni utilizzate nelle International Tables for X-Ray Crystallography.
Ad esempio le proprietà di simmetria dell’intera famiglia di strutture OD della wollastonite sono rappresentate dalla seguente simbologia:
P (1) 21 {(1) 2
/m 1
1/2/a2 1}
Tale simbologia indica che il singolo strato ha vettori di traslazione b e c (il terzo vettore, a0, non è vettore di traslazione dello strato strutturale). Esso presenta i seguenti elementi di simmetria: una elicogira binaria parallela a b (la componente traslatoria è ovviamente b/2); piano di simmetria ortogonale ad essa, nonché un centro di inversione. Il singolo strato va in ricoprimento con quello adiacente attraverso una elicogira binaria parallela a b, con componente traslatoria di b/4; con uno slittopiano a2 ortogonale a b, avente componente traslatoria pari all’intero vettore a0. È inoltre presente, tra le σ-POs, anche un centro di inversione.
4.2 Inosilicati idrati di calcio con catene tipo wollastonite
Foshagite, hillebrandite, jennite e xonotlite sono inosilicati idrati di calcio caratterizzati dalla presenza di catene tipo wollastonite; come già evidenziato in tab. 4.1, nelle prime tre specie tali catene sono singole mentre diventano doppie nella xonotlite.
Questi minerali compaiono tipicamente nelle fasi idrotermali tardive legate alla messa in posto di rocce magmatiche all’interno di complessi carbonatici; soltanto la xonotlite compare anche in altri ambienti geologici, solitamente in associazione a rocce ultrabasiche serpentinizzate.
Il rapporto Ca/Si è sempre ≥ 1: esso vale 2 nella hillebrandite, 1.5 nella jennite, 1.33 nella foshagite e 1 nella xonotlite. Nelle fasi appartenenti al gruppo della tobermorite, invece, tale rapporto è sempre inferiore a 1, eccezion fatta per l’oyelite, il cui assetto strutturale è ancora ignoto. I minerali
64
che andremo a considerare nei prossimi paragrafi formano generalmente cristalli aciculari minutamente fibrosi, di difficile utilizzo nello studio con tecniche di cristallo singolo. Inoltre, come conseguenza delle relazioni metriche fra le catene tipo wollastonite e le dimensioni degli spigoli dei poliedri di Ca essi presentano spesso un carattere OD, con la comparsa di fenomeni di disordine strutturale più o meno estesi.
4.2.1 Foshagite
La foshagite fu descritta per la prima volta da Eakle (1925) su campioni provenienti da Crestmore (California, USA). La struttura di questa fase è stata risolta e raffinata da Gard & Taylor (1960) in
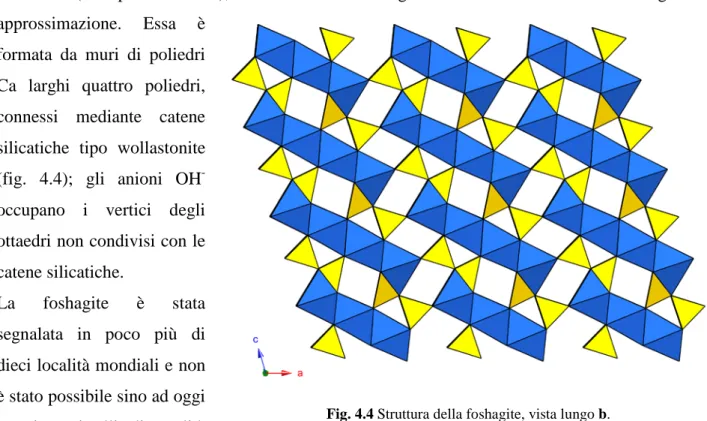
una cella triclina (gruppo spaziale P1), con parametri a 10.32, b 7.36, c 7.04 Å, α 90°, β 106.4°, γ 90°. I riflessi con k dispari sono deboli e diffusi, ad indicare una probabile natura OD di questo silicato; la struttura è stata determinata partendo da un modello strutturale ed utilizzando i soli riflessi h0l. Pertanto, considerato il basso di numero di riflessi utilizzati e l’alto valore del fattore di accordo R (0.23 per 58 riflessi), la struttura della foshagite è conosciuta solo con un certo grado di approssimazione. Essa è
formata da muri di poliedri Ca larghi quattro poliedri, connessi mediante catene silicatiche tipo wollastonite (fig. 4.4); gli anioni OH
-La foshagite è stata segnalata in poco più di dieci località mondiali e non è stato possibile sino ad oggi reperire cristalli di qualità
sufficiente per tentare un nuovo studio strutturale con tecniche di cristallo singolo. La principale difficoltà consiste nel fatto che questo silicato tende a formare aggregati di fibre con orientazione casuale attorno all’asse b; a questo si aggiunge la natura OD dei cristalli. Ferraris et al. (2004) sottolineano come i cristalli di foshagite siano sempre altamente disordinati, con riflessi deboli e diffusi lungo a* per k dispari, e derivano i due possibili politipi MDO. Nei diffrattogrammi di questo minerale sono presenti massimi sugli streaks continui compatibili con il politipo MDO occupano i vertici degli
ottaedri non condivisi con le catene silicatiche.
1 da
65
loro derivato; i cristalli ottenuti per sintesi mostrano invece la compresenza di domini di entrambi i politipi MDO (Ferraris et al., 2004).
In natura, la foshagite si rinviene quale prodotto di alterazione idrotermale tardiva a carico dei silicati di calcio formatisi nelle aureole termometamorfiche. È questa l’origine della foshagite della località tipo californiana, così come quella di Kilchoan, Ardnamurchan, Scozia (Agrell, 1965) e di Kushiro, Hiroshima Prefecture, Giappone (Kusachi et al., 1971). La foshagite è stata descritta anche in uno skarn a melilite sulla Dupezeh Mountain, Iraq, con monticellite, wollastonite, fassaite e baghdadite (Al-Hermezi et al., 1986); nella Bingham Canyon mine, Utah, USA (Wilson, 1995); nello skarn a cuspidina di Chesney Vale, Victoria, Australia, con vesuvianite e calcite (Henry, 1999); in venette tardive negli skarn a spurrite, tilleyite e gehlenite di Cornet Hill, Apuseni Mountains, Romania (Marincea et al., 2001) e, in analoga giacitura, negli skarn a spurrite di Fuka, Okayama Prefecture, Giappone (Satish-Kumar et al., 2004); in relazione a rocce skarnoidi nel deposito cuprifero di Gumeshevsk, Urali, Russia (Grabezhev et al., 2007); in xenoliti nel massiccio di Yoko-Dovyrenski, Siberia, Russia (Zadov et al., 2004; Gałuskin et al., 2007). Recentemente Zordan et al. (2008) hanno identificato mediante spettroscopia IR e diffrattometria di raggi X la presenza di questo silicato, in stretta associazione con xonotlite e calcite, anche nell’affioramento cornubianitico di Contrada Molini (Laghi, Vicenza). La foshagite compare inoltre nelle paragenesi a silicati di calcio della Wessels mine, con orientite e calcite (Kalahari Manganese Field, Repubblica Sudafricana; Von Bezing et al., 1991). Fibre di questo minerale sono state descritte da Gross (1977) nella Hatrurim Formation (Israele), in paragenesi con afwillite e minerali del gruppo della tobermorite.
I campioni studiati nel corso di questa tesi provengono da Crestmore (California, USA) e dalle aureole metamorfiche di Fuka e Mihara (Okayama Prefecture, Giappone). La loro identificazione è stata condotta mediante diffrattogrammi di polvere con camera Gandolfi (Tab. 4.3); i parametri di cella sono stati raffinati con il programma UNIT CELL (Holland & Redfern, 1997), utilizzando solo i riflessi univocamente indicizzati (Tab. 4.4). Infine, le analisi chimiche sono state condotte in modalità EDS (Tab. 4.5).
Tab. 4.3 – Diffrattogrammi di raggi X su campioni di foshagite Crestmore (diffr. n° 179) Mihara (diffr. n° 54) Fuka (diffr. n° 58) dhkl Ihkl dhkl Ihkl dhkl Ihkl h k l 9.8 vw 100 6.63 m 001 5.48 vw 4.94 m 4.97 w 2 0 0 4.69 vw 1 1 1 3.636 m 3.530 w 3.552 w 3.544 w 2 0 1
66
Tab. 4.4 – Parametri di cella dei campioni di foshagite studiati Crestmore Fuka Mihara Gard &
Taylor (1960) a (Å) 10.349(3) 10.305(4) 10.367(3) 10.32 b (Å) 7.342(2) 7.349(2) 7.350(2) 7.36 c (Å) 7.032(1) 7.067(2) 7.054(1) 7.04 α (°) 90.26(3) 90.62(3) 90.26(3) 90 β (°) 106.53(2) 106.41(2) 106.47(2) 106.4 γ (°) 89.99(3) 89.76(3) 90.47(3) 90
Tab. 4.5 – Analisi chimiche dei campioni di foshagite Crestmore (n = 2) Fuka (n = 1) wt.% wt.% SiO2 45.00 43.41 CaO 55.00 54.53 Na2O n.d. 2.05 totale 100.00 100.00
Il campione di Crestmore presenta una superficie formata da fibre disorientate di dimensioni sub-millimetriche e di colore bianco. Le osservazioni in microscopia elettronica a scansione hanno evidenziato la natura finemente fibrosa di questo campione (Fig. 4.5a), non trattabile pertanto con tecniche di cristallo singolo. La formula chimica, ricalcolata sulla base di 10 O pfu, assumendo la
presenza di 2 OH-, è
Ca3.95Si3.02O9(OH)2, in buon accordo con la formula teorica Ca4Si3O9(OH)2
Gli esemplari giapponesi sono stati estratti da alcuni campioni di skarn a spurrite provenienti da Fuka e Mihara (Okayama Prefecture); si tratta di vene di potenza millimetrica nelle quali la
. 3.357 mw 3.357 m 3.370 m 0 0 2 3.255 mw 3.264 m 3.254 w 3 0 0 3.054 vw 3.054 m 3.035* m 0 1 2, 3 11 3.034* m 2.940 s 2.940 s 2.938 s 2 2 0 2.801 w 2.816 w 2.806 w 3 0 2 2.691 vw 2.699 w vw 3 0 1 2.628 vw 2.469 w 2.489 w 2.489 mw 0 2 2 2.448 vw 3 2 0 2.312 mw 2.312 w 2.311 m 2 0 3 2.234 vw 1 1 3, 3 2 2 2.164 mw 2.164 w 2.164 m 3 2 1 2.114 w 2.114 mw 4 21 2.098* vw 2.097* w 3 0 2 2.050 w 2.045 w 2.050 mw 2 2 2 1.974 vw 1.950 w 5 0 0, 4 2 2 1.916 vw 1.913 w 1.914 mw 0 2 3 1.877* vw 1.873* mw 1.837 vw 1.837 m 1.830 mw 0 4 0 1.806 vw 1.770 vw 1.771 mw 1 2 3, 5 21 1.745 m 1.745 m 1.745 ms 5 2 2 , 4 2 3 1.591 w 1.604 w 1.604 w 4 2 2, 5 2 1
I riflessi indicati con il simbolo * sono attribuibili alla calcite o possono avere il contributo di questa fase. Le intensità sono stimate visivamente: s = forte, m = medio, w = debole, vw = molto debole.
67
foshagite è strettamente associata a calcite, da cui è difficilmente separabile. I diffrattogrammi Gandolfi raccolti su questi campioni sono di qualità peggiore rispetto a quello ottenuto sul materiale di Crestmore e presentano costantemente righe attribuibili alla calcite. Lo studio SEM-EDS è stato condotto esclusivamente sui campioni provenienti da Fuka, per i quali era presente maggior materiale. Anche in questo caso è stata evidenziata la notevole fibrosità di questi cristalli (Fig. 4.5b) anche alla scala di pochi μm. Le analisi EDS sui cristalli di Fuka hanno evidenziato la presenza di piccole percentuali di Na; il ricalcolo delle analisi porta alla formula (Ca3.97Na0.27)Σ=4.24Si2.95O9(OH)2. Si osserva inoltre che mentre il rapporto Ca/Si è prossimo al valore ideale, la somma dei cationi (Ca+Na) è significativamente superiore a 4. Ciò va a bilanciare elettrostaticamente, da un punto di vista formale, il deficit di Si ma richiede, da un punto di vista cristallochimico, l’esistenza di qualche meccanismo in grado di bilanciare la sostituzione Ca2+ → Na+. Naturalmente queste considerazione potranno essere più correttamente svolte una volta che siano disponibili dati chimici più accurati e campioni di migliore qualità.
Fig. 4.5 – Immagini SEM dei campioni di foshagite studiati nel corso di questa tesi di dottorato. a) cristalli fibrosi, Crestmore (California, USA); b) cristalli aciculari minutamente fibrosi, Fuka (Giappone).
4.2.2 Hillebrandite
La hillebrandite è una specie descritta per la prima volta da Wright (1908) su campioni provenienti da Velardeña (Durango, Messico), dove è associata a hibschite e wollastonite (Heller, 1953). Questo silicato è un minerale rarissimo, sino ad oggi identificato in poco più di dieci località al mondo; oltre che nella località tipo messicana, l’hillebrandite è stata infatti descritta in altri siti ben noti per la presenza di fasi C-S-H fra cui possiamo citare la Hatrurim Formation (Israele; Gross, 1977), Fuka (Okayama Prefecture, Giappone; Kusachi et al., 1989), Kushiro (Hiroshima Prefecture, Giappone; Kusachi et al., 1971), Crestmore (California, USA; Woodford et al., 1941), la Kombat mine (Namibia; Peacor et al., 1988; Dunn, 1991), Carlingford (Irlanda; Nockolds & Vincent, 1947);
68
Tunguska (Siberia; Reverdatto, 1964), Flekkeren (Oslo rift, Norvegia; Jamveit et al., 1997), il massiccio di Yoko-Dovyrensky (Zadov et al., 2004) e la Upper Chegem volcanic structure (Repubblica Kabardino-Balkaria, Russia; Gałuskina et al., 2009).
La struttura media della hillebrandite è stata descritta da Dai & Post (1995), facendo riferimento ad una cella con a 3.639, b 16.311, c 11.829 Å, gruppo spaziale Ccm21; questi autori hanno utilizzato soltanto i riflessi di famiglia, netti ed intensi, trascurando i riflessi deboli e diffusi indicativi di una periodicità a’ = 2a, pari a 7.28 Å, traslazione tipica delle catene tipo wollastonite. La struttura della hillebrandite (fig. 4.6) può essere descritta come
formata da una impalcatura tridimensionale di poliedri Ca, aventi coordinazione VI e VII, con la formazione di tunnel lungo [100]; in tali tunnel trovano posto le catene silicatiche tipo wollastonite. La determinazione strutturale ha mostrato la presenza di siti Si con occupanza 0.5; ciò è stato spiegato da Xu & Buseck (1996) i quali, studiando in microscopia elettronica a trasmissione ed in diffrazione elettronica cristalli di hillebrandite della località tipo messicana, hanno rilevato un raddoppio del parametro di cella a, evidenziato dalla presenza di streaks continui paralleli a b*. Essi pertanto ipotizzano l’esistenza di due possibili posizioni delle catene tipo wollastonite e quindi l’insorgere di un esteso
disordine strutturale. Merlino (1997), considerando le evidenze rappresentate dalla contemporanea presenza di riflessi netti e streaks diffusi e le assenze sistematiche non di gruppo spaziale, ha interpretato la struttura della hillebrandite utilizzando la teoria OD, derivando così i possibili politipi, le relative celle elementari ed i corrispondenti gruppi spaziali.
4.2.3
Jennite
La jennite è un raro silicato descritto fino ad oggi in circa quindici località al mondo. Carpenter et al. (1966) descrissero per la prima volta questo minerale su campioni provenienti da Crestmore (California, USA), in associazione a plombièrite e, occasionalmente, a scawtite e calcite. Successivamente la jennite è stata identificata nella Hatrurim Formation (Israele; Gross, 1977), a Zeilberg (Baviera, Germania; Wittern, 2001) e nella regione vulcanica dell’Eifel (Renania
Fig. 4.6 – Struttura della hillebrandite vista lungo [100].
69
Palatinato, Germania; Schüller, 1990), a Fuka (Kusachi et al., 1989), nella Wessels mine (Repubblica Sudafricana; Von Bezing et al., 1991) ed in due località italiane, la cava Campomorto presso Montalto di Castro (Viterbo; Passaglia & Turconi, 1982) e Colle Fabbri (Perugia; Stoppa et al., 2010).
L’importanza della jennite risiede nelle sue possibili relazioni con il già ricordato gel C-S-H, il principale agente legante dei cementi. Secondo vari autori, infatti, nel C-S-H sarebbero presenti domini tipo tobermorite e domini tipo jennite, con questi ultimi tendenti a diventare, con il passare del tempo, progressivamente più abbondanti (Taylor, 1997).
La struttura della jennite è stata risolta da Bonaccorsi et al. (2004) utilizzando un campione proveniente dalla località giapponese di Fuka. Come per numerose altre fasi di questo gruppo, anche i diffrattogrammi della jennite mostrano evidenze di una natura OD: i riflessi con k = 2n+1 sono sistematicamente deboli e possono essere netti oppure diffusi lungo a*, indicando che il disordine ha luogo lungo tale direzione. Trascurando questo insieme di riflessi, è possibile individuare una cella di famiglia monoclina A-centrata, con costanti di cella a 9.947, b 3.642, c 21.37 Å, β 101.90°. Invece, prendendo in considerazione anche i riflessi caratteristici, la cella reale è a 10.593, b 7.284, c 10.839 Å, α 99.67, β 97.65, γ 110.11°. La struttura della jennite (Fig. 4.7) è stata descritta da Bonaccorsi et al. (2004) in termini di unità modulari; i tre moduli che la compongono sono rappresentati da nastri tipo tilleyite formati da ottaedri Ca, catene silicatiche singole tipo wollastonite ed ottaedri Ca addizionali posti nella posizione di interstrato. Lo studio strutturale ha consentito di conoscere con sufficiente accuratezza il complesso sistema di legami a
70
idrogeno di questa fase C-S-H, potendo così formulare, per la jennite, la composizione Ca9Si6O18(OH)6·8H2
A temperature di 70-90°C, la jennite si trasforma in “metajennite”, con la perdita di 4 molecole di H
O. Il campione proveniente da Fuka e studiato da Bonaccorsi et al. (2004) presentava una completa occupanza degli ottaedri Ca addizionali presenti nell’interstrato; le jenniti sintetiche, al contrario, mostrano un deficit di Ca in queste posizioni strutturali, deficit che viene bilanciato mediante la presenza di legami Si-OH.
2O; questo processo è chiaramente identificabile nei diffrattogrammi di polveri per il differente valore della d002, pari a 10.45 Å nella jennite e 8.69 Å nella “metajennite”. La struttura della fase disidratata non è stata finora risolta ma la conoscenza dell’assetto strutturale della jennite consente di ipotizzare che la perdita di molecole di H2O che coordinano i siti Ca nella posizione di interstrato abbia serie conseguenze sulla stabilità della struttura; in aggiunta vengono meno anche alcuni legami a H formati fra un O del tetraedro di bridging delle catene silicatiche e queste molecole di H2O. La struttura pertanto deve riorganizzarsi per giungere ad una configurazione più stabile, ottenendo una appropriata coordinazione dei siti Ca precedentemente posti nell’interstrato. La formula chimica della “metajennite” è Ca9[Si6O16(OH)2](OH)8·2H2O.
4.2.4 Xonotlite
La xonotlite è stata descritta da Rammelsberg (1866) su campioni provenienti da Tetela de Xonotla (Messico); essa corrisponde ad altri due minerali descritti negli anni seguenti con i nomi di eakleite (Larsen, 1917; Foshag & Larsen, 1922) e jurupaite (Eakle, 1921). Larsen (1923) dimostrò, sulla base delle proprietà chimiche ed ottiche, l’identità fra xonotlite ed eakleite mentre Taylor (1954), ricorrendo alla diffrazione di raggi X, evidenziò l’uguaglianza fra xonotlite e jurupaite.
La xonotlite è probabilmente uno dei più comuni silicati idrati di calcio presenti in natura. Questa specie compare generalmente in due differenti ambienti geologici:
1) rocce termometamorfiche di alta T, spesso attorno ad intrusioni basiche; 2) vene associate a rocce basiche ed ultrabasiche.
In entrambi questi ambienti la xonotlite è il prodotto dell’attività idrotermale tardiva.
La xonotlite della località tipo messicana è stata raccolta in calcari termometamorfosati; giaciture analoghe presentano i campioni descritti da Smith (1954) e da Brown (1978) in rocce termometamorfiche del complesso magmatico di Bay of Islands (Newfoundland, Canada). In queste località la xonotlite è associata a prehnite, calcite e clinozoisite quale prodotto di alterazione idrotermale di una cornubianite pirossenica. In giacitura analoga la xonotlite è descritta anche da Shannon (1925) a Goose Creek (Virginia, USA).
71
In associazione a basalti idrotermalizzati, la xonotlite è stata descritta da Schwarz (1924, 1925) nel Minnesota (USA), in associazione a pectolite e prehnite (nel lavoro del 1924 quest’ultima fu descritta come diopside), assieme a vari solfuri (calcopirite, sfalerite, pirrotina e polydimite). Kaye (1953) ha descritto la presenza di xonotlite lungo il contatto tettonico fra serpentiniti e metavulcaniti nell’isola di Puerto Rico; la presenza di questo silicato idrato di calcio era limitata alla zona di taglio lungo la quale avevano potuto circolare i fluidi idrotermali.
In vene all’interno di serpentiniti, la xonotlite è stata descritta anche da Bonaccorsi et al. (1996) su campioni conservati nel Museo di Storia Naturale e del Territorio dell’Università di Pisa e provenienti da Montecastelli (Pomarance, Pisa). Tali campioni, descritti seppur con qualche dubbio come pectolite da A. D’Achiardi (1872/73), sono risultati alle moderne indagini diffrattometriche xonotlite. Una ulteriore tipologia di giacitura è descritta da O’Brien & Rodgers (1973) i quali hanno osservato lenti di xonotlite al passaggio fra xenoliti gabbrici e corpi rodingitici, nelle rocce ultrabasiche di Wairee (Nuova Zelanda).
Per deidratazione, la xonotlite si trasforma topotatticamente in wollastonite (Dent & Taylor, 1956; Shaw et al., 2000). L’importanza tecnologica della xonotlite è legata alla sua presenza nei cementi di alta T ed alle sue applicazioni in numerosi ambiti (dall’isolamento termico all’immobilizzazione di scorie nucleari).
Come gli altri silicati idrati di calcio caratterizzati dalla presenza di catene tipo wollastonite, la xonotlite mostra pattern di diffrazione caratterizzati da riflessi netti ed intensi (per k = 2n) e riflessi deboli e diffusi (per k = 2n+1). La diffusione dei riflessi è lungo le direzioni di a* e c*; si tratta pertanto di un caso di disordine lungo due direzioni.
La struttura della xonotlite, il cui primo modello è stato proposto da Mamedov & Belov (1955), vede la presenza di strati di poliedri Ca e doppie catene silicatiche (fig. 4.8) . Gli strati di poliedri Ca sono formati da due tipi di poliedri: un poliedro a
72
coordinazione ottaedrica e due poliedri a coordinazione VII. Questi ultimi sono descrivibili come prismi trigonali monocappati. I poliedri a coordinazione VI si uniscono condividendo spigoli e formano una colonna A che corre lungo [010]; essa ha, su ambo i lati, due colonne B e B’ formate da poliedri a coordinazione VII, generando quindi una sequenza di colonne BAB’ che va a creare, per condivisione di spigoli, strati di poliedri Ca. Le relazioni geometriche fra lo strato di poliedri Ca e le doppie catene silicatiche tipo wollastonite sono alla base dei fenomeni di disordine e della comparsa di più politipi (Gard, 1966; Hejny & Armbruster, 2001). La prima risoluzione strutturale di uno dei politipi ordinati di xonotlite si deve a Kudoh & Takéuchi (1979).
I quattro politipi ordinati (oltre a due politipi disordinati) della xonotlite possono essere derivati sulla base della teoria OD o essere ricavati per via geometrica partendo da una ipotetica cella tipo protoxonotlite (Kudoh & Takéuchi, 1979), con a 8.516, b 7.363, c 7.012 Å, β 90.37°. Differenti impilamenti della cella tipo protoxonotlite lungo [100] e [001] danno origine ai vari politipi. Lungo [100] si può avere un continuo spostamento della cella di +b/4 o –b/4 o un’alternanza delle due traslazioni. Nella direzione [001] le celle tipo protoxonotlite possono essere giustapposte o traslate di b/2. La combinazione di queste differenti modalità di impilamento origina i politipi riportati in tabella 4.6 (Hejny & Armbruster, 2001).
Il simbolo M indica la simmetria monoclina della cella della protoxonotlite; le tre lettere, con gli eventuali valori numerici, indicano la periodicità nello spazio in accordo con la notazione modificata di Gard (Guinier et al., 1984).
Tab. 4.6 – Parametri di cella e simmetria dei politipi ordinati della xonotlite (da Hejny & Armbruster, 2001).
a (Å) b (Å) c (Å) α (°) β (°) γ (°) G.S. proxonotlite 8.516 7.363 7.012 90.00 90.37 90.00 M2a2bc 8.712 7.363 7.012 89.99 90.36 102.18 P1 M2a2b2c 8.712 7.363 14.023 89.99 90.36 102.18 A1 Ma2bc 17.032 7.363 7.012 90.00 90.36 90.00 P2/a Ma2b2c 17.032 7.363 14.023 90.00 90.36 90.00 A2/a
I gruppi OH sono legati ai vertici liberi dei poliedri CaO6. Nei politipi M2a2bc e Ma2bc, metà dei poliedri CaO6 presenta due gruppi OH (posti sui due lati dello strato di poliedri Ca) mentre l’altra metà è priva di gruppi OH; viceversa, nei politipi M2a2b2c e Ma2b2c ogni poliedro CaO6 è legato a un gruppo OH. Quest’ultimo tipo di distribuzione dei gruppi OH è più bilanciata e sembrerebbe pertanto rappresentare una configurazione favorevole; le evidenze sperimentali confermano parzialmente questo dato. Infatti il politipo M2a2b2c è stato descritto da Kudoh & Takéuchi (1979) nei campioni di Heguri (Giappone) e da Hejny & Armbruster (2001) nei cristalli della N’Chwaning
73
II mine (Repubblica Sudafricana); questo politipo è sempre presente anche nei campioni studiati da Bernstein et al. (2009) benché talvolta non rappresenti il politipo più abbondante. Il motivo per cui il politipo M2a2b2c sia generalmente più abbondante del politipo Ma2b2c suggerisce comunque che, oltre alla distribuzione dei gruppi OH, altri meccanismi favoriscano la cristallizzazione di un politipo piuttosto dell’altro.
Nel corso di questa tesi di dottorato la xonotlite è stata identificati in campioni provenienti dagli skarn a spurrite di Mihara (Giappone) e da tre località italiane: Groppo di Gorro (Berceto, Parma), Montecastelli (Pomarance, Pisa) e Cava Grolla (Spagnago, Vicenza). Il campione giapponese presentava soltanto sottili venette fibrose in cui la xonotlite, identificata mediante un diffrattogramma con camera Gandolfi, era intimamente associata a calcite. L’eseguità del materiale non ci ha consentito un più attento studio, studio che è invece stato rivolto ai campioni italiani. Oltre ad essere stati caratterizzati per via diffrattometrica e chimica, si è tentato di effettuare studi strutturali e, laddove fosse presente una sufficiente quantità di materiale, studi termo-gravimetrici. I risultati di tali studi sono riportati in Appendice B.






![Fig. 4.6 – Struttura della hillebrandite vista lungo [100].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7531242.107077/10.892.471.796.352.740/fig-struttura-della-hillebrandite-vista-lungo.webp)
![Fig. 4.7 – Struttura della jennite vista lungo [010].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7531242.107077/11.892.201.713.737.1092/fig-struttura-della-jennite-vista-lungo.webp)
![Fig. 4.8 – Struttura della xonotlite vista lungo [010].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7531242.107077/13.892.379.796.640.1089/fig-struttura-della-xonotlite-vista-lungo.webp)
