vomito, diarrea, alopecia e dermatiti. Tutte queste ragioni spiegano la necessità di derivati del 5-FU più efficaci e meno tossici [22].
La comparsa dell’Acyclovir, 9-[(2-idrossietossi)metil] guanina, che si è dimostrato un eccellente agente antivirale ha stimolato la sintesi di una grande varietà di nucleosidi aciclici ( o seco-nucleosidi) modificati sia sulla base che sulla porzione aciclica.
L’azione antivirale di queste sostanze o loro metaboliti deriva generalmente dall’inibizione della DNA polimerasi. E’ noto che il Cidofovir, il pronucleotide carboaciclico derivante dalla citosina, possiede potente attività in vitro ed in vivo contro un ampio spettro di herpes virus, incluso l’HCMV, ed è stato approvato per il trattamento delle retiniti da citomegalovirus umano (HCMV) in pazienti malati di AIDS.
I nucleosidi aciclici pirimidinici 5-(1-azidovinyl)-sostituiti hanno dimostrato potente attività contro il virus dell’epatite B (HBV) senza una significante tossicità. Alcuni autori hanno riportato di recente l’attività antivirale di
pirimidinici sono risultati molto interessanti per la loro attività antitumorale, visto che è stato dimostrato che vari derivati del 5-FU sono attivi contro alcune linee cellulari maligne per via dell’inibizione della timidilato sintetasi attraverso la formazione della 5-fluorodesossiuridinamonofosfato o attraverso l’incorporazione della 5-fluorouridinamonofosfato nell’RNA. In alcuni tumori maligni l’attività delle uridine fosfato è aumentata; per questo, gli aciclonucleosidi del 5-FU potrebbero mostrare maggiore attività antitumorale e minore tossicità del 5-FU stesso.
Sono stati infatti sintetizzati e saggiati per l’attività antitumorale alcuni derivati aciclonucleosidici del 5-FU di formula generale 42. questi aciclonucleosidi sono interessanti per due motivi:
a. la presenza del gruppo ossidrilico sulla catena laterale che può essere fosforilato e
b. costituiscono una nuova classe di agenti antitumorali, in quanto il 5-FU è legato ad una aldeide citostatica come l’acroleina o ad alcuni suoi omologhi e quindi due sostanze attive sono combinate in un unico composto.
Preparazione dei composti di tipo 42
La reazione del 2,4-bis-o-trimetilsilil-5-fluorouracile (44) generato in situ (da esametildisilozano, MDS, e clorotrimetilsilano, CTS) con i cicloacetali di tipo 1 in acetonitrile, ha fornito i desiderati composti (42) (Tabella 4, a-g) dopo l’aggiunta di 1,2,5 equivalenti di cloruro stannico (SnCl4) [22].
ATTIVITA’ BIOLOGICA
Attività antitumorale in vitro
L’attività antitumorale dei composti 42b-g è stata testata in vitro su cellule umane HEp (Tabella 4).
In generale, si può dire che:
a) il composto più attivo (IC50: 18 µM) è il 42c, la struttura più lipofila per la
porzione ciclopentossi, e
b) il composto meno attivo (IC50: 4300 µM) è il 42g, che non può portare
all’acroleina per divisione enzimatica del frammento animale, in condizioni non ossidative.
Tossicità: Prove in vivo. Metodo A
Il test della tossicità è stato condotto su topi della specie Mus Musculus, ratti albini svizzeri, sani, di 10-12 cm di lunghezza (coda esclusa), che raggiungono un peso di 30-35 g da adulti. Ad ogni ratto sono state somministrate dosi singole da 50, 100, 200, 500 e 1000 mg/kg di peso per rappresentare statisticamente lotti di animali. Topi con un peso medio di 26 g sono stati utilizzati per i dosaggi 25-200. Lotti di animali con peso medio di 27 g sono stati utilizzati per i dosaggi più alti da 500 e 1000 mg/kg. La crescita dei topi è stata seguita controllandone il peso. Non è stata valutata la tossicità né a breve termine né a lungo termine in quanto gli animali morirono al termine del loro ciclo biologico.
Metodo B
E’ stato condotto uno studio controllo con 5-FU commerciale, iniziando con un gruppo di 10 femmine di topo OF1, di circa 6 settimane. Dopo averle pesate ed aver calcolato il peso medio in grammi (24 g), fu stabilita la dose giornaliera che ciascuna doveva ricevere:
4,8 mg/topo equivalenti a 200 mg/kg, una concentrazione vicina alla DL50 del 5-FU (180 mg/Kg). Ciascun topo fu inoculato con 0,5 ml di 5-FU in dosi cumulative, i.e., dal giorno 0 al giorno 6. La stessa procedura fu eseguita sui composti 42b, 42c, 42d (miscela diastereomerica) e 42e (miscela
diastereomerica), tenendo in mente sia la percentuale di 5-FU contenuto nelle nostre molecole che la quantità di 5-FU presente nel modello di DL50 che fu stabilito corrispondere a 200mg/kg.
I topi trattati con 5-FU dimostrarono chiaramente una perdita di peso giornaliera in confronto con i controlli. Questa perdita di peso fu accompagnata da un cambiamento nell’aspetto degli animali, come ad esempio cambiamenti nel pelo e sua perdita ed irritazione della zona anale. Allo stesso modo, fu osservata una riduzione delle attività vitali negli animali trattati. Al settimo giorno e dopo l’ultima iniezione di 5-FU, otto dei dieci topi trattati morirono e i due restanti morirono il giorno dopo. Come nei controlli, il cambiamento di peso non seguì un tendenza ben definita, non c’era variazione nell’aspetto dei topi ne furono osservati segni di una apparente tossicità.
In conclusione, la morte non giunse per via dei trattamenti. Gli animali morirono quando giunse a termine il loro ciclo vitale.
MODIFICAZIONI CHIMICHE NELLA PORZIONE ACICLICA DELLE 3-(2-IDROSSIETOSSI)-1-ALCOSSI-PROPILNUOCLEOBASI
Le modificazioni chimiche includono:
- il blocco irreversibile della funzione ossidrilica come nei composti (46) e (47),
- la sostituzione dell’ossidrile con un atomo di cloro (48), - la ossidazione a gruppo metossicarbonilico (49),
- l’aumento di lipofilicità come nei composti (50-53), nei quali la catena aciclica è legata a diverse nucleosidi,
- l’aumento di idrofilicità (54) e l’inclusione della cloridrica sui due atomi di carbonio della catena aciclica (55).
In questo studio è stato incluso anche il composto (56) che contiene, come sostituenti, il 5-fluorouracile.
I composti (46-56) possono essere visti come profarmaci delle nucleobasi, contenenti una porzione di acroleina.
Dopo idrolisi chimica o enzimatica, possono essere generate dallo stesso farmaco due sostanze biologicamente attive, come conseguenza dell’attivazione del farmaco.
- L’acroleina stessa inibisce la crescita delle cellule ovariche nel criceto (IC50: 50 µM). Tutti questi composti (46-56) sono stati inoltre
saggiati per l’attività citotossica nei confronti del carcinoma del colon HT-2P ed il composto più attivo è stato inserito per studiare le modificazioni nella proliferazione e nel grado di differenziazione nelle linee cellulari RD di rabdomiosarcoma umano.
- Tabella 5
-
a
Ad-: adenina
Attività biologica
Evidenze considerevoli indicano che il fenotipo maligno nei tumori non è uno stato irreversibile, ma rappresenta una malattia dovuta ad alterazioni nella maturazione. Recentemente è stato sviluppato il concetto di terapia
di differenziazione che si basa sulla conversione di cellule maligne ad un fenotipo più benigno attraverso differenziazione indotta da sostanze chimiche. La ricerca ha dimostrato che le cellule del rabdomiosarcoma possono essere indotte ad uno stadio differenziato privo di potenziale proliferativo da farmaci antineoplastici come ad esempio la citarabina e l’antibiotico actinomicina D. I rabdomiosarcomi sono la forma più comune di tumori maligni del tessuto molle nei bambini e costituiscono il 5% di tutti i tumori maligni pediatrici.
citarabina actinomicina D
Il grado di differenziazione cellulare nel rabdomiosarcoma è stato determinato con il marker classico desmina; più di recente sono state valutate l’espressione dell’alfa-actinina e della tropomiosina per determinare il grado di organizzazione citoscheletale.
N N NH2 O O HOH2C HO HO
Citotossicità in vitro verso l’HT-29
L’attività citotossica dei composti (46-50) e (52-56) fu valutata con il saggio del 3-(4,5-dimetiltiazol-2-yl)-2,5-difeniltetrazolio bromuro verso la linea cellulare HT-29 di tumore umano del colon. L’unico composto attivo risultato essere il composto (55) (IC50: 70 µM), che è 8 volte meno attivo
del 5-FU (9 µM), il resto della serie invece mostrò valori di IC50 superiori a
100 µM ( attività insufficiente a queste concentrazioni per determinare la IC50).
Differenziazione ed inibizione della crescita in vitro nella linea cellulare RD
La linea cellulare del rabdomiosarcoma (RD) fu ottenuta dalla “American Type Culture Collection”.
Quattro beute (75 cm2) di culture replicate con ciascuna una quantità di cellule di 2x106 furono esposte ad una soluzione 90 µM del composto (55) insieme al controllo. Le cellule furono indotte con il composto (55) fino al sesto giorno, quando furono raccolte le cellule corrispondenti al trattamento di ciascun giorno [22].
Analisi di citofluorimetria (CFM) hanno fornito dati quantitativi sui cambiamenti nell’espressione di desmina, α-actina e tropomiosina in linee cellulari RD in linee cellulari RD parenterali (controllo), dopo 6 settimane di
trattamento con 5-FU (90 µM) e dopo 6 giorni di trattamento con il composto (55) (90 µM).
Il 5-FU porta a chiari segni di differenziazione nelle linee cellulari RD. Questa azione è stata dimostrata anche nella linea cellulare HT-29 di carcinoma del colon. Tuttavia, il 5-FU ha mostrato un’elevata tossicità cellulare ed è stato usato per brevi trattamenti, dopo i quali le cellule era indotte a differenziarsi utilizzando classici agenti induttori.
Una tale risposta differenziativa potrebbe implicare un’importante morbosità sui tessuti sani. Quindi, l’uso del composto (55) presenta vantaggi sul 5-FU quali:
a) minore tossicità
b) incremento significativo nei marker di differenziazione .
La relazione inversa osservata tra la proliferazione e la differenziazione in cellule tumorali e normali suggerisce un approccio alternativo nella terapia contro il cancro che non coinvolge l’uccisione di cellule, ma al contrario induce le cellule maligne e differenziarsi in forme benigne prive di potenziale proliferativo.
(b/-)-1-[2-(2-IDROSSIMETILFENOSSI)-1-METOSSIETIL]-5-FLUOROURACILI 40B-G: ATTIVITA’ ANTIPROLIFERATIVA,
DISREGOLAZIONE DEL CICLO CELLULARE ED INDUZIONE APOPTOTICA CONTRO LE CELLULE DEL CANCRO AL SENO
tra donne di età compresa tra 35 e 54 anni e la seconda causa di morte da cancro in donne di età tra 55 e 74 anni.
La natura disseminata del tumore al seno e lo sviluppo di tumori incrociati resistenti sono la principale causa del fallimento delle terapie attuali. Da quando un tumore viene individuato, esiste un’elevata probabilità che siano presenti lesioni sospette di metastasi, e molte di queste conterranno già una resistente subpopolazione di cellule.
Non sorprendentemente, c’è un sostanziale interesse nell’identificazione di nuovi agenti antitumorali per il trattamento del cancro del seno. L’apoptosi o morte cellulare programmata è un meccanismo innato attraverso il quale cellule non necessarie, difettose o danneggiate vengono eliminate rapidamente e selettivamente dal nostro corpo.
Avviene durante rimodellamenti tissutali, sviluppo embrionale, e modellamenti della regolazione immunitaria. L’apoptosi è il meccanismo principale utilizzato dal sistema immunitario e dai farmaci chemioterapici per eliminare cellule tumorali.
Le cellule tumorali resistenti evadono l’azione degli agenti antitumorali aumentando la propria soglia apoptotica. Questo ha spronato lo sviluppo di nuovi composti chimici capaci di indurre l’apoptosi in cellule tumorali chemio/immuno-resistenti.
Nonostante che il cancro al seno sia spesso curato con agenti citotossici convenzionali è stato dimostrato che è difficile indurre apoptosi in cellule di questo tipo di cancro utilizzando questi farmaci.
L’identificazione di terapie particolarmente efficaci nell’indurre apoptosi, può portare a grandi successi clinici. In particolare sono stati sintetizzati analoghi ciclici del 5-FU O,N acetalici di formula generale (57), per valutare la loro attività biologica contro il cancro al seno [22].
Chimica
a) preparazione degli intermedi aciclici O,O-acetali (58 a-f), e
b) reazione di questi composti aciclici con la base pirimidinica per dare le strutture desiderate.
I 5-cloro-2-idrossibenzil alcoli sono stati ottenuti per riduzione della corrispondente aldeide salicilata [24].
Gli idrossiacetali (58 d-f) sono stati preparati in modo simile alla preparazione dei composti (58 a-c) [25] per alchilazione degli alcoli salicilici (schema*) con bromoacetaldeide dimetil acetale, usando come base sodio idruro in dimetilformammide anidra. La sostituzione del gruppo metossilico con il FU negli O,O-acetali (58) a-f è stata ottenuta per reazione con 5-FU in presenza di HMDS (esametildisilazono) e triclorosilano (TCS) in catalisi acida di cloruro tannico.
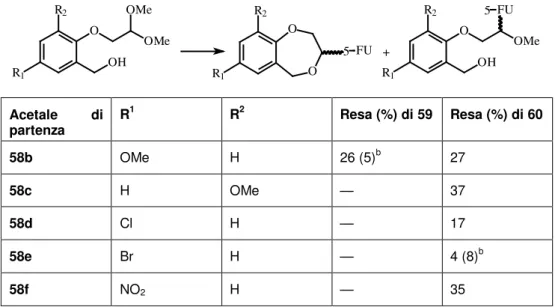
La reazione di condensazione tra gli O,O-acetali acilici (58) ed il 5-FU da luogo ai composti ciclici (59) e/o agli O,N-acetali aciclici (60), rispettivamente attraverso un processo la cui regioselettività dipende dalla presenza e dalla natura dei sostituenti R1 ed R2 dell’anello benzenico
(Tabella 6)[22].
In generale, nella maggior parte dei casi l’attacco della porzione del 5-FU avviene sull’N-1 e, raramente, sull N-3 dell’anello uracilico.
Questo è stato confermato dai dati di 1H NMR [22].
In relazione con la reazione di condensazione tra gli O,O-acetali aciclici (58) ed il 5-FU, è stato possibile affermare quanto segue:
1. la natura e la posizione dei sostituenti R1 ed R2 ha mostrato una
chiara influenza sia sulle rese che sulla regioselettività del processo. Infatti, la sostituzione in posizione 5 di (58) incide nel modo seguente (è stato considerato solo R2=H):
1.1 i sostituenti elettron-attrattori (Cl, Br e NO2) inducono
preferenzialmente la formazione degli O,N-acetali aciclici (60) 1.2 il gruppo OMe, l’unico sostituente elettron-donatore utilizzato,
arresta la regioselettività della reazione formando sia gli O,N-acetali ciclici (59) che gli aciclici (60) in rapporto 1/1.
1.3 In alcuni casi, la presenza dei sostituenti nell’uno o nell’altro tipo di O,O-acetali ha permesso di isolare I derivati nei quali la porzione del 5-FU è legata attraverso l’N-3 della base pirimidinica.
2. Infine, la sostituzione sul C-3 di (58) con il solo gruppo studiato (OMe) chiaramente ha favorito la formazione dell’O,N-acetale aciclico.
Il nitrogruppo di (60) è stato ridotto con SnCl2 diidrato in etanolo a riflusso
per dare (60g) (Figura 6)
O OH OMe FU R2 R1 5 57 O OH OMe R2 R1 OMe OH OH R2 R1 a R1= R2 = H b R1= OMe; R2 = H c R1= H; R2 = OMe d R1= Cl; R2 = H e R1= Br; R2 = H f R1= NO2; R2 = H 58
Tabella 6. Formazione di O,N-acetali ciclici e aciclici a partire dall’O,O-acetale
aciclico 58, con tempo di reazione 24 ha. [22]
O OH OMe R2 R1 OMe O OH OMe FU R2 R1 5 + R2 R1 O O 5 FU Acetale di partenza R1 R2 Resa (%) di 59 Resa (%) di 60 58b OMe H 26 (5)b 27 58c H OMe — 37 58d Cl H — 17 58e Br H — 4 (8)b 58f NO2 H — 35 a 5-FU,HMDS, TCS, SnCl4/CH2Cl2, MeCN.
bTutte le rese si riferiscono ai composti nei quali la porzione del 5-FU è legata attraverso l’N1 ad eccezione di 60e e 59b nei quali il legame è attraverso l’N3 e le relative rese sono riportate fra parentesi.
Figura 6. Reagenti: (a) SnCl2 ·2H2O, EtOH.
Lo scopo dello studio era identificare composti capaci di indurre la differenziazione cellulare in cellule di cancro del seno umano MCF-7. Cellule di cancro del seno differenziate mostrano proprietà che sono associate alla lattazione compresa la formazione di depositi di grasso a livello del citoplasma.
a O OH OMe FU R2 R1 5 R1= NO2 60f R1= NH2 60g
La percentuale di cellule positive ai lipidi è stata determinata con il marcante Nile Red in un citometro a flusso. Il Nile Red è una specifica sonda fluorescente usata per quantificare il contenuto di lipidi intracellulari, con citometri a flusso, nelle cellule mammarie.
Tutti i composti studiati [22] (escluso il 61a) hanno causato un incremento dei contenuti lipidici rispetto al controllo dopo 3 giorni di trattamento. Il composto (Z)-62 ha incrementato la percentuale di lipidi per valori di più di sei volte maggiori rispetto a quelli delle cellule non trattate. Inoltre, (Z)-62 ha incrementato la percentuale di cellule nella fase S in maniera significativa, che potrebbe contraddire l’alto livello di differenziazione.
OH Y X OMe 5 FU (Z)-62 X=Y: CH=CH
Sebbene l’arresto in G1 era stato al centro dell’attenzione nella differenziazione, alcune ricerche sono incentrate sul coinvolgimento dell’arresto delle fasi G2/M ed S in questi fenomeni. Studi più recenti hanno dimostrato che nucleotidi e nucleosidi purinici causano arresto della fase S e differenziazione in cellule umane di leucemie mielogene croniche. La duplicazione del genoma cellulare durante la fase S del ciclo cellulare è critica perché durante questo processo le cellule sono molto suscettibili all’induzione della differenziazione.
L’introduzione degli atomi di cloro e bromo causa un incremento dei livelli di differenziazione da 2 a 4 volte rispetto al controllo. L’arresto delle cellule nella fase G0/G1 da parte di questi composti mostra il passaggio ad uno stato quiescente non proliferativo che implica l’ingresso nel processo di differenziazione. Altri hanno mostrato recentemente che nuovi composti chinolonici causano l’accumulo di goccioline di grasso come un marker
fenotipico di differenziazione, perdita dell’espressione dell’antigene K167, un marker del ciclo cellulare indicativo dell’entrata nella fase G0, e riducono i livelli proteici della fase G1.
Questi dati dimostrano che il meccanismo attraverso il quale questi composti esercitano il loro effetto antiproliferativo è l’induzione di differenziazione della linea cellulare MCF-7 del cancro al seno umano, invece che l’induzione di apoptosi o i loro effetti citotossici. I composti in esame hanno portato le cellule tumorali a rientrare nei normali canali di sviluppo ed hanno costituito una nuova categoria di agenti sperimentali contro il tumore al seno.
Regolatori del proteasoma: attivatori ed inibitori
Esistono due grossi sistemi di proteolisi cellulare: lisosomi e proteasomi[26].
La degradazione delle proteine è finemente regolata e si svolge attraverso due vie principali.
La prima è rappresentata dai lisosomi, orfanelli vescicolari contenenti idrolisi acide con diversa specificità, deputati allo smaltimento di proteine endogene costitutive o esogene internalizzate per endocitosi e pinocitosi[27]. L’altra via proteolitica coinvolge l’ubiquitina, un polipeptide di 76 amminoacidi, che funge da marcante per la degradazione, ed un complesso multienzimatico, il proteasoma.
Il sistema ubiquitina-proteasoma non richiede compartimentazione ed il complesso catalitico agisce ubiquitariamente (Figura 7) (nucleo, citoplasma e in associazione al reticolo endoplasmatico), a pH neutro, con meccanismi talvolta ATP dipendenti che coinvolgono numerose molecole adiuvanti e che sono state accorpate nel cosiddetto “Signalosoma” [28]. Mentre i lisosomi sono specializzati nella rottura delle proteine introdotte nelle cellule tramite endocitosi, i protesomi costituiscono il meccanismo