13
2. SISTEMI ALLIL OSSIRANICI E ALLIL AZIRIDINICI
DERIVATI DA IMMINOGLICALI
2.1 Stato dell’arte.
Nel laboratorio in cui ho svolto la presente tesi sperimentale era stato trovato come le reazioni degli allil epossidi diastereoisomeri 2.1α e 2.1β derivati, rispettivamente dal D-allale e dal D-galattale con O-nucleofili, quali gli alcooli e monosaccaridi parzialmente protetti, conducessero ai corrispondenti O-glicosidi e disaccaridi attraverso un processo completamente 1,4-regio- e stereoselettivo con formazione di soli α-O-glicosidi da 2.1α e soli β-O-glicosidi da 2.1β, in un processo di glicosilazione stereospecifico, non catalizzato e direttamente substrato-dipendente (Schema 2.1).5,6
O BnO ROH (3 equivalenti) benzene O O H O R O BnO O BnO ROH (3 equivalenti) benzene O O H O R O BnO 2.1αααα 2.1ββββ α α α α-O-glicosidi β β β β-O-glicosidi O O OBn OBn HO HO OR OR
Schema 2.1. Meccanismo del processo stereospecifico di glicosilazione degli alcooli mediante gli epossidi 2.1α e 2.1β.
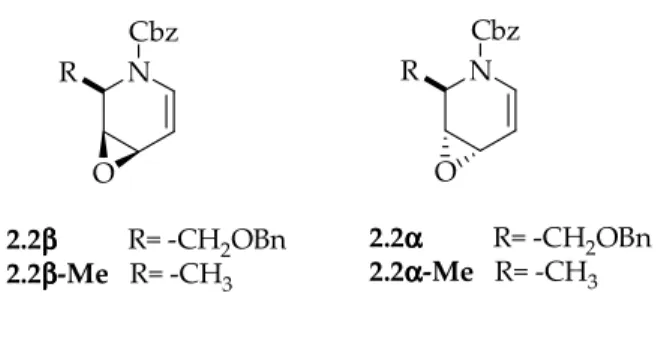
Sulla base di questi precedenti risultati e a seguito dell’interesse sviluppatosi nel gruppo di ricerca verso la sintesi di azazuccheri2 facendo uso di imminoglicali, era stato inizialmente deciso di verificare se gli allil epossidi diastereoisomeri N-Cbz sostituiti 2.2α e 2.2β, gli azaanaloghi degli epossidi 2.1α e 2.1β, derivati rispettivamente dal D,L-imminoallale e D,L
-14
imminogalattale, , si sarebbero comportati allo stesso modo nelle loro reazioni con gli alcooli.7 N BnO O OBn N BnO BnO N OBn O N O OBn O BnO O 2.2ααα'α 2.2αααα 2.2ββββ 2.2βββ'β OBn O O O
Schema 2.2. Aspetto conformazionale degli epossidi 2.2α e 2.2β.
Calcoli teorici preliminarmente ed opportunamente condotti avevano indicato come gli epossidi 2.2α e 2.2β esistessero in soluzione esclusivamente nei corrispondenti conformeri 2.2α’ e 2.2β’ con il gruppo CH2OBn in posizione assiale, come mostrato nello Schema 2.2. Questo risultato indicava come questi epossidi, per quanto apparentemente riconducibili a sistemi mobili, in realtà si comportassero da sistemi praticamente rigidi.7
L’interesse verso questo tipo di substrati azotati derivava anche dalla osservazione che niente era presente in letteratura, non solo per quanto riguarda gli epossidi 2.2α e 2.2β, ma anche relativamente alla glicosilazione di alcooli da parte di imminoglicali e/o loro derivati. A questo riguardo era invece noto come imminoglicali opportunamente sostituiti fossero in grado di reagire con C-nucleofili in presenza di acidi di Lewis in condizioni di reazione tipo-Ferrier. In queste condizioni, ad esempio il tri-O-acetil-imminoglucale 2.4 conduceva al corrispondente β-anomero 2.5 con stereoselettività da buona a elevata (24-72% de). Il risultato ottenuto era stato attribuito ad un preferenziale attacco pseudoassiale del nucleofilo sul preferenziale conformero 2.5β dello ione N-acilimminio intermedio 2.5 con la catena laterale sostituente - CH2OBn in posizione assiale (Schema 2.3).8
Nu BF3. Et2O attacco pseudoassiale N AcO OR O Nu N AcO OR O N RO O OAc OAc N O OAc RO OAc Nu OAc AcO AcO 2.4 2.5αααα 2.5ββββ 2.6
Schema 2.3. Attacco preferenziale pseudoassiale del nucleofilo sulla conformazione 2.5β dello ione N-acilimminio intermedio.
15
Sulla base di questi risultati ricavati dalle recente letteratura, appariva così interessante andare a verificare se i nuovi allil epossidi 2.2α e 2.2β derivati da imminoglicali fossero capaci non solo di glicosilare gli alcooli, ma anche se la selettività faciale associata al processo di glicosilazione fosse indipendente o meno dalla natura, pseudoassiale o pseudoequatoriale, dell’attacco nucleofilo.
Il diolo trans 2.13 necessario per la sintesi dell’epossido 2.2β era stato preparato in modo completamente stereoselettivo attraverso una applicazione racema della procedura di sintesi enantioselettiva di Comins (Schema 2.4).9
CbzCl THF -25°C N Cbz OMe Cl BnOCH2Cu(CN)Li2 2-(Th) -78 °C N Cbz OMe H2C2O4 sat.aq. 65% N Cbz BnO O N OMe N Cbz O N Cbz BnO O N Cbz BnO OH Pb(OAc)4 toluene riflusso HCl/EtOH 50 °C 94% MeNBH(OAc)3 acetone/AcOH 72% BnO 2.7 2.8 2.9 2.10 2.11 2.12 2.13 BnO HO HO AcO
Schema 2. 4. Sintesi del diolo trans 2.13.
Il protocollo sintetico si basa sulla addizione regioselettiva (-78ºC) del cuprato 2.8 al carbonio C(2) del cloruro di piridinio 2.7, generato in situ per reazione della 4-metossi piridina con benzil cloroformiato (CbzCl).
Il cuprato 2.8 (non commerciale) è ottenibile solo in situ e subito fatto reagire con il cloruro di piridinio 2.7 e la sua preparazione è alquanto laboriosa. Per la sua sintesi è necessario, infatti, disporre inizialmente del (benzilossimetil)-tributil stannano 2.16, preparato per aggiunta a -78°C del benzilclorometil etere commerciale ad una soluzione di Bu3SnLi ottenuta per reazione del Bu3SnH con LDA a 0°C (Schema 2.5) . Lo stannano 2.16 grezzo viene poi purificato mediante cromatografia flash o distillazione a pressione ridotta. Successivamente, lo stannano 2.16 viene trattato a –78 °C con BuLi: lo scambio Sn/Li è
16
rapido e porta alla formazione del corrispondente reagente α-alcossiorganolitio 2.17 che, per aggiunta di litio 2-tienil(ciano)cuprato viene trasformato nel cuprato 2.8 [(PhCH2OCH2)[(2-Th)Cu(CN)]Li2], desiderato (Schema 2.5).
Schema 2.5. Sintesi del cianocuprato misto di ordine superiore 2.8.
La reazione del diolo 2.13 con TBS-Cl (1.1 equiv) sfortunatamene non era risultata regioselettiva e conduceva ad una miscela 40:23:37 del corrispondente 3-OTBS (2.19), 4-OTBS (2.20), e 3,4-di-4-OTBS (2.21) derivati, che erano stati separati mediante flash cromatografia. Il successivo trattamento di 2.19 con MsCl conduceva al mesilato 2.22 poi deprotetto con TBAF per dare il trans idrossi mesilato 2.23. La ciclizzazione base-catalizzata (t-BuOK/MeCN) di 2.23 conduceva al desiderato epossido 2.2β (Schema 2.6).7
Ph O Sn(Bu)3 Bu3SnH LDA THF, 0°C Bu3SnLi PhCH2OCH2Cl -78°C t.a. 2.16 2.14 2.15 n-BuLi THF, -78°C Ph O Li 2.17 Ph O Cu(CN)Li2 S S Cu(CN)Li 2.8 - 78°C 2.18
17 N Cbz HO OH BnO TBDMS-Cl DMF imidazole TsCl 36% Py N Cbz R1O OR2 BnO t-BuOK MeCN N BnO Cbz O N BnO R2O OR1 MsCl/Py 82% N Cbz Cbz OTBS MsO BnO 2.13 2.19. R1= TBS; R2= H (40%) 2.20. R1= H; R2= TBS (23%) 2.21.R1= R2= TBS (37%) 2.22 TBAF THF 75% N OH MsO BnO Cbz t-BuOK MeCN 2.2ββββ 2.23 2.24. R1= Ts; R2= H 2.24-al. R1= H; R2= Ts 2.24- Ac. R1= Ts; R2= Ac
Schema 2.6. Sintesi dell’epossido 2.2β a partire dal diolo trans 2.13.
Alternativamente, il trattamento del diolo trans 2.13 con TsCl (1.5 equiv) conduceva in modo completamente regioselettivo al trans idrossitosilato 2.24, quale unico prodotto di reazione, ma la reazione era caratterizzata da una resa poco soddisfacente (36%), ragionevolmente a causa della concomitante formazione del molto più reattivo regioisomero allil tosilato 2.24-al, che si decompone rapidamente nell’ambiente di reazione, determinando la perdita di buona parte del diolo 2.13 di partenza.
Per la sintesi dell’epossido 2.2α, il mesilato 2.25 ottenuto per mesilazione (MsCl/Py) dell’α-idrossichetone 2.12, era stato isomerizzato parzialmente con AcONa a riflusso in MeCN per dare una soddisfacente miscela 6:4 del mesilato di partenza 2.25 e dell’epimero 2.26. Dopo separazione mediante flash cromatografia, mentre il prodotto di partenza veniva riciclato, il mesilato 2.26 veniva ridotto in modo diastereoselettivo mediante NaBH4 in presenza di CeCl3 per dare il trans idrossimesilato 2.27 in modo completamente stereoselettivo. La ciclizzazione di 2.27 in ambiente alcalino (t-BuOK/MeCN) conduceva all’epossido desiderato 2.2α (Schema 2.7).
18 N Cbz O BnO N Cbz O BnO N Cbz OH BnO N Cbz BnO AcONa MeCN riflusso 34% NaBH4 CeCl3 -40°C 88% t-BuOK MeCN RO MsO MsO 2.26 2.27 2.2αααα 2.12. R=H 2.25. R=Ms O MsCl Py
Schema 2.7. Sintesi dell’epossido 2.2α .
In un esame preliminare del comportamento regio- e stereoselettivo di questi nuovi allil epossidi 2.2α e 2.2β, derivati da imminoglicali con O-nucleofili, era stato presa in considerazione la reazione con il semplice MeOH facendo uso delle condizioni di reazione previste dal protocollo A e dal protocollo B.
In condizioni di protocollo A (alcool come solvente) il trattamento di una soluzione metanolica del trans idrossimesilato 2.27 con MeONa (1 equiv) conduceva, attraverso la formazione intermedia del corrispondente epossido 2.2α, ad una miscela 75:25 degli anomerici metil α-O-glicoside 2.28α e β-O-glicoside 2.28β (Schema 2.8).
N Cbz N Cbz N Cbz N Cbz N Cbz N Cbz MeONa (1 equiv) MeOH MeONa (1 equiv) MeOH 2.28αααα (75%) 2.28βββ (25%)β 2.29αααα(15%) 2.29ββββ (85%) HO OH MsO HO HO OMe OMe OMe OH TsO 2.27 2.2αααα 2.24 N Cbz HO OMe BnO BnO O N Cbz 2.2ββββ O BnO BnO BnO BnO BnO BnO
Schema 2.8. Reazione degli allil epossidi 2.2α e 2.2β con MeOH in condizioni di protocollo A.
Nelle medesime condizioni l’epossido 2.2β (dall’ idrossitosilato trans 2.24) conduceva ad una miscela 85:15 dei corrispondenti metil β-O-glicoside 2.29β e α-O-glicoside 2.29α. Le coppie dei glicosidi 2.28α-2.28β e 2.29α-2.29β erano state separate e indipendentemente caratterizzate dal punto di vista strutturale e stereochimico.
19
Il risultato indicava come in entrambi i casi la reazione di glicosilazione fosse completamente regioselettiva con esclusivo attacco del nucleofilo alcool (MeOH) sul carbonio C(1) del sistema allilico. Le reazioni non erano però completamente stereoselettive in quanto veniva ottenuta in entrambi i sistemi una miscela dei corrispondenti metil glicosidi α e β diastereoisomeri. Tuttavia, e questo risultava particolarmente importante, in entrambi i casi il glicoside presente in maggior quantità risultava avere la stessa configurazione dell’epossido di partenza: l’α-O-glicoside 2.28α dall’epossido 2.2α ed il β-O-glicoside 2.29β dall’epossido 2.2β (Schema 2.8).
Quando le stesse reazioni venivano poi ripetute facendo uso del protocollo B (3 equiv di MeOH, in MeCN come solvente), le reazioni di metanolisi degli epossidi 2.2α e 2.2β, diventavano completamente regio- e stereoselettive con esclusiva formazione del metil α-O-glicoside 2.28α da 2.2α e del metil β-O-α-O-glicoside 2.29β dall’epossido 2.2β.
Successivamente, altri O-glicosil accettori diversi dal MeOH quali l’EtOH, i-PrOH e t-BuOH sono stati esaminati nella loro reazione con gli epossidi 2.2α e 2.2β (Schema 2.9).
N Cbz N Cbz N Cbz N Cbz N Cbz N Cbz t-BuOK (1 equiv) ROH t-BuOK (1 equiv) ROH OH MsO HO OR OH TsO HO OR 2.27 2.24 2.2αααα 2.2ββββ 2.30. R=Et 2.31. R=i-Pr 2.32. R=t-Bu 2.33. R=Et 2.34. R=i-Pr O O BnO BnO BnO BnO BnO BnO
Schema 2.9. Reazione degli allil epossidi 2.2α e 2.2β con O-glicosil accettori diversi dal MeOH in condizioni di protocollo A.
Con la sola esclusione del t-BuOH nella sua reazione con l’epossido 2.2β che conduceva ad una miscela complessa di reazione, la reazione degli epossidi 2.2α e 2.2β erano risultate completamente regio- e stereoselettive già in condizioni di reazione di protocollo A, con esclusiva formazione del corrispondente alchil O-glicoside (2.30, 2.31 e
20
2.32 da 2.2α e 2.33, 2.34 da 2.2β) avente la stessa configurazione dell’epossido di partenza (Schema 2.9). Un simile comportamento in condizioni di protocollo A non era mai stato osservato con gli allil epossidi 2.1α e 2.1β, derivati dai glicali.
La completa 1,4-regio- e stereoselettività osservata nelle reazioni di 2.2α e 2.2β con gli alcooli è stata razionalizzata attraverso l’intervento di un legame a idrogeno tra l’ossigeno ossiranico e l’O-nucleofilo. In questo modo il nucleofilo (alcool) viene ad essere trasportato sulla faccia α nel caso dell’epossido 2.2α, che reagisce attraverso l’unico conformero possibile 2.2α’, come mostrato in 2.35a, o sulla faccia β nel caso dell’epossido 2.2β, che reagisce attraverso l’unico conformero possibile 2.2β’, come mostrato in 2.36a. In questo modo il nucleofilo viene a trovarsi appropriatamente disposto per un suo attacco entropicamente favorito sul corrispondene carbonio C(1) dalla stessa parte verso cui si dirige l’anello ossiranico: un attacco α diretto in 2.35a (via a, attacco pseudoequatoriale) ed un attacco β-diretto in 2.36a (via b, attacco pseudoassiale), (Schema 2.10).
N BnO Cbz N OBn O BnO O H O R O R H N O OBn O BnO O OR H R H N BnO Cbz a b HO OR HO OR 2.35a 2.36a a b alchil α-O-glicale alchil β-O-glicale
Schema 2.10. Conformazioni reattive degli allil epossidi 2.2α e 2.2β nelle reazioni di glicosilazione di O-nucleofili.
Questo indicava chiaramente come con gli epossidi 2.2α e 2.2β la selettività della O-glicosilazione degli alcooli non dipenda dalla natura pseudoassiale o pseudoequatoriale dell’attacco nucleofilo sul C(1) del corrispondente immino glicale, come poteva essere atteso sulla base dei precedenti risultati riportati in letteratura su questi sistemi con i C-nucleofili (selettività prodotto-dipendente, Schema 2.3). Al contrario, con gli epossidi 2.2α e 2.2β la selettività risultava dipendere solo dalla configurazione dell’epossido di partenza e dalla corrispondente direzione della coordinazione ossigeno ossiranico-nucleofilo, in una sorta di selettività substrato-dipendente, precedentemente mai osservata su sistemi analoghi.
A questo riguardo, un particolare significato assumeva il risultato della reazione di glicosilazione dell’epossido 2.2α. Infatti, una selettività prodotto-dipendente avrebbe portato al corrispondente β anomero invece dell’osservato α anomero. Al contrario nell’ epossido
21
2.2β sia la selettività prodotto-dipendente, che quella substrato-dipendente, conduceva al medesimo composto, il corrispondente β anomero.
Nel contesto di questa razionalizzazione, solo nel caso del più nucleofilo MeOH e comunque solo in presenza di un eccesso di questo (protocollo A), poteva verificarsi che anche un attacco da parte di molecole di nucleofilo non coordinate diventasse competitivo e conduca a quantità, anche se minoritarie, di metil O-glicosidi aventi configurazione opposta rispetto all’epossido di partenza (Schema 2.10). Nel caso di tutti gli altri alcooli, probabilmente la loro ridotta nucleofilicità, associata alla evidente più marcata tendenza degli epossidi 2.2α e 2.2β alla coordinazione, ma evidentemente è sufficiente a determinare una completa stereoselettività direttamente substrato dipendente, anche in presenza di un forte eccesso di nucleofilo (protocollo A).7
2.2 Sintesi dei 6-desossi epossidi 2.2α-Me e 2.2β-Me
I risultati descritti nel paragrafo precedente, avevano indicato come gli epossidi 2.2α e 2.2β fornissero, in reazioni di addizione nucleofila con alcooli, esclusivamente i corrispondenti O-glicosidi (β O-glicosidi dall’epossido 2.2β ed α-O-glicosidi dall’epossido 2.2α) secondo un nuovo processo di glicosilazione direttamente substrato-dipendente (Schema 2.9). Nella razionalizzazione proposta per giustificare la completa 1,4 regio- e stereoselettività non era stato attribuito alcun ruolo alla O-funzionalità, -OBn, presente sul C(6) della catena laterale (Schema 2.10). Tuttavia, nell’epossido 2.2β la O-funzionalità sul C(6) avrebbe potuto prendere parte all’ancoraggio del nucleofilo sulla faccia β del sistema allil-ossiranico mediante un processo di chelazione come rappresentato ipoteticamente, nella struttura chelata bidentata 2.37 (Fig. 2.1) che avrebbe potuto giocare un ruolo decisivo nel determinare la β-stereoselettività osservata.
OBn O N Cbz O H R 2.37
22
Nell’epossido 2.2α, per semplici ragioni strutturali, ovviamente un processo analogo di chelazione non può realizzarsi, ma il gruppo –OBn in C(6) avrebbe potuto, comunque, influenzare il processo di addizione nucleofila mediante il proprio effetto induttivo elettronattrattore (-I).
Era quindi risultato interessante introdurre una modifica strutturale negli allil epossidi 2.2α e 2.2β, relativamente alla catena sul C(5), andando a sostituire il gruppo –CH2OBn con un semplice gruppo metilico –CH3, che non presenta più alcuna O-funzionalità. Questa modifica strutturale si concretizzava nella sintesi dei nuovi 6-desossiepossidi 2.2α-Me e 2.2β-Me (Fig.2.2). In questo modo, se nei nuovi epossidi metil sostituiti 2.2α-2.2β-Me e 2.2β-2.2β-Me, il processo di O-glicosilazione fosse risultato ancora completamente regio- e stereoselettivo e direttamente substrato-dipendente, avremmo potuto escludere l’intervento della catena laterale –CH2OBn in C(5) nel dirigere la regio- e stereoselettività del processo di addizione nucleofila. N Cbz R O N Cbz R O 2.2ββββ R= -CH2OBn 2.2ββββ-Me R= -CH3 2.2αααα R= -CH2OBn 2.2αααα-Me R= -CH3
Figura 2.2. Allil epossidi di tipo imminoglicale 2.2α, 2.2β e corrispondenti 6-desossi derivati 2.2α-Me, 2.2β-Me.
Qualora potesse essere esclusa l’influenza di tale sostituente, gli allil epossidi 2.2β-Me e 2.2α-Me, decisamente più semplici da sintetizzare rispetto agli analoghi –CH2OBn sostituiti, potevano essere presi a modello ed utilizzati per studiare il comportamento di questi sistemi ossiranici derivati da imminoglicali anche con altri nucleofili (C-, N- ed S-nucleofili) e per la sintesi di nuovi sistemi eterociclici quali le aziridine, con la garanzia che tutto quanto avessimo ottenuto, osservato e ricavato, in termini di reattività, regio- e stereoselettività, con i sistemi 5-metilderivati, avrebbe potuto essere ragionevolmente esteso ai corrispondenti sistemi –CH2OBn sostituiti in C(5). In aggiunta a queste considerazioni l’interesse verso i sistemi 2.2β-Me e 2.2α-Me era motivato anche dall’osservazione che i sistemi 6-desossi con
23
struttura riconducibile agli imminoglicali, sono presenti in molti composti biologicamente attivi.
Come nel caso degli epossidi 2.2α e 2.2β, gli epossidi 2.2α-Me e 2.2β-Me erano stati sintetizzati in forma racema e non in forma enantiomericamente pura. Questa scelta era determinata dal fatto che per i nostri studi, basati sulla regio- e diastereoselettività delle reazioni di addizione, non era assolutamente necessario disporre di materiale di partenza enantiomericamente puro (della serie D o della serie L), tra l’altro non proprio agevolmente disponibile, ed il cui utilizzo avrebbe reso tutto quanto lo studio inutilmente molto più complesso e costoso. E’ comunque evidente che qualunque risultato fosse stato ottenuto sul sistema racemo corrispondente agli epossidi 2.2α-Me e 2.2β-Me, questo avrebbe potuto essere trasferito sul corrispondente sistema chirale.
La sintesi degli epossidi 2.2α-Me e 2.2β-Me procede attraverso un precursore comune, l’ α-idrossichetone 2.38 (Schema 2.11), ottenuto mediante un’applicazione racema del protocollo enantioselettivo di Comins,9,10 come sarà successivamente discusso in questa tesi. N Cbz O Me HO N Cbz Me O N Cbz Me O 2.2ββββ-Me 2.38 2.2ααα-Meα
Schema 2.11. Idrossichetone 2.38, il precursore comune per la sintesi di 2.2α-Me e 2.2β-Me.
2.3 Reazioni di glicosilazione di alcooli con gli epossidi 2.2α-Me e 2.2β-Me
Le reazioni di glicosilazione di semplici alcooli bassobollenti quali EtOH, i-PrOH e alcool allilico mediante gli epossidi 2.2α-Me e 2.2β-Me sono risultate completamente 1,4-regio- e stereoselettive in modo direttamente substrato-dipendente, già in condizioni di protocollo A, come già visto con gli epossidi 2.2α e 2.2β: viene in tutti casi ottenuto solo il corrispondente α-O-glicoside da epossido 2.2α e solo il corrispondente β-O-glicoside dall’epossido 2.2β . Nel caso de MeOH si osserva un diverso comportamento tra i due epossidi. Mentre con l’epossido 2.2β la reazione di glicosilazione condotta sotto protocollo A
24
non è stereoselettiva e porta alla formazione di entrambi i possibili metil α- e β-O-glicosidi, la corrispondente reazione dell’epossido 2.2α è già completamente α stereoselettiva in condizioni di protocollo A. Comunque ripetendo la reazione di glicosilazione del MeOH con l’epossido 2.2β in condizioni di protocollo B, un risultato direttamente substrato-dipendente con esclusiva formazione del corrispondente metil β-glicoside, viene anche in questo caso ottenuto (Schema 2.12). N Cbz O Me N N HO OR HO Me Me OR Cbz Cbz protocollo A: ROH(solvente)
protocollo B: ROH(3 equiv) in MeCN N Cbz O Me N N HO OR HO Me OR Me Cbz Cbz 2.3ααα-Meα 2.3βββ−β−−Μ−ΜΜeΜ ROH ROH protocollo A (80%) protocollo B (>99%) protocollo A (>99%) R= Me R= Me
R= Et, i-Pr, t-Bu
(20%) (<1%) (<1%) α αα α-O-glicoside ββββ-O-glicoside α αα α-O-glicoside β β β β-O-glicoside
protocollo A (>99%) R= Me, Et, i-Pr, t-Bu (<1%)
Schema 2.12 Glicosilazione di alcoolicon gli epossidi 2.2α-Me e 2.2β-Me.
Il comportamento degli epossidi 2.2α-Me e 2.2β-Me è stato esaminato anche con un con l’alcool benzilico e quello del solo epossido 2.2α-Me con il diaceton-D-glucosio, ovvero con O-nucleofili per i quali, data la loro natura fisica (altobollente il primo, solido il secondo) condizioni di protocollo A non sono possibili e che quindi devono necessariamente essere utilizzati in condizioni di protocollo B. Anche in queste condizioni, che prevedono l’uso di soli 3 equiv di O-nucleofilo come glicosil accettore, la reazione di glicosilazione da parte degli epossidi 2.2α-Me e 2.2β-Me è risultata completamente regio- e stereoselettiva con esclusiva formazione del corrispondente benzil α-O-glicoside, da 2.2α-Me, e benzil β-O-glicoside da 2.2β-Me, e del solo disaccaride con legame β-glicosidico da 2.2β-Me, secondo il concetto di glicosilazione direttamente substrato-dipendente.
25
Dall’insieme dei risultati ottenuti con gli O-nucleofili è stata ricavata l’importante informazione secondo cui il comportamento degli epossidi 2.2α-Me e 2.2β-Me con O-nucleofili è del tutto analogo a quello già ottenuto con i corrispondenti epossidi 2.2α e 2.2β. E’ così possibile realizzare anche in questi sistemi 6-desossi un interessante processo di O-glicosilazione substrato-dipendente in cui la configurazione dell’epossido di partenza viene trasferita non solo al C(4), ma anche, e cosa più importante, al C(1) del prodotto di reazione determinando di fatto la configurazione dell’ O-glicoside ottenuto: α-O-glicoside dall’epossido 2.2α-Me e β-O-glicoside dall’epossido 2.2β-Me. Questa completa 1,4 regio- e stereoselettività osservata nelle reazioni degli epossidi 2.2α-Me e 2.2β-Me con gli O-nucleofili considerati, è stata razionalizzata ammettendo, anche in questo caso l’intervento di un’efficace coordinazione, in forma di legame a idrogeno, tra l’ossigeno ossiranico e l’idrogeno dell’alcool nucleofilo. La coordinazione del nucleofilo con l’ ossigeno ossiranico degli epossidi 2.2α-Me e 2.2β-Me, reagenti attraverso il rispettivo unico conformero 2.2α’-Me e 2.2β’-2.2α’-Me, con il nucleofilo, guida sia la regioselettività del processo [il nucleofilo è “forzato” a reagire sul C(1)], sia la stereoselettività dello stesso in quanto la coordinazione sopra ricordata determina l’attacco stereoselettivo del nucleofilo dalla stessa parte verso cui si estende l’anello ossiranico, come mostrato nello Schema 2.13.
N O OBn O N O OBn O N Cbz HO OR Me O H R Me a a Me O R H b b N Cbz HO OR Me
αααα-O-glicoside 2.2αααα'-Me 2.2ββββ'-Me ββββ-O-glicoside Schema 2.13. Razionalizzazione della regio- e stereoselettività osservata nelle reazioni di addizione di alcooli agli epossidi 2.2α-Me e 2.2β-Me.
I risultati ottenuti nelle reazioni di addizione nucleofila degli epossidi 2.2β-Me e 2.2α-Me con gli alcooli sono quindi del tutto paragonabili a quelli precedentemente ottenuti, con gli stessi nucleofili, con gli epossidi 2.2β e 2.2α e dimostrano chiaramente come la catena – CH2OBn sul C(5) non svolga alcun ruolo nel dirigere la regio- e stereoselettività del processo di addizione nucleofila. Questo risultato autorizzava pure a ritenere fosse corretto utilizzare gli epossidi 2.2β-Me e 2.2α-Me, più facilmente accessibili, quali efficaci modelli per studiare
26
la reattività dei sistemi ossiranici imminoglicali con altri nucleofili. Poi i risultati ottenuti con gli epossidi 2.2β-Me e 2.2α-Me avrebbero potuto essere correttamente trasferiti, se necessario, ai corrispondenti epossidi 2.2β e 2.2α.
2.4 Sintesi delle 6-desossi-N-nosil aziridine 2.3α-Me e 2.3β-Me
Come naturale proseguimento dello studio, sia sintetico che comportamentale di questi sistemi imminoglicali, era stato ritenuto interessante individuare una procedura che fosse in grado di trasferire in modo completamente regio- e stereoselettivo una funzionalità amminica o ammino protetta sul carbonio C(4) di un sistema immino glicale. Sulla base delle informazioni acquisite nei lavori precedenti e delle considerazioni a suo tempo fatte relativamente alla più facile accessibilità sintetica dei sistemi 6-desossi, era stato ritenuto utile ed efficace, verificare questa possibilità facendo uso delle 6-desossi allil N-nosil aziridine 2.3α-Me e 2.3β-Me (Schema 2.14). Infatti, come nelle reazioni di glicosilazione degli alcooli, gli epossidi 2.2β-Me e 2.2α-Me avevano trasferito sul C(4) un gruppo ossidrilico in modo completamente regio- [solo sul C(4)] e stereoselettivo (configurazione dell’O-glicoside uguale a quella dell’epossido), così le aziridine 2.3α-Me e 2.3β-Me, qualora si fossero comportate allo stesso modo nelle medesime condizioni, avrebbero trasferito un gruppo ammino protetto sul C(4), in modo ancora completamente regio- e stereoselettivo, come desiderato. Resta inteso comunque che tutto quanto fosse stato possibile ottenere dalle aziridine 2.3α-Me e 2.3β-Me, avrebbe potuto poi essere correttamente trasferito alle corrispondenti aziridine 6-OBn sostituite 2.3α e 2.3β, obiettivamente più complesse da sintetizzare e che quindi in questo primo approccio non erano state considerate (Schema 2.14).11
27 N Cbz Me N Cbz Me N Cbz Me N Cbz Me HO OR HO OR N Cbz N Cbz Me N Cbz Me N Cbz Me OR OR ROH ROH ROH ROH β β β β-Glicoside α α α α-Glicoside β ββ β-Glicoside α αα α-Glicoside Me NsHN NsHN O O N Ns 2.2ββββ−−−Μ−ΜΜeΜ 2.2αααα−−−−ΜΜΜΜe 2.3ββββ-Me 2.3αααα-Me 4 4 4 4 N Ns N Cbz Me OR H2N 4 N Cbz Me OR H2N 4 N N O BnO BnO O 2.1ββββ 2.1αααα Cbz Cbz PhSH PhSH K2CO3 K2CO3
Schema 2.14. Glicosilazione di alcooli con epossidi 2.2α-Me e 2.2β-Me e con le aziridine 2.3α-Me e 2.3β-Me.
L’uso di aziridine attivate, ovvero di aziridine portanti sull’azoto gruppi acilici (RCO-) o solfoni lici (RSO2-), era assolutamente necessario per permettere loro di reagire in un ambiente neutro o solo leggermente alcalino tipico dei nostri protocolli A e B. Infatti solo un gruppo di questo tipo è in grado di delocalizzare la parziale carica negativa che si sviluppa sull’azoto aziridinico nello stato di transizione di un processo di apertura a seguito di un attacco nucleofilo. Aziridine non attivate (ovvero non portanti alcun gruppo sostituente o portanti un gruppo alchilico sull’azoto aziridinico) rimarrebbero insensibili alle nostre condizioni di reazione in quanto capaci di reagire solo in ambiente acido o in presenza di un acido di Lewis ovvero in condizioni che non sono compatibili con la stabilità dei sistemi immino glicali da noi utilizzati e/o degli eventuali prodotti di reazione.
La scelta del gruppo attivante nosile (o-nitro-benzensolfonile) era dovuta al fatto che, oltre a possedere tutte le caratteristiche elettroniche richieste, è anche un gruppo che, a differenza del gruppo tosile e mesile, parimenti utilizzabili, può essere facilmente allontanato attraverso una semplice reazione SNAr facendo uso del semplice protocollo PhSH/K2CO3, come qui sotto mostrato inizialmente (Schema 2.15). Questo avrebbe permesso di
28
trasformare i prodotti 4-N-ammino protetti ottenuti nella reazione di O-glicosilazione, nei corrispondenti composti 4-ammino derivati (Schema 2.14).12
K N S O O O NHR N SPh O O S O O NHR N SPh O O S O O NHR N SPh O O S O O NHR SPh O2N SO2 RNH -SPh O2N SO2 RNH2 SNAr H2O PhS RNH-Ns O
Schema 2.15. Deprotezione del gruppo –NHNs mediante reazione SNAr.
Si deve inoltre aggiungere che il gruppo nosile, al pari del gruppo tosile e mesile, ma differentemente dal gruppo acetile e simili, conferisce al legame –NH- adiacente, una acidità sufficiente, tale da poter essere deprotonato in ambiente alcalino, condizione necessaria nella fase di sintesi delle aziridine stesse, come vedremo quando esamineremo la sintesi delle aziridine 2.3α-Me e 2.3β-Me.
Le due aziridine 2.3α-Me e 2.3β-Me erano state preparate in forma racema a partire dal corrispondente epossido di opposta configurazione, rispettivamente 2.2α-Me e 2.2β-Me, a loro volta preparati, come abbiamo anticipato nelle pagine precedenti, a partire da un comune intermedio sintetico, l’idrossichetone 2.38 (Schema 2.16).
N Cbz N Cbz N Cbz N Cbz N Cbz Me Me Me Me Me O N Ns N O Ns O HO 2.38
2.3αααα-Me 2.2ββββ−−−−ΜΜeΜΜ 2.2αααα−−−−Me 2.3βββ-Meβ Schema
29
2.5 Reazioni di glicosilazione degli alcooli con le N-nosil aziridine 2.3α-Me e
2.3β-Me
Il comportamento regio- e stereochimico delle nuove allil N-nosil aziridine 2.3α-Me e 2.3β-Me con O-nucleofili, esaminato nelle reazioni di addizione in condizioni di protocollo A (nucleofilo come solvente) con semplici alcooli bassobollenti come MeOH, EtOH, i-PrOH, t-BuOH e alcooli allilici, si è rilevato completamente 1,4-regio- e stereoselettivo con attacco nucleofilo al carbonio C(1) del sistema allilico con l’esclusiva formazione dei corrispondenti alchil 4-(N-nosilammino)-N-Cbz-immino-O-glicosidi-2,3-insaturi α dall’aziridina 2.3α-Me e β dall’aziridina 2.3β-Me aventi la stessa configurazione dell’aziridina di partenza. La sola eccezione è data dalle reazioni di metanolisi che portano a miscele dei metil O-glicosidi anomerici corrispondenti α e β sia dall’aziridina 2.3α-Me che dall’aziridina 2.3β-Me. Dalla reazione di metano lisi dell’aziridina 2.3β-Me, l’anomero β che presenta la stessa configurazione dell’aziridina di partenza, è il maggiore prodotto (β:α = 85:15), mentre dall’aziridina 2.3α-Me il risultato ottenuto è invertito ed il metil β-O-glicoside, che presenta configurazione opposta all’aziridina di partenza costituisce il prodotto maggioritario (β:α = 65:35) (Schema 2.17).11
30 N Cbz N Ns Me N N NsHN OR NsHN Me Me OR Cbz Cbz protocollo A: ROH(solvente)
protocollo B: ROH(3 equiv) in MeCN N Cbz N Ns Me N N NsHN OR NsHN Me Me OR Cbz Cbz 2.3αααα-Me 2.3ββββ−−−−ΜΜΜΜe ROH ROH protocollo A (35%) protocollo B (>99%) protocollo A (>99%) R= Me R= Me
R= Et, i-Pr, t-Bu
(65%) (<1%) (<1%) α αα α-O-glicoside ββ-O-glicosideββ α αα α-O-glicoside ββββ-O-glicoside protocollo A (15%) protocollo B (<1%) protocollo A (<1%) R= Me R= Me
R= Et, i-Pr, t-Bu
(85%) (>99%) (>99%)
Schema 2.17. Regio- e stereoselettività delle reazione di glicosilazione degli alcooli con le aziridine 2.3α-Me e 2.3β-Me secondo protocolli A e/o B.
Comunque, in entrambi i casi, quando le reazioni venivano ripetute utilizzando pochi equivalenti di MeOH in MeCN anidro (condizioni di reazione di protocollo B), veniva osservata una completa stereoselettività ed il corrispondente metil O-glicoside avente la stessa configurazione dell’aziridina di partenza (α da 2.3α-Me e β da 2.3β-Me) costituisce l’unico prodotto di reazione. Le condizioni di protocollo B sono necessariamente usate nella glicosilazione del benzil alcool con le aziridine 2.3α-Me e 2.3β-Me e diaceton-D-glucosio (monosaccaride parzialmente protetto) con l’aziridina 2.3β-Me, presi come esempi di O-nucleofili per i quali le condizioni di reazione di protocollo A non sono chiaramente applicabili. In ogni caso, il corrispondente 4-(N-nosilammino)-α-O-glicoside da 2.3α-Me e il 4-(N-nosilammino)-β-O-glicoside da 2.3β-Me sono i soli prodotti di reazione.
Analogamente ai risultati ottenuti con gli epossidi 2.2α-Me e 2.2β-Me, il completo regio- e stereocontrollo osservato nella glicosilazione di alcooli da parte delle aziridine 2.3α-Me e 2.3β-2.3α-Me, può essere spiegato ammettendo l’intervento di un processo di coordinazione (legame ad idrogeno) tra l’azoto dell’aziridina e l’O-nucleofilo. In questo caso, il nucleofilo
31
(alcool) viene di fatto ad essere trasportato, rispettivamente, sulla faccia α, nel caso di 2.3α-Me reagente attraverso il conformero 1A, o sulla faccia β, nel caso di 2.3β-2.3α-Me reagente attraverso il suo corrispondente conformero 1B, venendosi così a trovare opportunamente disposto per un attacco, entropicamente favorito, sul carbonio C(1) dalla stessa parte dell’anello aziridinico (routes a e b, Schema 2.18).
N Ο OBn CH3 N a H Ο R N CH3 N O R Η Ο OBn 1 b O R H route a Me N route b Cbz NsHN OR 2.3ααα-Meα 2.3ββββ−−−−ΜΜΜΜe Ν Me Cbz NsHN OR O H R Ns Ns
R = Me, Et, i-Pr, t-Bu, Bn, allile, diaceton-D-glucosio α
αα
α-O-glicoside ββ-O-glicosideββ
1A 1B
Schema 2.18. Razionalizzazione della selettività substrato dipendente nella glicosilazione di alcoli dalla allil aziridine 2.3α-Me e 2.3β-Me.
In questo modo, i corrispondenti 4-(N-nosilammino)-N-Cbz-immino-O-glicosidi-2,3-insaturi (α da 2.3α-Me e β da 2.3β-Me) aventi la stessa configurazione dell’aziridina di partenza, venivano ottenuti con completa regio- e stereo selettività direttamente substrato-dipendente.11
2.6 Reazione di deprotezione degli alchil N-nosil-O-glicosidi ottenuti dalle
aziridine 2.3α-Me e 2.3β-Me
Per deproteggere selettivamente la funzionalità N-nosilammino degli α- e β-glicosidi ottenuti per glicosilazione degli alcooli da parte delle aziridine 2.3α-Me e 2.3β-Me era necessario applicare un protocollo che fosse compatibile con il mantenimento del gruppo uretanico (N-Cbz). Per far questo è stato utlizzato il protocollo PhSH/K2CO3, che già aveva avuto successo sul sistema glicale.12 L’applicazione di questo protocollo all’i-propil α-O-glicoside, derivato dall’aziridina 2.3α-Me, e al diasteroisomero i-propil β-O-α-O-glicoside, derivato dall’aziridina 2.3β-Me (R = i-Pr) conduce in modo semplice ed attraverso una reazione particolarmente pulita ai corrispondenti 4-ammino-α- e β-O-glicosidi che vengono ottenuti secondo brevi tempi di rezione (2 h) e con resa soddisfacente (65-68%, calcolata dopo purificazione dall’eccesso di PhSH) (Schema 2.19).
32 N NsHN OR Me Cbz N NsHN OR Me Cbz α α α α-O-glicoside ββββ-O-glicoside N H2N OR Me Cbz N H2N OR Me Cbz
4-ammino-ααα−α−−−O-glicoside 4-ammino-βββ−β−−−O-glicoside
PhSH PhSH
K2CO3 K2CO3
(65%) (68%)
Schema 2.19. Deprotezione dei 4-(N-nosilammino)-O-glicosidi ai corrispondenti 4-ammino-O-glicosidi.
In conclusione, era così stato possibile dimostrare la possibilità di introdurre un ammino gruppo libero sul C(4) di un N-Cbz-immino-O-glicoside-2,3-insaturo, facendo uso di reazioni di glicosilazione di alcooli da parte di corrispondenti allil N-nosil aziridine. Nel solo 4-ammino-O-glicoside ottenuto, la configurazione attorno al C(4) e all’anomerico C(1) è la stessa e corrisponde a quella dell’aziridina di partenza, in una nuova versione di glicosilazione direttamente substrato-dipendente applicata ad un sistema N-Cbz-immino glicale.11,12