3. Materiali e metodi
3.1. Soggetti inseriti nello studio
Nel presente studio sono stati inseriti 30 volontari sani, 15 soggetti con diagnosi psichiatrica di disturbo bipolare in cura da almeno quattro settimane con antipsicotici tipici:
o Aloperidolo: 2-8 mg/die
o Clorpromazina: 20-250 mg/die
o Perfenazina: 12 mg/die
o Pimozide: 2-6 mg/die
e 12 soggetti con diagnosi psichiatrica di disturbo bipolare, in cura da almeno quattro settimane con antipsicotici atipici:
o Clozapina: 50-200 mg/die
o Risperidone: 3 mg/die
3.2. Separazione di piastrine dal sangue
Materiali
Anticoagulante (AC: Sodio citrato 2.2% e Acido citrico 1.2%) da utilizzare 1:6 e conservare a 4°C.
Metodi
Il sangue prelevato viene fatto scivolare molto lentamente lungo le pareti di una falcon in modo da rompere meno globuli rossi possibili ed unito ad un’aliquota di anticoagulante.
Si centrifuga a 150-200 g per 15 minuti a temperatura ambiente separando così il plasma ricco (PRP); il PRP viene trasferito con una pipetta Pasteur in falcon da 15 ml.
Successivamente il PRP viene centrifugato a 1500 g per 15 minuti a temperatura ambiente e si ottiene cosi il pellet di piastrine.
3.3. Preparazione di lisati di membrane da un pellet di piastrine per i saggi di Immunoblotting
Materiali
Tampone T50A (Tris- HCl 50mM EDTA 20Mm, NaCl 150mM) con
inibitori delle proteasi benzamidina 16mg/100ml, bacitracina 20mg/100ml, inibitore della tripsina 2mg/ml).pH 7.4 a 4° C;
tampone T5 b ipotonico (Tris -HCl 5mM, EDTA 5mM) con inibitori delle proteasi, pH 7.4 a 4° C; PBS (NaH2PO4 9.1mM, Na2HPO4 1.7mM, NaCl 150mM) pH 7.4 a 4° C, RIPA (PBS fino a 10 ml, Deossicolato 0.5%, NP40 1ml stock 10%, SDS 1ml stock 1% con inbitori delle proteasi x il blot da aggiungere ai 10ml di RIPA al momento dell’uso, PMSF 30µl stock 0.2M, Aprotinina 20µl stock 2µg/ml, Ortovanadato 100µl stock 0.1M.
Metodo
Il pellet vieno sospeso in 4ml di T50A più inibitori (il volume totale è circa 2/3 del PRP) e centrifugato a 17.500 g per 15 minuti a 4° C. eliminato il sovranatante si risospende in circa 2ml di T5b ipotonico con gli inibitori e si omogeneizza tramite potter in ghiaccio. Dopo aver portato il volume fino a 4ml con lo stesso tampone, si centrifuga a 30.000 g per 15 minuti; si risospende il pellet in circa 300µl di RIPA (1µg in 1µl) con gli inibitori, si ripete il passaggio con il potter in ghiaccio e si trasferisce in eppendorf. Si
centrifuga a 13.500 rpm per 45 minuti a 4° C. dopo aver tolto il precipitato che è costituito dai lipidi di membrana insolubili, si procede al proteico sul sovranatante che è formato da proteine di membrana solubilizzate).
3.4. Dosaggio del contenuto proteico con il metodo BIO-RAD
Per determinare il contenuto delle proteine totali presenti nei nostri campioni, si impiega il reattivo commerciale Biorad, il quale si basa sul sistema colorimetrico di Bradford (1976).
Il dosaggio proteico Biorad è basato sul cambiamento del colore del colorante Comassie Brillant Blue G-250, in risposta a varie concentrazioni di proteine. Il reattivo si lega principalmente ai residue aminoacidici aromatici o basici, come ad esempio l’arginina. Come standerd viene utilizzata una soluzione di ? globulina a concentrazione nota.
Si segue il seguente schema per la determinazione del contenuto proteico del campione in esame.
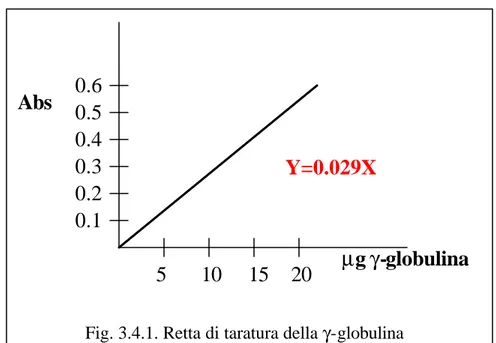
Viene poi effettuata la lettura allo spettrofotometro alla lunghezza d’onda di 595nm. in quanto si sviluppa una colorazione tendente al blu, la cui intensità è direttamente proporzionale alla concentrazione proteica presente nei campioni. Con il valore di lettura ottenuto si ricava la concentrazione di
taratura costruita utilizzando 5 soluzioni contenenti quantità note di γ -globulina comprese tra 2.5µg e 20µg.
Per determinare la concentrazione di proteine presenti viene eseguito il seguente calcolo, dal quale possiamo vedere se la concentrazione risulta troppo alta o troppo bassa così da poter effettuare le opportune diluizioni.
( M – B ) x 5
0,029 M: media dei campioni
B: media dei bianchi 5: fattore di diluizione 5 10 15 20 0.1 0.2 0.3 0.4 0.5 0.6 µg γ-globulina Abs Y=0.029X
Per la determinazione quantitativa del contenuto proteico è stato applicato il metodo Bio-Rad secondo il seguente protocollo:
Reagente A* (Dc Protein Assay Bio -Rad): 20µl di reagente S (Dc Protein Assay Bio-Rad) per ogni ml di reagente A (se i campioni da dosare non contengono detergenti non aggiungere il reagente S, il reagente A* è stabile per una settimana, se si forma del precipitato, scaldare la soluzione ed agitare con il vortex).
Standard: soluzione di albumina bovina (soluzione stock).
La soluzione “stock” viene diluita con 5 ml di H2O distillata ad una concentrazione di 5.76 mg/ml.
Diluizioni dell’albumina per effettuare la retta di taratura (tab. 3.4.1):
Campione H2O Tampone A B F 20 µl - - 20 µl 100 µl 800 µl E 20 µl - - 20 µl 100 µl 800 µl D 20 µl - - 20 µl 100 µl 800 µl C 20 µl - - 20 µl 100 µl 800 µl B 20 µl - - 20 µl 100 µl 800 µl A 20 µl - - 20 µl 100 µl 800 µl Campione 20 µl 20 µl - 100 µl 800 µl Bianco - 20 µl 20 µl 100 µl 800 µl
3.5. Immunoprecipitazione dei recettori A2A AR e D2 DR
Materiali
Per i recettori D2: anticorpo anti-D2R mouse, S. Cruz, Biotechnology Germania; per il recettore A2A: anticorpo anti-A2AR, rabbit, Alpha Diagnostoc, USA. RIPA lysing-buffer (PBS, Na-deossicolato 0.5%, NP-40 10%, SDS 10%, PMSF 0.2M, aprotinina 20µg/ml, ortovanadato 0.1M, pH 7.4), LAEMMLY (1ml di glicerolo, 0.5ml di ß-mercaptoetanolo, 3ml di SDS al 10%, 1.25ml di Tris-HCl 1M, 1-2mg di bromofenolo, pH 6.7).
Metodo
Un milligrammo di proteine solubili viene immunoprecipitato mediante l’aggiunta di anticorpo diretto contro il recettore A2A o D2 e lasciato in agitazione a 4° C per tutta la notte. Successivamente, vengono aggiunti 50µl di proteina A Sepharose (Pharmacia Biotech) e si tiene in agitazione a 4° C per 2h, poi si centrifuga a 8000 g per 1 minuto. Il pellet viene poi lavato con 1ml di RIPA e solubilizzato in LAEMMLY.
3.6. Elettroforesi su gel di poliacrilammide con sodio dodecilsolfato (SDS-PAGE)
L’elettroforesi consiste nella migrazione di macromolecole cariche indotta dalla presenza di un campo elettrico; quella effettuata su gel di poliacrilammide è uno dei metodi maggiormente impiegati per l’analisi qualitativa di miscele di proteine. I gel di poliacrilammide agiscono come setacci molecolari, rallentando la migrazione delle proteine in modo proporzionale alla loro massa molecolare. Prima di essere caricati sul gel, i lisati cellulari vengono bolliti per 5 minuti in presenza di SDS e ß-mercaptoetanolo, al fine di ottenere la denaturazione. Le molecole di SDS, a carica negativa, sono dotate di catene idrocarburiche in grado di interagire con le regioni idrofobiche delle proteine, annullandone la carica intrinseca e conferendo loro una densità di carica negativa uniforme. Il ß-mercaptoetanolo, inoltre, scinde i legami disolfuro destabilizzando la struttura terziaria delle proteine. In queste condizioni si applica un campo elettrico che fa migrare le proteine esclusivamente in base al loro peso molecolare in direzione dell’estremità del gel collegata al polo positivo. L’apparecchio per l’elettroforesi è composto da un alimentatore, che fornisce una corrente diretta tra gli elettrodi nella cella elettroforetica e da una cella elettroforetica parzialmente riempita con tampone di corsa o running buffer.
Materiali
Tampone pH=8.8 (90.75 g di tris base 1.5 M ,2 g di SDS 0.4% per 500 ml di acqua bidistillata millipore); Lower Gel 12% (16 ml di acrilammide al 30%, 10 ml di tampone pH=8.8 , 4 ml di glicerolo, 10 ml di acqua bidistillata millipore); tampone pH =6.8 (15 g di tris base 0.5M, 2g di SDS 0.4% per 500 ml di acqua bidistillata millipore); Upper Gel 4% (1.33 ml di acrilammide al 30% , 2.5ml di tampone a pH =6.8, 1 ml di glicerolo , 5.2 ml di acqua bidistillata millipore ); Running Buffer 10x pH =6.8 (30g di Tris, 1.44 g di Glicina, 10 g di SDS per 1L di acqua bidistillata millipore, diluito 1:10 al momento dell’uso).
Preparazione del gel
Il gel è costituito da due parti a diversa concentrazione di poliacrilammide: l’ “Upper Gel” (a concentrazione minore) , in cui i campioni vengono caricati , e una porzione rimanente in cui avviene la separazione , “Lower Gel”. Quest’ultimo viene colato tra due lastre di vetro mantenute separate e parallele da due sottili spaziatori. A polimerizzazione ultimata, tra i due vetrini, viene posizionato un pettine e messo l’upper gel; i denti del pettine lasceranno nell’upper gel polimerizzato degli spazi a forma di pozzetto nei quali vengono caricati i campioni. Al Lower Gel, degassato per circa 5 minuti, si aggiungono sul momento 33.3 µl di Ammonio persolfato al 10% e
del caricamento sul gel a ciascuna alquota di lisato, contenente 30µg di proteine, viene aggiunta la soluzione di LAEMMLY (4 x). Sul gel viene caricata anche un’aliquota di standard a peso molecolare noto, per la valutazione della proteina di interesse al termine della corsa elettroforetica. Successivamente si procede all’applicazione del voltaggio secondo la formula : 135 mA x N° di gel / N° ore di corsa = mA da impostare
3.7. Blotting
Terminata la corsa elettroforetica, il gel viene posto in un secondo apparecchio, che permette di trasferire il tracciato elettroforetico delle singole proteine separate nel gel su un foglio di nitrocellulosa . Questo metodo è conosciuto come “Protein blotting” o “Western blotting”.
Materiali
Transfert Buffer (56.7 g di Glicina, 12 g di Tris, 4 g di SDS e 400 ml di metanolo per 4 L di acqua bidistillata); tampone TBS pH =8.8 (Trizma base 10M, NaCl 150M); Blotto A (TBS, Milk 5%, Tween-20 0.05 %); TBS-T (TBS,Tween-20 0.05%).
carta da filtro previamente bagnati con tampone di trasferimento. Il “sandwich” è posizionato in una cella provvista di due elettrodi paralleli ed immerso completamente nel tampone di trasferimento. Impostando una corrente pari a 480 mA in direzione perpendicolare al gel, le proteine vengono trasferite sul foglio di nitrocellulosa in circa 2 ore. A questo punto la nitrocellulosa viene tolta delicatamente dal “sandwich”, posizionata in un contenitore idoneo e tenuta in agitazione con Blotto A per 45 minuti a temperatura ambiente. Questo lavaggio ha la funzione di bloccare i siti di interazione idrofobica aspecifici presenti sulla nitrocellulosa. Dopo il lavaggio viene aggiunto l’anticorpo primario e si lascia il tutto in incubazione per 2 ore a temperatura ambiente, successivamente si eseguono tre lavaggi da 5 minuti ciascuno con Tampone TBS-T prima di procedere a 2 ore di incubazione con l’anticorpo secondario. L’anticorpo secondario è coniugato con la perossidasi di rafano, HPR (Horseradish peroxidase) che, in presenza di acqua ossigenata, ossida il luminolo (ECL, Amersham Pharmacia Biotech), con lo sviluppo di luce che può essere rivelata esponendo la nitrocellulosa ad una lastra fotografica (fig.3.7.2). La quantificazione delle bande immunoreattive è realizzata mediante l’utilizzo di un densitometro (GS- 670, Biorad).
Nitrocellulosa Gel Fig. 3.7.1 Carta Spugna Polo negativo (lastra bucata nera) Polo positivo
Substrato incolore Prodotto chemioluminescente
Proteina bersaglio Anticorpo secondario coniugato con enzima (nel nostro caso HRP)
Anticorpo primario specifico BSA (riduce il legame per la proteina bersaglio non specifico alle IgG)
Fig. 3.7.2
Sviluppo chemioluminescenza HRP + ECL (H2O2 + luminolo)
3.8. Stripping e Re-Blotting
Una volta che le bande immunoreattive sono state visualizzate su lastra fotografica è possibile rimuovere gli anticorpi dalla nitrocellulosa mediante la metodica dello “stripping ”, rendendone possibile un suo ulteriore utilizzo.
Materiali
Tampone di STRIPPING pH = 6.7 (2-mercaptoetanolo 100 mM, SDS 2%, Tris 62.5 mM); Tampone TBS pH =8.8 (Trizma base 10M, NaCl 150M); Blotto A (TBS, Milk 5%, Tween-20 0.05%); (TBS, Tween-20 0.05%).
Metodi
La nitrocellulosa viene messa a contatto con la soluzione di STRIP e tenuta in agitazione in bagnetto riscaldato ad una temperatura di 50°C per 30 minuti. Successivamente si procede a tre lavaggi con TBS-T da 5 minuti ciascuno; dopo segue un periodo di agitazione per 45 minuti con Blotto A a temperatura ambiente in modo da bloccare i siti di interazione idrofobica aspecifici. Si aggiunge l’anticorpo primario e si lascia ad incubare per 2 ore a temperatura ambiente, successivamente si fanno tre lavaggi da 5 minuti ciascuno con TBS-T prima di procedere a 2 ore di incubazione con l’anticorpo secondario. Grazie alla reazione che avviene tra perossidasi di rafano e luminolo in presenza di acqua ossigenata, si ha sviluppo di luce
3.9. Preparazione di omogenato di membrane da un pellet di piastrine per lo studio di binding del recettore A2A
Materiali
Tampone T50a (Tris-HCl 50mM EDTA 20mM, NaCl 150mM) con
inibitori delle proteasi (benzamidina 16mg/100ml, bacitracina 20mg/100ml, inibitore della tripsina 2mg/100ml) pH=7,4 a di 4°C; Tampone T50b ipotonico (Tris-HCl 5mM EDTA 5mM) con inibitori delle proteasi pH=7.4 a 4°C; Tampone T50c (Tris-HCl 50mM, EDTA 0.8mM) con inibitori delle proteasi pH=7.4 a 4°C.
Metodo di preparazione dell’omogenato
Il pellet piastrine viene scongelato e sospeso in 4 ml di T50a con inibitori (il volume totale è circa 2/3 del PRP).
Si centrifuga a 17.500 g (12.100 rpm) per 10 minuti a 4°C, si sospende il pellet in 3 ml di T50b ipotonico e si potterizza in ghiaccio.
L’omogenato viene centrifugato a 35.000 g (17.000 rpm) per 10 minuti a 4°C.
Il pellet viene sospeso in 2 ml di T50c con ADA (adenosina deaminasi). Si lascia ad incubare per 15 minuti a 37°C,si centrifuga a 35.000 g (17.000 rpm) per 10 minuti a 4°C. Si aggiunge 1ml di T50c senza ADA ed il tutto viene omogeneizzato con il potter, il contenuto delle proteine presenti viene valutato quantitativamente mediante dosaggio proteico con il metodo Bio-Rad.
3.10. Studio dei recettori con la metodica di binding radioattivo
La tecnica di binding è un metodo biochimico che permette di ottenere l’identificazione di siti recettoriali (binding sites) in cellule bersaglio o frazioni subcellulari, tramite l’utilizzo di radioisotopi.
I siti recettoriali, al contrario dei recettori, sono siti accettori senza alcuna azione di competizione agonista-antagonista e quindi indipendente da una risposta fisiologica.
Solo quando sono soddisfatte le correlazioni, da un punto di vista sperimentale, tra l’affinità del ligando in vitro e la potenza farmacologica in vivo,la specificità tissutale, la stereospecificità, la saturabilità, la reversibilità e l’alta affinità, il binding site è relazionato al sito recettoriale. La fomazione del complesso ligando-binding site è una reazione reversibile che segue la legge di azione di massa ed è caratterizzata da una propria costante di associazione e di dissociazione (kd) del complesso recettore-ligando, che ha le dimensioni di una concentrazione. La kd rappresenta la concentrazione del ligando che all’equilibrio occupa il 50% dei siti recettoriali. Essa è inversamente correlata all’affinità del ligando per il recettore, quindi ad un aumento della kd corrisponde una diminuizione dell’affinità. La densità dei siti recettoriali (Bmax) è indicata come numero massimo di molecole che si possono legare nel tessuto, è correlata el numero dei binding-sites presentied è espressa come fmoli/mg di proteine presenti in quel dato tessuto. La concentrazione dei recettori, nella maggior parte dei tessuti, è nel range di fentomoli o picomoli, quindi nella tecnica del binding vegono utilizzati ligandi con una alta affinità, alta selettività, alta attività specifica (legata alla quantità di molecole che sono marcate radioattivamente). La metodica del
contenente il recettore in esame, con un ligando del recettore, marcato con un isotopo radioattivo, in condizioni di tempo, di temperatura e di pH controllate, sufficienti al raggiungimento dell’equilibrio. Durante il tempo di incubazione una certa quantità del ligando (X*) forma con il recettore (R) il complesso recettore-ligando (RX*). La quantità in eccesso di X* che è rimasta lib era viene tolta dal complesso RX* tramite una successiva filtrazione sottovuoto. Tale procedura deve essere effettuata velocemente e con l’utilizzo di tampone freddo, in modo da ridurre al minimo la possibilità di dissociazione del complesso, che è rimasto trattenuto nel filtro. Dalla misurazione della radioattività residua sul filtro, effettuata mediante uno scintillatore a fase liquida, si ricava il legame specifico del ligando interessato. L’esistenza di un compartimento, a cui il ligando si lega in modo non saturabile, fornisce il legame non specifico o aspecifico, che viene misurato valutando il legame residuo di ogni concentrazione di radioligando in presenza di un forte eccesso di ligando non marcato (X), il quale spiazza il ligando marcato da tutti i siti saturabili, compresi quelli recettoriali. Il binding specifico viene quindi calcolato come differenza tra il legame totale (misurato in assenza di agente competitore) ed il legame aspecifico (misurato in presenza di agente competitore) e rappresenta il 70-80% del legame complessivo. Il legame aspecifico deve essere il più basso possibile (20-30% del legame totale).
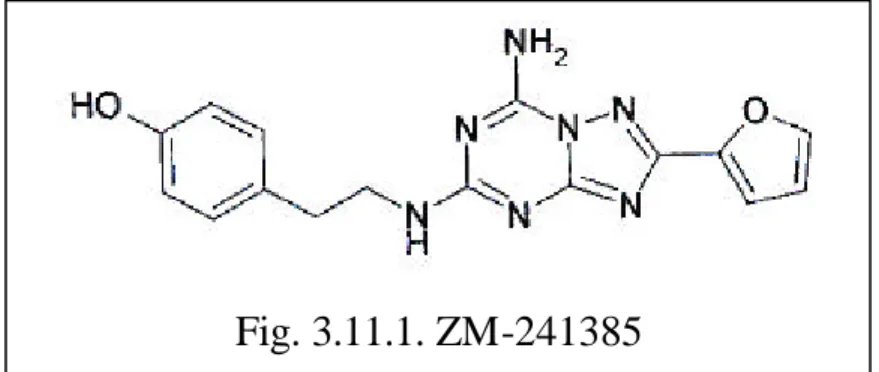
3.11. Determinazione del legame del radioligando [3H]-ZM-241385 al
recettore adenosinico A2A su omogenato di membrane di piastrine umane.
Materiali
Tampone T50c (Tris-HCl 50mM, EDTA 0.8mM), [ 3
H]-ZM-241385 ([3H] -4-(2-[7-amino-2-(2-furyl)[1,2,4] triazolo [2,3-a][1,3,5] triazin 5-yl-amino]ethyl)phenol), filtri in fibra di vetro Whatmann GF/C, strumento Millipore per la filtrazione.
Metodi
L’omogenato, ottenuto secondo la metodica descritta nel paragrafo 2.2, viene diluito con un opportuno volume di tampone T50c a seconda del contenuto di proteine presenti nel campione. Aliquote di tale omogenato , contenenti circa 50µg di proteine, determinate mediante dosaggio proteico,
vengono incubate con [3H]-ZM-241385 (attività specifica 23.47µCi/nmole) a concentrazioni crescenti secondo lo schema per 1 ora a 4°Cin un volume finale di 250µl di T50c. Il legame aspecifico viene determinato incubando un’aliquota delle membrane in presenza di ZM-241385 80µM. Trascorso il tempo di incubazione i campioni vengono sottoposti a filtrazione, utilizzando i filtri in fibra di vetro Whatmann GF/C, effettuando tre lavaggi in provetta con 3 ml di tampone freddo ciascuno. La filtrazione viene effettuata utilizzando uno strumento millipore, costituito da un supporto analitico a
consente la filtrazione contemporanea di 12 campioni. Mediante il processo descritto, il radioligando rimasto “libero” (X*) è separato da quello “legato”. Successivamente, ciascun filtro viene posto in vials di plastica, in cui sono stati versati 4ml di liquido di scintillazione. La radioattività residua sul filtro viene letta mediante uno scintillatore a fase liquida ed è espressa in disintegrazioni per minuto (dpm). Dal valore dei dpm ottenuti è possibile risalire ai µCi presenti nelle vials , dividendo i dpm per 2.220.000, essendo: 1µCi = 2.220.000 dpm. Conoscendo il valore dell’attività specifica del radioligando è possibile risalire alle nanomoli di recettore presenti nel campione. Schema 3.11.1 T50c ZM-241385 (1 mM) Membrane [3H]ZM-241385 M(proteine) 190 µl - 10 µl 12,5 nM M’ 190 µl - 50 µl 10 µl 12,5 nM F 170 µl 20µl 50 µl 10 µl 12,5 nM F' 170 µl 20µl 50 µl 10 µl 12,5 nM Controllo - - - 10 µl 12,5 nM M 180 µl - 50 µl 20 µl 12,5 nM M' 180 µl - 50 µl 20 µl 12,5 nM F 160 µl 20µl 50 µl 20 µl 12,5 nM F' 160 µl 20µl 50 µl 20 µl 12,5 nM Controllo - - - 20 µl 12,5 nM M 150 µl - 50 µl 50 µl 12,5 nM M' 150 µl - 50 µl 50 µl 12,5 nM F 130 µl 20µl 50 µl 50 µl 12,5 nM F' 130 µl 20µl 50 µl 50 µl 12,5 nM Controllo - - - 50 µl 12,5 nM M 190 µl - 50 µl 10 µl 125 nM M' 190 µl - 50 µl 10 µl 125 nM F 170 µl 20µl 50 µl 10 µl 125 nM F' 170 µl 20µl 50 µl 10 µl 125 nM Controllo - - - 10 µl 125 nM
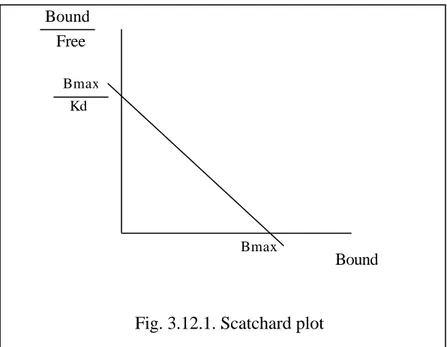
3.12. Analisi Scatchard: determinazione della Kd e della Bmax
Una delle trasformazioni più usate per l’analisi dello studio di binding è lo Scatchard plot, che permette di identificare i parametri cinetici (Kd e Bmax) del legame di un radioligando al proprio sito recettoriale.
Lo Scatchard plot si ottiene riportando in ascissa il valore della radioattività associata al filtro espressa come bound (B), cioè come la concentrazione del rad ioligando complessato con il recettore in fmol/mg di proteine, ed in ordinata il rapporto tra la concentrazione di radioligando legato (B) e il radioligando libero (F) espresso in nM.
Nel caso di un singolo ligando che interagisca con una sola popolazione di siti recettoriali ad un’unica affinità per il ligando,il plot è dato da una retta in cui la pendenza è –1/Kd, l’intercetta sull’asse x rappresenta la Bmax, mentre l’intercetta sull’asse y rappresenta il rapporto Bmax/Kd.
Fig. 3.12.1. Scatchard plot
L’equazione di Scatchard è:
=
Spesso i dati di legame non possono essere trasformati in una retta; si può ottenere una curva concava, caratteristica di una popolazione recettoriale con cooperatività positiva (l’affinità del ligando per il recettore aumenta all’aumentare dell’occupazione dei sitirecettoriali) oppure convessa.
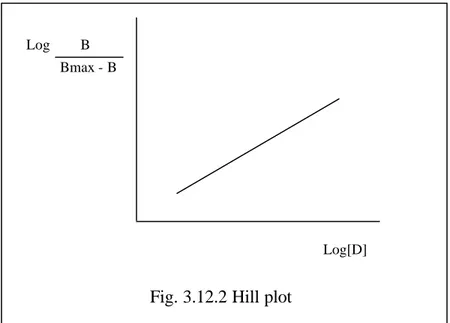
Un’altra trasformazione usata per lo studio di binding consiste nell’Hill plot. Il diagramma di Hill viene costruito riportando in ordinate il rapporto log B/Bmax-B, ed in ascisse il log [F], dove [F] rappresenta la concentrazione del ligando libero. Il grafico risultante è una retta la cui pendenza è il coefficiente di Hill (n ) che dà informazioni sulla presenza di uno o più siti di interazione
Bound – Bound Free Kd + Bmax Bound Free Bound Bmax Kd Bmax
Se nH = 1, il ligando interagisce con una singola popolazione recettoriale tramite una reazione bimolecolare, seguendo la legge di azione di massa, oppure con più siti recettoriali con uguale affinità.
Se nH > 1, probabilmente c’è cooperatività positiva tra i siti recettoriali, mentre se nH < 1, siamo di fronte a siti recettoriali con cooperatività negativa.
Fig. 3.12.2 Hill plot Log B
Bmax - B
3.13. Trattamento “in vitro” delle piastrine con farmaco antipsicotico tipico
Un pool di piastrine ottenuto da quattro volontari sani, è stato trattato con un farmaco antipsicotico tipico, l’aloperidolo, ad una concentrazione di 0.006 µg/ml o 0.03µg/ml per 24 ore. La concentrazione del farmaco è stata decisa in base al dosaggio medio somministrato ai pazienti psicotici e valutando, inoltre, la farmacocinetica dell’antipsicotico. Dopo incubazione, sono state isolate le piastrine e sono stati determinati i parametri di binding all’equilibrio tramite saggi di binding con radioligando come descritto precedentemente.