Stereoselective synthesis of a Glc-NAc conjugate as
potential modulator
1. INTRODUCTION
1.1 O-GlcNAc in cancer biology
[1]O-linked β-N-acetylglucosamine is the covalent addition of an N-acetylglucosamine
sugar moiety to hydroxyl groups of serine and/or threonine residues of cytosolic and nuclear proteins. Modification of proteins by O-GlcNAc (O-GlcNAcylation) is a post-translational modification, which is catalyzed by the O-GlcNAc transferase (OGT); indeed, this enzyme catalyzes the addition of O-GlcNAc from the high energy donor substrate UDP-GlcNAc, that is an end-product of the hexosamine biosynthetic pathway (HBP).
O-GlcNAc removal is catalyzed by O-GlcNAcase (OGA) and the balance of activity of
this two enzymes (OGT and OGA) determines the rate of GlcNAc on proteins.
O-GlcNAcylation plays a crucial regulatory role on many proteins and it impacts
several cellular processes, such as cell cycle progression, transcription, cellular stress responses and epigenetic control of gene expression.
Recently, hyper-O-GlcNAcylation has been thought to be a general feature of cancer cells, because several research groups have noticed that elevated levels of O-GlcNAcylated proteins occur in human malignancies. A lot of typical hallmarks of cancer can be linked with hyper-O-GlcNAcylation.[2] Besides, reducing hyper-O-GlcNAcylation blocks tumor
growth in a variety of cancer models.
1.1.1 Cancer cell metabolic reprogramming and hyper-GlcNAcylation
One of the most interesting features of cancer cells is a phenomenon, also known as Warburg effect,[3] in which cancer cells depend mostly on glycolysis instead of oxidative
phosphorilation as the main energy sources, even in the presence of high oxygen tension. This fact can appear unusual, because glycolysis is much less efficient than oxidative phosphorilation in producing ATP, which is the energy source of the cells, and cancer cells have got increased energy needs. So, how can we explain this metabolic reprogramming of cancer cells? It was initially proposed that the metabolic reprogramming balanced bio-energetic and bio-synthetic needs of cancer cells. As they divide very quickly, they increase
their glucose uptake, but the excess glucose taken up may also serve to synthesize nucleotides and lipids. The switch to glycolysis in cancer cells is due in part to tumor cell hypoxia, oncogenes and mutant tumor soppressors, which activate some pathways to promote Warburg-like metabolism. One of the main consequences is up-regulation of glucose transporters, especially GLUT-1, which increases glucose import into tumor cells. The abundance of glucose not only contributes to increased glycolytic flux, but also increases pentose phosphate pathway, which participates in nucleotides synthesis. Besides, about 2-5% of glucose fluxes through the HBP, so increased glucose concentration in cancer cells determines an increased HBP flux.
Cancer cells are also addicted to glutamine,[4] that is they require a high concentration of
glutamine to survive and proliferate. Oncogenes can up-regulate glutamine uptake. Glutamine is the donor substrate in the conversion of fructose-6-phosphate to glucosamine-6-phosphate in the HBP; thus excess glutamine uptake in cancer cells can contribute to increased flux through the HPB and, consequently, can increase levels of HBP end-product UDP-GlcNAc.
Such metabolic reprogramming in cancer cells meets energetic needs for rapid cell proliferation and supplies intermediates for cancer cell biosynthesis. Cancer cell metabolic changes (increased glucose uptake due to Warburg effect and increased glutamine uptake) cooperate to increase HBP flux. As we have already seen, the end-product of HBP is UDP-GlcNAc, which is the donor substrate used by OGT in enzymatic addition of GlcNAc. Increased HBP flux is linked to elevated levels of GlcNAc[5] and this fact leads to the
hypothesis that cancer metabolism is a cause of the observed hyper-GlcNAcylation, which is a common feature of a lot of cancer types.
1.1.2 Altered expression levels of enzymes involved in O-GlcNAc in
cancer cells
There are several evidences that OGT and OGA expressions are deregulated in cancer cells, in fact it has been demonstrated that OGT is overexpressed, while OGA levels are
suggests that the limiting factor for O-GlcNAcylation is not polypeptides but UDP-GlcNAc: increasing cellular concentration of UDP-GlcNAc results in increased O-GlcNAcylation by enhancing the activity of OGT. Thus, both increased flux through the HBP and the increased expression of OGT (accompanied by a reduced expression of OGA) in cancer cells likely contribute to hyper-O-GlcNAcylation.
Besides, it has been explained that there are negative feedback mechanisms which cells use to buffer large changes in levels of O-GlcNAc; in fact, it has been demonstrated that elevation of O-GlcNAc by pharmacological inhibition of OGA leads to decreased OGT expression and increased OGA expression; conversely, pharmacologically lowering O-GlcNAc levels results in higher OGT expression and lower OGA expression. These changes are consistent with feedback signals to normalize abnormal increases or decreases in O-GlcNAc levels, although the mechanisms underlying such a feedback mechanism are not known. Despite this negative feedback mechanism which attempts to dampen changing
O-GlcNAc levels, cancer cells maintain an elevated level of O-GlcNAcylation, elevated
OGT and decreased OGA compared to non-transformed cells. Therefore, cancer cells succeed to bypass this negative feedback mechanism which attempts to normalize O-GlcNAc levels. Besides, it appears that cancer cells are selectively addicted to hyper-O-GlcNAcylation, as knocking down levels of OGT in transformed cells can selectively cause their death.
1.1.3 Links between O-hyper-GlcNAcylation and specific hallmarks of
cancer
Two typical hallmarks acquired by cancer cells are sustaining proliferative signaling and evading growth suppressors; these features give cancer cells an infinite replicative potential. In fact, oncogenic changes often promote cell cycle progression and bypass cell cycle checkpoints.[2] For example FoxM1 acts as a key positive regulator of cell cycle
progression and its overexpression is linked to oncogenesis; O-GlcNAcylation is implicated in the stability of FoxM1, so that reducing hyper-O-GlcNAcylation decreases levels of FoxM1 and consequently decreases cell cycle progression.[6]
stresses, proteotoxic stresses, hypoxia) which may trigger cell death in non-transformed cells. However, cancer cells evolve mechanisms to cope with these stresses and to resist cell death.[2] This is another hallmark of cancer and there are several evidences that
GlcNAcylation in cancer may play an anti-apoptotic role. Besides,
hyper-O-GlcNAcylation can contribute to face the stress generated by reactive oxygen species (ROS); ROS can be generated either endogenously by normal aerobic metabolism or derived exogenously from the extracellular environment. Excessive ROS production leads to oxidative stress, which can lead to damages of DNA, lipids and proteins. The relationship between oxidative stresses and cancer is complex, because cancer cells increase oxidative stresses due to many reasons and oxidative stress contributes to oncogenesis; however, excessive oxidative stress in cancer cells must be combated to avoid cell death. One potential connection between cancer cell protection against oxidative stress and hyper-O-GlcNAcylation is PFK1, the rate limiting enzyme in glycolisis. Reduced glutathione (GSH) is very important to combat oxidative stress and the co-factor NADPH plays a critical role to maintain pools of reduced GSH. O-GlcNAcylation of PFK1 inhibits its activity in cancer cells, decreasing rates of flux through glycolysis and increasing shunt of fructose-6-phosphate into the PPP, which has as direct consequence the increased generation of NADPH. Thus, hyper-GlcNAcylation contributes, through the increased production of NADPH, to maintain a pool of reduced GSH to combat ROS-induced cell death.
Another typical feature of cancer tissues is the induction of angiogenesis during tumorigenesis.[2] Angiogenesis is not only essential for delivery of oxygen and nutrients to
the interior of a tumor, but also contributes to allowing cancer cells to invade surrounding tissues and to disseminate to distant organs. Proliferation and differentiation of endothelial cells are linked to a lot of pro-angiogenic factors, such as Vascular Endothelial Growth Factors (VEGFs) and Fibroblast Growth Factor (FGFs). Recent evidence indicates that tumor angiogenesis may in part be supported by O-GlcNAcylation: reducing
hyper-O-GlcNAcylation in cancer cells by knocking down OGT expression inhibits the
enhancing the activity of the metalloproteinases.
One of the most important hallmark of cancer cells is their ability to invade other tissues and to form metastasis, which are the cause of over 90% of cancer mortality.[2] Emerging
evidences suggest that hyper-O-GlcNAcylation in cancer may be involved in tumor invasion and metastasis. Until now the mechanisms of how hyper-O-GlcNAcylation may regulate tumor invasion and metastasis are not completely understood, also because different cancer types may utilize distinct and/or overlapping tumor invasion mechanisms.
At the end, we can't forget that hyper-O-GlcNAcylation can contribute to metabolic reprogramming in cancer, in fact this post-translational modification can modify several glycolytic enzymes, determining their up-regulation.
1.2 A natural glyconiugate: Bleomycin
On the basis of these experimental evidences, we can affirm that N-acetylglucosamine is a very interesting sugar moiety, which can be added to new potential anticancer agents. In fact, it has been widely demonstrated that cancer cells have got increased energetic needs, so that they catch more glucose in respect to non-transformed cells. Therefore there is an always increasing interest towards new potential anticancer drugs, which present a sugar moiety, because they result more selective for cancer tissues and consequently they cause less side-effects. Besides, we can find a higher concentration of these anticancer drugs in cancer cells rather than in normal tissues, because the sugar moiety is recognized as an energy source by transformed cells. These aspects are not negligible, because current cancer therapies are often limited by side effects resulting from a lack of tumor cell selectivity. Thus, there is a great interest in molecules that can distinguish between healthy and cancerous tissues. A few such compounds have been described, but they are generally polypeptides or polysaccharides; however, their size can limit their pharmacological utility and the prospect for successful utilization in classic therapeutic strategies would increase if the size of these tumor-targeting molecules could be reduced. Indeed smaller compounds tend to be more readily amenable to use in prodrug conjugates.
In the literature we can find several examples of molecules with anticancer activity that are characterized by a sugar moiety. One example is Bleomycin:[8] the bleomycins are a
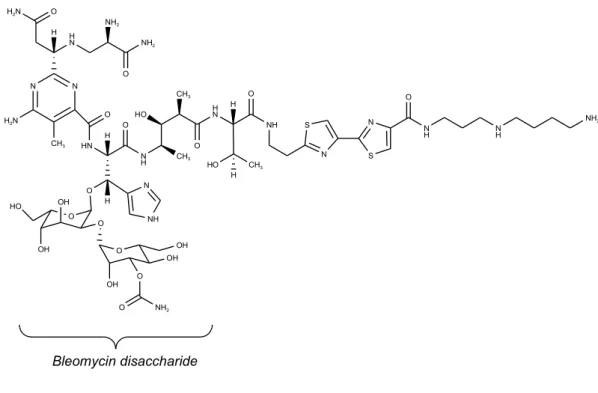
family of glycopeptide-derived antitumor antibiotics used clinically for the treatment of squamous cell carcinoma and malignant lymphomas. In addition to its antitumor activity, Bleomycin has been recognized for its ability to target tumors and it has been demonstrated that the disaccharide moiety of Bleomycin is critical for the selective tumor cell targeting of this molecule. The identification of the molecular elements in Bleomycin responsible for tumor cell targeting allows not only the exploration of analogues with improved properties but also the selective delivery of other drugs to tumor cells. The disaccharide moiety of Bleomycin consists of L-gulose linked to 3-carbamoylmannose, as depicted in Figure 1.1.
This anti-cancer molecule was studied in cell culture: firstly it was shown that Bleomycin selectively targeted human breast carcinoma cells, but not the normal breast cell line. Furthermore, it was found that the analogue deglycobleomycin, which lacks of the disaccharide moiety, did not target either cell line. Then, the disaccharide of Bleomycin was synthesized and coupled to a commercially available linked (the cyanine dye Cy5); Bleomycin and deglycobleomycin were conjugated to the cyanine dye Cy5 too. It was
Figure 1.1: Structure of Bleomycin
N N H2N H2N O CH3 H H N NH2 O NH2 HN O N H O H O O H N NH O CH3 HO CH3 H CH3 HO H S N N S N H O N H NH2 O OH HO OH O O OH OH O OH O NH2 H N NH Bleomycin disaccharide
bound selectively to cancer cells and were internalized. These results establish that the Bleomycin disaccharide is both necessary and sufficient for tumor cell targeting. This experiment has revealed the importance of this uncomplicated dysaccharide, which can selectively target a variety of cultured cancer cells. The implications of these findings for cancer therapy are clear: incorporation of this sugar moiety into preexisting or novel cancer therapeutics could increase drug delivery directly to the tumor cells, lowering the necessary dosage and potentially reducing side effects.
1.3 Dual targeting of the Warburg Effect using a
Glucose-conjugate of LDH-A inhibitor
1.3.1 Warburg effect
The research group where I carried out my thesis has taken the case of Bleomycin as a model in order to synthesize a glico-conjugate as a potential anticancer compound; this new potential anticancer agent has been planned on the basis of some observations about solid tumors. In fact, nearly a century ago, German scientist Otto Warburg observed that solid tumors deviate from most normal tissues because they are characterized by an enormous consumption of glucose and high rates of aerobic glycolysis.[3] These features
represent an advantage for the survival of cancer cells, because they allow them to survive in normoxic or hypoxic environments. Hypoxia is a condition characterized by the insufficient oxygenation of the tissues; this condition happens when the oxygen tension is low, but in cancer cells hypoxia can became a physiologic condition, even in presence of an elevated oxygen tension. Indeed the rapid cell proliferation typical of malignancies is not accompanied by a equally rapid growth of blood vessels. Thus, the tissues which are further from the blood vessels, do not receive the amount of oxygen they need to survive and to proliferate. Tumor hypoxia can seem an advantage in order to find an anticancer therapy, as cancer cells have got high energy needs to survive and to proliferate and the low oxygen concentration can not supply enough energy. Actually, cancer cells located in hypoxic tissues are highly resistant to the most common therapies, such as radiotherapy and chemotherapy; they also increase their aggressiveness and invasion capacity. The
resistance to chemotherapy is due to the lacking blood flux through tumor tissues, since drugs are generally spread by blood vessels; instead the resistance to radiotherapy depends on the low O2 concentration in the tissues. The low oxygen concentration in cancer tissues
is one of the principal causes of the Warburg effect, which explains the switch of cancer cells to a glycolytic metabolism. This metabolic reprogramming is kept, even if the normal levels of oxygen are replaced, and this fact demonstrates that the glycolitic process is not only a temporary answer to the situation of hypoxia, but it is a real genic alteration which makes cancer cells more aggressive and invasive.
1.3.2 LDH-A inhibitors as new potential anticancer agents
The molecular mechanism underlying the Warburg effect has explained that tumor cells overexpress the glucose transporter GLUT-1[9] and enzymes of glycolysis, including
Lactate Dehydrogenase Isoform A (LDH-A),[10] which has emerged as a new potential
anticancer target. This enzyme plays a very important role in the glycolytic process, indeed it catalyzes the conversion of piruvate to lactate, with the contemporary oxidation of NADH to NAD+ and new molecules of NAD+ are necessary in order to let glycolysis
happen. Much of the lactate produced in this reaction is excreted into the tumor microenvironment, acidifying it and limiting immune system access to tumor tissue. Overexpression of LDH-A has been noted in numerous solid tumors; thus, recently, a research group of our Department reported the discovery of N-hydroxyindole (NHI) based LDH-inhibitors as anticancer agents. NHI inhibitors are attractive candidates because of their facile synthesis, selective toxicity towards cancer cells and in vitro and in cell-culture efficacy.[11]
NHIs inhibitors constitute for these reasons an outstanding compound class to demonstrate the concept of dual targeting of the Warburg effect by linking glucose to a glycolytic enzime inhibitor. The presumed mechanism is illustrated in Figure 1.2.
The considered NHIs inhibitors are reported below:
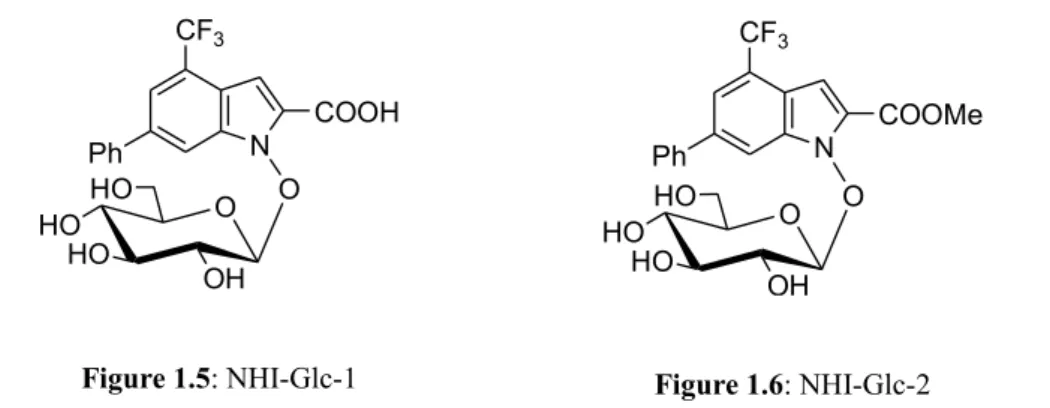
In effort to enhance tumor cell selectivity and efficacy of NHI-1 and NHI-2, glucose conjugates NHI-Glc-1 and NHI-Glc-2 were synthesized and evaluated:[12]
In vitro evaluation with LDH-A revealed that non-conjugated NHI-1 and NHI-2 and their glucose-conjugated derivatives are competitive inhibitors of LDH-A by binding to the NADH binding pocket; conjugation with the sugar moiety lowered inhibitory potency twofold for NHI-Glc-1 and sevenfold for NHI-Glc-2. Evaluation of these compounds
Figure 1.5: NHI-Glc-1 O HO HO HO OH N COOH CF3 Ph O Figure 1.6: NHI-Glc-2 O HO HO HO OH N COOMe CF3 Ph O Figure 1.3: NHI-1 N COOH CF3 OH Ph Figure 1.4: NHI-2 N COOMe CF3 Ph OH
Figure 1.2: Dual targeting of the Warburg effect by glucose conjugated
against different types of cancer cells, which generally overexpress LDH-A and GLUT-1, demonstrated that NHI-Glc-1 was inactive; however NHI-Glc-2 had three- to fivefold and six- to ninefold enhanced potency over NHI-2 and NHI-1 respectively.
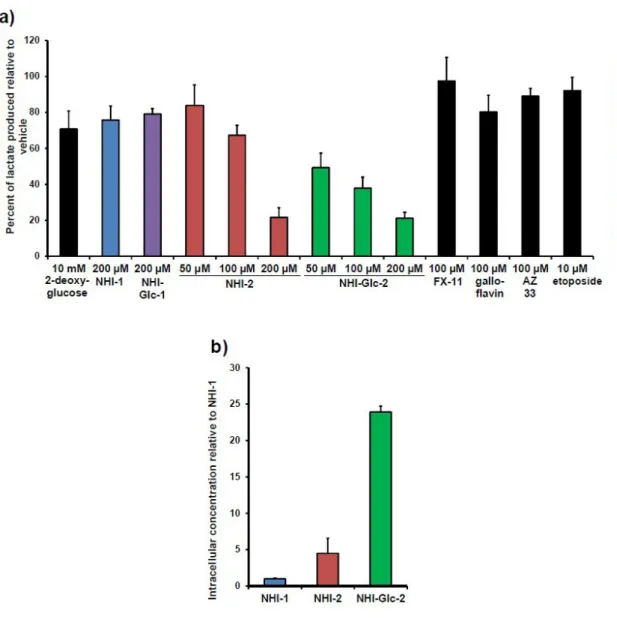
To assess efficacy in inhibiting lactate production in cells, HeLa cells were treated with varying concentration of NHIs and other reported LDH-A inhibitors; after 8 hours of treatment, the lactate content in cell culture was quantified by GC-MS. According to the proliferation assay results, NHI-Glc-1 had only modest effect at 200 μM, similar to its aglycone NHI-1. In contrast, treatment with NHI-Glc-2 led to significant, dose-dependent reduction in cellular lactate production (more potent than the aglycone NHI-2).
To test if the enhanced cancer cell toxicity and lactate production inhibition by NHI-2 and NHI-Glc-2 was due to enhanced cell uptake, the ability of these compounds to penetrate A549 cells was evaluated. Thus A549 cells were treated with compound or vehicle for 4 h and cell lysates were subjected to LC-MS analysis. In lysate from A549 cells, NHI-2 was present in about 4-5-fold higher concentration than NHI-1 and NHI-Glc-2 was approximately 24-fold higher:
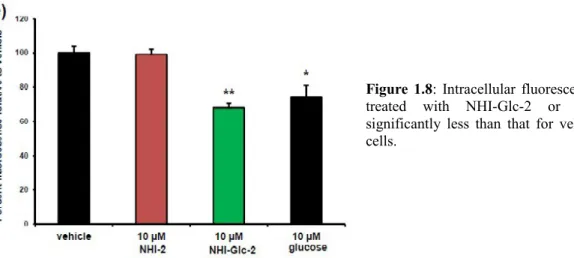
To examine whether NHI-Glc-2 enters cells via GLUT-transporters, a competition assay was performed between NHI-Glc-2 and GB2-Cy3, a fluorescent Cy3-linked glucose bioprobe. During this assay, A549 cells, which highly express GLUT-1, were treated for 30 minutes with 2.5 μM GB2-Cy3 in the presence of either vehicle or 10 μM 2, NHI-Glc-2 or glucose. Treatment with NHI-NHI-Glc-2 and glucose caused a statistically significant decreased in fluorescence, whereas treatment with NHI-2 did not. This evidence suggests that NHI-Glc-2 competes with GB2-Cy3 for cellular entry through GLUT- transporters.
Figure 1.7: A) Results of treatment of HeLa cells with various compound, showing that NHI-Glc-2 reduces
lactate production more efficiently that NHI-2. B) Intracellular concentration of NHIs inhibitors in A549 cells.
In summary, NHI-Glc-2 is the first compound with a dual targeting of the Warburg effect; NHI-Glc-2 has improved potency against cancer cells and shows increased cell permeability compare to the aglycone NHI-2. The modest reduction in inhibition potency on isolated enzyme is highly compensated for by improved cellular uptake via GLUT-transporters.
1.4 Reactivity of ManNAc-oxazoline with phenols
On the basis of our knowledge about GlcNAcylation, we have tried to synthesize O-GlcNAc-NHI-2. In the synthetic strategy we followed (see Results and Discussion, Chapter 2), the glycosyl-donor of the glycosylation reaction is the GlcNAc-oxazoline 1.15, which is easily obtained by a commercially available precursor, the 2-deoxy-2-amino-α-D
-glucopyranose chloridrate. The reactivity of this oxazoline has been widely studied: this compound reacts with nucleophiles exclusively on C(1) carbon, according to a SN2
mechanism, and the ideal condition of the reaction is using an acid catalyst (especially a Lewis acid) in a solvent as CH2Cl2 or DCE and at high temperature (Scheme 1.1).
Oxazolines are often used as glycosylating agent in the preparation of glycoproteins, various O-glycosides and oligosaccharides. Indeed, in the literature a variety of examples of this glycosylation of alcohols are described: in presence of different catalysts the oxazoline gives the desired product in reactions with aliphatic, little-sized alcohol.[13]
Figure 1.8: Intracellular fluorescence of cells
treated with NHI-Glc-2 or glucose is significantly less than that for vehicle-treated cells.
Conversely, glycosylation of phenolic -OH has not been accomplished so far; this kind of reaction is very interesting for our aim, because phenols have some features which make their reactivity similar to the reactivity of the NHI based inhibitors. A rare example of reaction with a phenol has recently been reported:[14] in this particular case the
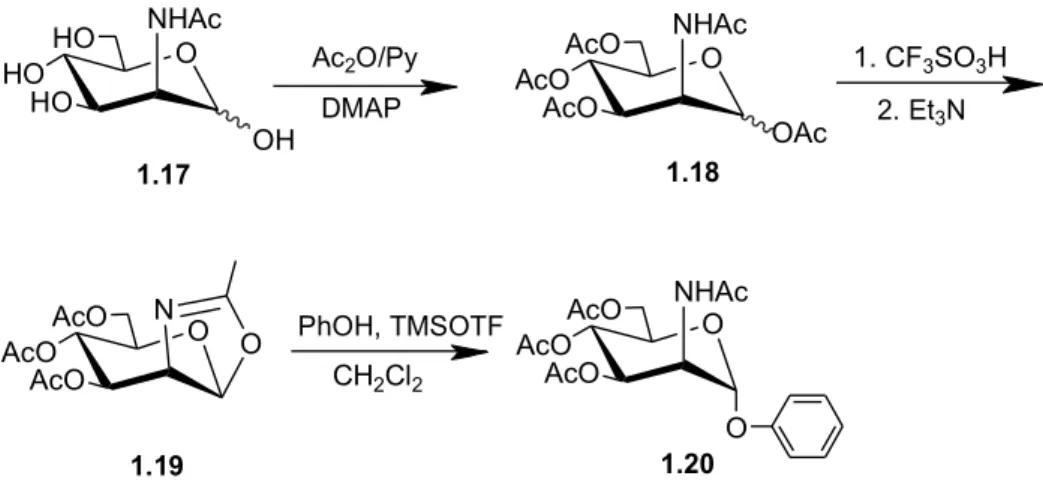
glycosylation of phenol employs ManNAc-oxazoline, which is synthesized starting from the commercially available 2-acetamido-2-deoxy-D-mannopyranoside, that is firstly
converted to its peracetate.
The glycosylation of phenol with oxazoline with β-manno configuration was extensively tested by the authors of the research we have considered; in fact they tested several reaction conditions, including variation of the catalyst and solvent, without success. At the end, they found that trimethylsilyl trifluoromethanesulfonate in dichloromethane was able to catalyze the oxazoline ring opening with phenol, in order to afford α-phenyl glycoside in 56% yield (Figure 1.9). The same reaction was also tested with p-nitrophenol, but the desired α-nitrophenyl glycoside was produced under these conditions only in trace amounts. This result indicates that the deactivation of phenol by the electron-withdrawing nitro-group decreases its reactivity in the glycosylation reaction with oxazoline, while previously published results[15] showed that the presence of electron-donating groups
increases the yields.
Scheme 1.1: General reaction of an alcohol with Glc-NAc-oxazoline 1.15.
O AcO AcO AcO N O O AcO AcO AcO AcHN R-OH cat 1.15 Nucleophile conjugated with the N-Acetylglucosammine group. CH2Cl2
Figure 1.9: Synthetic pathway of the synthesis of phenyl 2-acetamido-2-deoxy-α-D -mannopyranoside 1.20. O HO HO HO NHAc OH O AcO AcO AcO NHAc O AcO AcO AcO N O O AcO AcO AcO NHAc O OAc CH2Cl2 1.17 1.18 1.19 1.20 Ac2O/Py DMAP PhOH, TMSOTF 1. CF3SO3H 2. Et3N
2. RESULTS AND DISCUSSION
The encouraging results about the anticancer activity of Glc-NHI-2 (Figure 1.6) incited us to undertake the synthesis of the Glc-NAc conjugate 1.12, in order to study if the presence of the N-acetylglucosamine group could increase the selectivity and the affinity of the drug for tumor cells.
The first approach followed to synthesize the desired conjugate 1.12 is illustrated in retrosynthetic Scheme 1.2.
Tri-O-PMB-glucal (1.8) can undergo an interesting process of acetamidoglycosylation[16]to obtain, in a one-pot procedure, the Glc-NAc N-hydroxyindole
(NHI) conjugate 1.11. Then the glycoconjugate 1.11 should be deprotected on PMB-O-functionalities, under oxidant conditions (DDQ), to give product 1.12.
Acetamidoglycosylation is an attractive procedure, for nitrogen transfer, that efficiently effects the directly installation of the native C(2)-N-acetylamino functionality onto glycal donors and, additionally, allows glycosidic coupling with various glycosyl acceptors, in an overall one pot acetamidoglycosylation procedure (Scheme 1.3).
Scheme 1.2: Rethrosyntetic approach to conjugate 1.12.
O HO HO HO NHAc N COOMe CF3 Ph O O PMBO PMBO PMBO NHAc N COOMe CF3 Ph O 1 .11 O PMBO PMBO OPMB 1.8 1.12
In this protocol a new solfonium reagent derived from combination of thianthrene-5-oxide and triflic anhydride, is employed for glycal activation.
Such a transformation, as carefully confirmed by 1H and 13C NMR studies at low
temperature, also using labeled isotopes as 15N and 18O,[17] proceeds by way of the reaction
pathway illustrated in Scheme 1.3, in which the initial step involves low-temperature activation of thianthrene-5-oxide with triflic anhydride to generate thianthrene bis(triflate)
1.2 in situ. Electrophilic activation of the enol ether functionality in 1.1 by 1.2 leads to the
formation of a pyranoside intermediate (1.3) incorporating an oxocarbenium triflate functionality at C(1) and a thianthrene solfonium moiety at C(2). The introduction of the
Scheme 1.3: Acetamidoglycosylation of generic glycal 1.1.
O RO RO RO S O Me N SiMe3 O OR RO RO N O Me + + TfO
-[PhEt2NSiMe3]+[TfO]
-O OR RO RO Tf2O, CH2Cl2/CHCl3 1.1 S S AcNHSiMe3, Et2NPh S O RO RO RO S H + 2TfO - S + 1.3 1.4 S S 1.5 OTf + -1.2 OTf O OR RO RO 1.60 Nu NHAc NuH acid
TMS-acetamide. A key function of the N-trimethylsilyl protecting group in this amide reagent is to provide steric shielding at the nitrogen atom to favour initial addition of the amide oxygen atom onto C(1) of the activated glycal, thereby generating the putative acetimidate intermediate 1.4. Subsequent intramolecular displacement of the C2-thianthrene-sulfonium moiety by the imidate nitrogen accompanied by loss of the N-TMS protective group (either as TMSOTf or more likely as its N,N-diethylaniline adduct) generates the bicyclic dihydrooxazole (oxazoline) intermediate 1.5. Acid-mediated oxazoline ring-opening in the presence of glycosyl acceptor affords then the desired glycoside 1.60 in the final stage of the acetamidoglycosylation.
With this background, we tried to synthesize the Glc-NAc-NHI-2 conjugate 1.12 as shown in Scheme 1.4.
Commercially available tri-O-acetyl-D-glucal 1.6 was fully saponified with MeONa/MeOH, affording D-glucal 1.7. Alkylation of 1.7 with p-methoxybenzyl chloride
and sodium hydride in DMF for 12 h at room temperature afforded compound 1.8, which
Scheme 1.4: Acetamidoglycosylation of glucal 1.7 with NHI-2 1.9. 1.6 O AcO AcO OAc MeONa MeOH 1.7 O HO HO OH NaH, DMF O PMBO PMBO OPMB 1.8 S S OTf 1.2 1) 2) TMSNHAc, PhNEt2 N COOMe CF3 Ph OH 3) 4) Acid catalyst NHI-2 1.9 O PMBO PMBO OPMB 1.10 O N CH3 O PMBO PMBO PMBO NHAc N COOMe CF3 Ph O 1.11 TfO DDQ compound 1.12 PMBCl
constitutes the starting material for our acetamidoglycosylation reaction. Protected glucal
1.8, in a mixture of CHCl3/CH2Cl2 in a 2:1 ratio, was treated with thianthrene and Tf2O at
-78 °C for 15 minutes. Then, N,N-diethylaniline and N-TMS acetamide were added sequentially and the reaction mixture was stirred at 23 °C for 30 minutes. Amberlyst-15 acidic resin and the glycosyl acceptor (NHI 1.9) were then added. Unluckily, 1H NMR of
the crude reaction mixture did not show any consistent glycosylation product.
Even when using different Lewis acids, such as MgI2, LiClO4 or TMSOTf, the
acetamidoglycosylation did not work.
Then we tried to isolate the stable oxazoline intermediate 1.10 prior to the introduction of the acid and the glycosyl acceptor, but only a very low yield (less then 5%) of the desired oxazoline product was obtained, thus demonstrating that the main problem was the formation of the PMB-O-protected oxazoline.
The necessity to introduce a PMB-ether functionality on the glucal system derives from the instability of the N-O bond under the reducing conditions used, for example, to remove benzyl ether protecting groups. On the other hand, ester groups (such as acetyl groups) are incompatible with the acetamidoglycosylation process.
In the light of these evidences we decided to change our synthetic approach.
Therefore, the synthetic pathway we decided to follow in order to synthesize the desired glyco-conjugate is depicted in retrosynthetic Scheme 1.5.
This approach is characterized by the formation of a key compound, the
Scheme 1.5: Rethrosynthetic approach to conjugate 1.12 by using oxazoline 1.15.
O HO HO HO NHAc N COOMe CF3 Ph O 1.12 O AcO AcO AcO NHAc N COOMe CF3 Ph O 1.16 O AcO AcO AcO N O 1.15
synthesis of 2-acetamido-2-deoxy-b-D-glucopyranosides. Therefore the first goal was the synthesis of Glc-NAc-oxazoline 1.15, which can quite easily be obtained using the synthetic pathway illustrated in Scheme 1.6.
The intermediate oxazoline 1.15 is obtained starting from a commercially available precursor, the D-Glucosamine hydrochloride 1.13, which was subjected to the conventional
peracetylation reaction, using Ac2O/Py in a ratio of 1:2. Actually, the peracetylated
glucopyranose we obtained at the end of this reaction is also a commercially available product, but it is a bit expensive; thus, since the peracetylation reaction proceeds with elevated yield, we decided to start from the cheaper D-Glucosamine hydrochloride 1.13
and then to convert it into the α-peracetyl-D-glucosamine 1.14.
Subsequently Glc-NAc-oxazoline 1.15 was efficiently prepared by means of a particularly good methodology, based on the use of TMSOTf as acid promoter of the reaction; the employed solvent was anhydrous DCE and the reaction mixture was heated at 50 °C. Also this reaction proceeds with elevated yields and can bring to the synthesis of multigrams of oxazoline. The reason of its efficiency bases on the interesting reaction mechanism: the acetamide group at C(2) is an excellent neighbouring group, so the rate of this reaction results enhanced because of an anchimeric assistance phenomenon. Anchimeric assistance (also called Neighbouring Group Participation or NGP) is defined as the interaction of a reaction centre with a lone pair of electrons in an atom; in the case we are examining anchimeric assistance performs in the way represented in Scheme 1.7.
Scheme 1.6: Synthetic pathway for synthesizing Glc-NAc-oxazoline 1.15.
O HO HO HO H3N OH 1.13 Cl O AcO AcO AcO AcHN 1.14 1.15 OAc O AcO AcO AcO N O TMSOTf DCE 50 °C Ac2O Py
The loss of acetyl group on the anomeric position of α-peracetyl-D-glucosamine 1.14 as
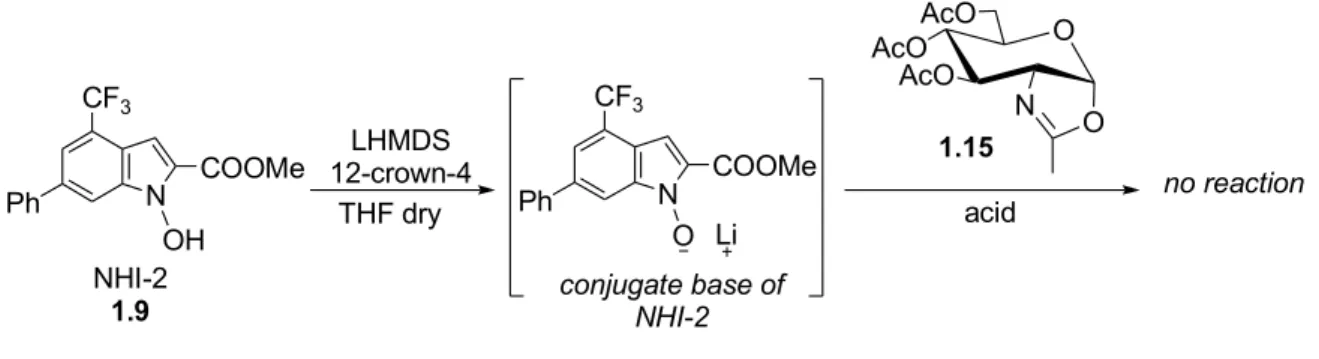
a consequence of the activation by TMSOTf is assisted through the mechanism of anchimeric assistance by the acetamide group at C(2); the cyclic carbocation that is formed following this activation loses a proton and it is converted into the stable α-oxazoline 1.15. As it has already been accurately explained (see Introduction, Chapter 1), compounds with α-oxazoline moiety play an important role in carbohydrate chemistry, because they are often used as glycosyl-donor in glycosydic-bond forming reactions. Indeed, the opening-reaction of α-oxazoline 1.15 with many nucleophiles (driven in chlorinated solvents, high temperatures and with participation of various catalysts) is regioselective at C(1) and affords 1,2-trans-glycosides in completely stereoselective way. The nucleophile we used in the ring-opening reaction is the Lactate Dehydrogenase Inhibitor with N-hydroxyindole-based structure NHI-2 1.9 (see Figure 1.3, Chapter 1). The crucial reaction of the synthetic pathway to Glc-NAc-NHI-2 is right the ring-opening reaction of α-oxazoline 1.15 with NHI-2 1.9; this synthetic step appeared problematic, especially for two reasons:
• NHI-2 1.9 has got a very low reactivity as nucleophile; furthermore the N-O bond is labile, so that in many reaction conditions the reduced product derived from NHI-2 1.9 is recovered.
Scheme 1.7: Mechanism of oxazoline's formation involving a process of anchimeric assistance.
O AcO AcO AcO AcHN 1.14 OAc TMSOTf DCE 50 °C O AcO AcO AcO HN O O AcO AcO AcO HN O O AcO AcO AcO N O H 1.15 O AcO AcO AcO N O -H+
Our first attempts to the synthesis of the 1,2-trans-glycoside 1.12 aimed to enhance the nucleophilic character of NHI-2 1.9, because the research group where I performed my thesis had successfully used this approach in order to synthesize the carba analogue of Glc-NHI-2.[18] Therefore, the first procedure we followed presupposed to convert NHI-2 1.9
into its conjugate base; the reaction conditions are depicted in Scheme 1.8.
NHI-2 1.9 is dissolved in dry THF, then it is treated with LHMDS and 12-crown-4; LHMDS is a strong bulky base and for this reason has not nucleophilic character, so that it can salificate the hydroxy group of NHI-2 1.9 without causing secondary undesired reactions. 12-crown-4 is a crown ether, that is a tetramer of ethylene oxide, which has high affinity for lithium cations; LHMDS forms in presence of crown ether 12-crown-4 an intimate ion pair, constituted by the chelate system ether-Li+ and by HMDS-, which gains
more basic character. The described chelate system is represented in Figure 1.10.
The obtained lithium salt of the nucleophile is not isolated, but directly treated with α-oxazoline 1.15. An interesting feature of this experimental procedure is that it did not
Scheme 1.8: First tested procedure to perform the ring-open reaction of oxazoline 1.15.
Figure 1.10: Intimate ion pair between 12-crown-4 and LHMDS.
N COOMe CF3 Ph OH LHMDS 12-crown-4 THF dry N COOMe CF3 Ph O Li conjugate base of NHI-2 NHI-2 1.9 1.15 acid no reaction O AcO AcO AcO N O O O O O LHMDS O O OLi O Si N Si
require the use of an excess of NHI-2, indeed α-oxazoline 1.15 and NHI-2 1.9 are in a 2:1 ratio. The reaction conditions presuppose the use of a catalyst. We tested two different types of acid-catalysis: in one case we added 0.2 equivalents of a Lewis acid, Cu(OTf)2, in
the other case, we used acidic resin Amberlyst 15. Both reaction conditions did not afford the desired product; besides, the crude mixture derived from these reactions was subjected to 1H NMR experiment and the obtained experimental data pointed out that glycosylation
had not occurred and that the only products we could recuperate were NHI-2 1.9 and its reduced derivative. These evidences confirmed an already known feature of NHI-2 1.9, that is the frailty of the N-O bond, which can be reduced in a light-hasted process.
Thus, we decided to change the synthetic strategy and to orient our attention towards the classical conditions of oxazolines opening-reactions. In the literature these reactions are reported to occur in chlorinated solvents and in presence of an acid-catalysis, especially at high temperatures.
The first reaction-conditions we tested are depicted in Scheme 1.9.
Firstly, in a schlenk, under argon atmosphere, we dissolved α-oxazoline 1.15 in anhydrous DCE, then 0.5 equivalents of CSA as catalyst and, finally, 2.5 equivalents of nucleophile were added to the resulting solution. The reaction mixture was heated to 80 °C for 24 h, then we observed the disappearance of the starting material; however, in the washed organic solution we did not found the desired Gluc-NAc-conjugate, as it was confirmed by 1H NMR data.
To obviate the low reactivity of the glycosyl acceptor, we tested another kind of
Scheme 1.9: Attempt of α-oxazoline 1.15 ring-opening reaction in presence of CSA as catalyst. 1.15 O AcO AcO AcO N O CSA NHI-2 DCE dry N COOMe CF3 Ph OH NHI-2 1.9 no reaction
The reaction was performed in anhydrous dichloroethane, in presence of molecular sieves 4 Å and using 0.5 equivalents of TMSOTf. In this case oxazoline 1.15 and NHI-2
1.9 are used in a 1:1.5 ratio, therefore these conditions appeared very promising because
they promoted glycosylation reaction without needing a large excess of nucleophile. The reaction was performed first of all at room temperature, without affording the desired compound 1.16; we observed that at room temperature NHI-2 1.9 did not completely dissolve in dichloroethane, thus we decided to heat reaction-mixture to 80 °C in order to favour the complete dissolution of NHI-2 1.9. After 24 h we verified the disappearance of the starting compound 1.15 and the formation of a new product with lower Rf in a very
polar eluant (CHCl3/acetone 9:1); the obtained crude mixture was subjected to flash
chromatografy to afford the peracetyl Glc-NAc-conjugate 1.16 (yield 40%). 1H NMR of
compound 1.16 shows the presence of four acetyl groups at δ 2.07, 2.03, 2.02 and 1.77, different from the chemical shifts reported for oxazoline 1.15 (2.07, 2.05, 2.04 and 2.00), and the presence of anomeric proton at δ 5.52 with J = 9.05.
Compound 1.16 was also synthesized as illustrated in Scheme 1.11.
Scheme 1.10: Ring-opening reaction of α-oxazoline 1.15 with TMSOTf (procedure a). 1.15 O AcO AcO AcO N O molecular sieves 4 TMSOTf Å 80 °C DCE dry O AcO AcO AcO NHAc N COOMe Ph O 1.16 peracetyl Glc-NAc conjugated N COOMe CF3 Ph OH NHI-2 1.9
α-oxazoline 1.15 was dissolved in freshly distilled diethyl ether, then an excess of NHI-2 1.9 (3.0 equiv) was added at 0 °C to the resulting solution. Finally we added BF3·Et2O as
Lewis acid. The reaction mixture was stirred at room temperature for 48 hours; purification of the crude mixture by preparative TLC afforded compound 1.16. These reaction conditions are characterized by the advantage of working at room temperature, but at the same time, we spotted two disadvantages, that is the use of an excess of nucleophile and a lower yield (10%) compared with the procedure employing TMSOTf. For these reasons we preferred using TMSOTf as catalyst, in order to obtain a greater amount of compound 1.16. Summarizing that, we tested various reaction conditions in order to obtain α-oxazoline
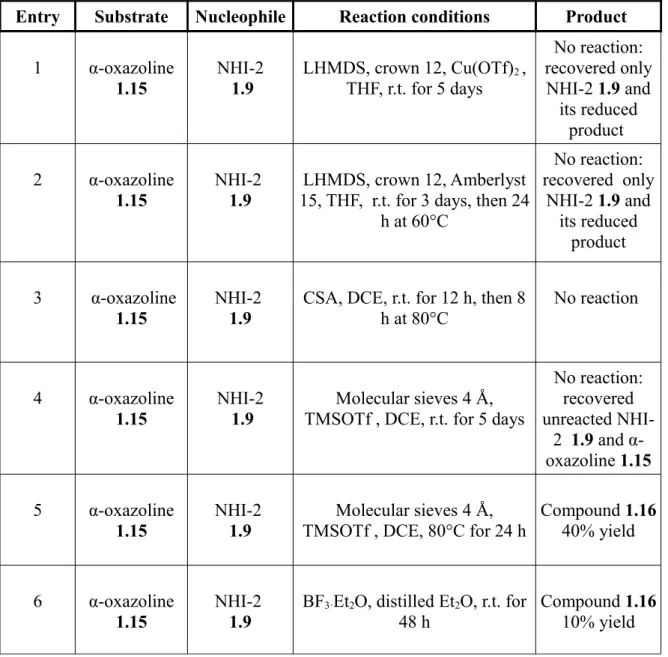
1.15 opening-reaction, which are illustrated in below reported Table 1.1.
Scheme 1.11: Ring-opening reaction of α-oxazoline 1.15 with BF3·Et2O(procedure b).
1.15 O AcO AcO AcO N O distilled Et2O O AcO AcO AcO NHAc N COOMe CF3 Ph O 1.16 peracetyl Glc-NAc conjugated N COOMe CF3 Ph OH NHI-2 1.9 BF3.Et2O r.t., 48 h
Entry Substrate Nucleophile Reaction conditions Product 1 α-oxazoline 1.15 NHI-2 1.9 LHMDS, crown 12, Cu(OTf)2 , THF, r.t. for 5 days No reaction: recovered only NHI-2 1.9 and its reduced product 2 α-oxazoline 1.15 NHI-2 1.9 LHMDS, crown 12, Amberlyst 15, THF, r.t. for 3 days, then 24
h at 60°C No reaction: recovered only NHI-2 1.9 and its reduced product 3 α-oxazoline 1.15 NHI-2 1.9
CSA, DCE, r.t. for 12 h, then 8 h at 80°C
No reaction
4 α-oxazoline
1.15 NHI-2 1.9 TMSOTf , DCE, r.t. for 5 daysMolecular sieves 4 Å,
No reaction: recovered unreacted NHI-2 1.9 and α-oxazoline 1.15 5 α-oxazoline
1.15 NHI-2 1.9 TMSOTf , DCE, 80°C for 24 hMolecular sieves 4 Å, Compound 1.1640% yield
6 α-oxazoline
1.15 NHI-2 1.9 BF3·Et2O, distilled Et48 h 2O, r.t. for Compound 1.1610% yield
Table 1.1: Reaction-conditions tested to obtain the peracetyl-Glc-NAc conjugate 1.16.
As reported, most of our attempts were unsuccessful: in two attempts we recovered NHI-2 1.9 and reduced NHI-2 (entries 1 and 2); in entry 4, NHI-2 1.9 and α-oxazoline did not react, so that we could found in the crude mixture both unreacted compounds. Finally, we found two different kinds of conditions (entries 5 and 6) which afforded the desired glycosylation product 1.16, even if with significantly different yields.
The last synthetic step we carried out was the alkaline hydrolysis of acetyl groups; acetyl groups are generally removed by a classical reaction with MeONa in methanol as
solvent. However, compound 1.16 presented a relevant problem, which determined saponification classic reaction conditions not to be ideal. Indeed compound 1.16 is poorly soluble in methanol (as in other protic solvents) at room temperature and this feature required reaction conditions to be modified. In particular, we dissolved compound 1.16 in a mixture of CH2Cl2/MeOH in 2:3 ratio, in order to obtain its complete dissolution. Then we
treated the resulting mixture with a freshly prepared solution 0.33 M of MeONa in MeOH. After 4 hours at room temperature, we observed the formation of the desired product 1.12; as depicted in Scheme 1.12.
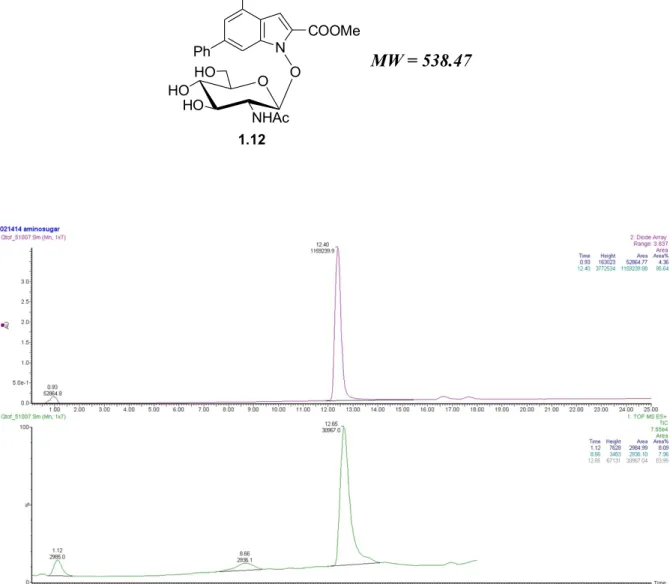
Compound 1.12 has a typical feature, in fact it is soluble in MeOH at high temperature, but at room temperature it tends to precipitate as pail yellow solid. Thus, it was purified by crystallization from MeOH, affording the Glc-NAc conjugate 1.12, chemically pure, as NMR data (1H NMR, 13C NMR, HSQC) confirm. The purity of compound 1.12 has been
further verified by UV analysis at 254 nm and LC-MS analysis (see Appendix, Chapter 5). Thus, the first part of my thesis ended with this important result, that is the completely stereoselective synthesis of Glc-NAc conjugate 1.12. This compound will now be subjected to biological assays at our Department and to cells assays at University of Urbana-Illinois in order to verify if the presence of the N-acetylglucosamine moiety determines an enhanced anticancer activity and a greater affinity and selectivity for cancer cells, compared to the previous derivatives NHI-2 1.9 and Glc-NHI-2.
Scheme 1.12: Alkaline hydrolysis of acetyl groups.
O AcO AcO AcO NHAc N COOMe CF3 Ph O 1.16 MeONa/MeOH CH2Cl2/ MeOH HO O HO HO NHAc N COOMe CF3 Ph O 1.12
3. EXPERIMENTAL
General procedures: All reaction were performed in flame-dried modified Schlenk
(Kjeldhal shape) flasks fitted with a glass stopper or rubber septa under a positive pressure of argon. Air/and moisture-sensitive liquids and solutions were transferred via a syringe. Organic solution were dried on MgSO4 and concentrated by a rotary evaporator below 40
°C at ca. 25 Torr. Flash chromatography was performed employing 230-400 mesh silica gel. Analytical TLC was performed on Alugram SIL G/UV254 silica gel sheets
(Macherey-Nagel) with detection by 0.5% phosphomolybdic acid solution in 95% EtOH. Preparative TLC was performed on 0.25 or 0.5-mm Machery-Nagel DC-Fertigplatten UV254 silica gel
plates.
Materials: MeONa, tri-O-acetyl-D-glucal, PMBCl, 60% mineral oil dispersion NaH,
Tf2O, N,N-diethylaniline, N-(TMS)acetamide, Amberlyst 15, D-Glucosamine
hydrochloride, TMSOTf, BF3·Et2O, acidic resin Amberlite, 1.0 M LHMDS in THF,
12-crown-4, Cu(OTf)2, anhydrous DMF over molecular sieves, MeOH for HPLC, anhydrous
pyridine, anhydrous CHCl3, anhydrous CH2Cl2 over molecular sieves were purchased from
Aldrich and used without purification. Molecular sieves 4 Å were purchased from Fluka and used without purification. Et3N was distilled from CaH2. THF, Et2O, toluene were
distilled from sodium/benzophenone. Anhydrous DCE was distilled by microdistillation at ordinary pressure.
Instruments: Infrared (IR) spectra were obtained using a FTIR spectrophotometer. Data
are presented as frequency of absorption (cm-1). Specific rotations were measured with a
digital polarimeter with a 1 dm cell. Proton and carbon-13 nuclear magnetic resonance (1H
NMR and 13C NMR) spectra were recorded at 250 and 62.5 MHz respectively; chemical
shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and refer to residual protium in the NMR solvent (CHCl3: δ 7.26; CD3OD: δ 3.31; DMSO-d6: δ
2.50). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet and/or multiple resonances), integration, coupling constant in Hertz (Hz). Melting points are determined by Kofler apparatus and they are uncorrected.
3.1 Synthesis of 2-acetamido-2-deoxy-α-glucopiranoside via
acetamidoglycosilation
D-glucal
MeONa (0.050 g, 0.92 mmol) was added to a solution of tri-O-acetyl-D
-glucal 1.6 (4.0 g, 14.7 mmol) in MeOH (40.0 mL) and the resulting reaction mixture was stirred at room temperature for 5 h under argon atmosphere. Evaporation of the organic solvent afforded a product (2.0 g, 93% yield) consisting of 1.7, practically pure as a syrup: Rf = 0.27 (9:1 CH2Cl2/MeOH);
FTIR (neat film) 3398, 2926, 1645, 1413, 1230, 1074, 1015, 824, 735 cm-1. 1H NMR
(CD3OD) δ 6.26 (dd, 1H, J = 6.0, 1.6 Hz), 4.60 (dd, 1H, J = 6.0, 2.2 Hz), 4.04 (dt, 1H, J =
7.0, 1.9 Hz), 3.85-3.58 (m, 3H), 3.48 (dd, 1H, J = 9.5, 7.0 Hz). 13C NMR (CD
3OD) δ 146.5,
106.1, 81.9, 72.5, 72.1, 63.8. Anal.Calcd for C6H10O4: C, 49.31; H, 6.90. Found: C, 49.14;
H, 6.78.
3,4,6-Tri-O-(p-methoxybenzyl)-D-glucal
A solution of D-glucal 1.7 (2.0 g, 13.7 mmol) in anhydrous DMF (40.0 mL) was added dropwise at 0 °C to a washed (hexane 3x50 mL) suspension of 60% NaH in mineral oil (2.74 g, 65.8 mmol, 5.0 equiv) in anhydrous DMF (40.0 mL). After stirring at room temperature for 30 min, the suspension was cooled at 0 °C and p-methoxybenzilchloride (PMBCl) (6.1 mL, 45.2 mmol, 3.3 equiv) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for further 3 h. Dilution with Et2O and evaporation of the
washed (saturated aqueous NaCl) and dried (MgSO4) organic solution afforded
3,4,6-Tri-O-(p-methoxybenzyl) (PMB) derivative 1.8 (5.87 g, 85% yield), practically pure as a
yellow oil, which was used in the next step without any further purification: Rf = 0.20 (8:2
Hexane/AcOEt); 1H NMR (CDCl 3) δ 7.27-7.24 (m, 4H), 7.16-7.12 (m, 2H), 6.88-6.82 (m, 6H), 6.40 (dd, 1H, J = 7.5, 2,5 Hz), 4.84 (dd, 1H, J = 7.5, 2.5 Hz), 4.74 (d, 1H, J = 12.5 Hz), 4.59-4.46 (m, 5H), 4.18-4.14 (m, 1H), 4.05-3.98 (m, 1H), 3.80 (s, 3H), 3.80 (s, 3H), 1.7 O HO HO OH O PMBO PMBO OPMB 1.8
68.33, 55.44.
Typical procedure for C(2)-acetamidoglycosylation
Trifluoromethanesulfonic anhydride (120 μL, 0.72 mmol, 2.0 equiv) was added to a solution of 3,4,6-Tri-O-(p-methoxybenzyl)-D-glucal (0.180 g, 0.36 mmol, 1.0 equiv) and thianthrene-5-oxide (0.168 g, 0.72 mmol, 2.0 equiv) in a mixture of chloroform and dichloromethane (9.0 mL; 2:1 v/v) at -78 °C, the reaction mixture was stirred at this temperature for 10 min, then N,N-diethylaniline (229.0 μL, 1.44 mmol, 4.0 equiv) was added, followed by N-(TMS)acetamide (0.142 g, 1.08 mmol, 3.0 equiv). The mixture was immediately warmed to 23 °C and stirred at this temperature for 30 min. Then solid NHI-2
1.9 (0.240 g, 0.72 mmol, 2.0 equiv) was added. Amberlyst-15 acidic resin (0.140 g) was
then added and reaction stirred for 30 h. The mixture was filtered and concentrated under vacuum but the residue analyzed by 1H NMR did not show any consistent glycosylation
product.
3.2 Synthesis of 2-acetamido-2-deoxy-α-
D
-glucopiranoside
via Glc-NAc oxazoline opening reaction
2-acetamido-1,3,4,6-tetra-O-acetyl-2-deoxy-α-D-glucopyranoside
Commercially available D-Glucosamine hydrochloride 1.13 (4.00 g,
18.6 mmol, 1.0 equiv) was suspended in a 2:1 mixture of pyridine and Ac2O and the suspension was stirred 20 hours at room
temperature. Dilution with toluene (3x50 mL) and evaporation of the organic solution afforded a crude product (11.5 g), which was subjected to flash chromatography. Elution with 9:1 hexane/AcOEt mixture yielded compound 1.14 (7.4 g, 97% yield), pure as a white solid with physical and chemical properties consistent with the properties described in the literature:[19] R
f = 0.30 (AcOEt); 1H NMR (CDCl3): δ 6.17 (d, 1H, J = 3.6 Hz), 5.95 (d, 1H, J = 9.1 Hz), 5.23 (m, 2H), 2.09 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 1.94 (s, 3H). 13C NMR (CDCl 3): δ 171.4, 170.6, 170.1, 169.0, 168.6, 90.5, 70.4, 69.7, 67.4, 61.4, 50.8, 22.8, 20.7, 20.5. O AcO AcO AcO AcHN 1.14 OAc
2-amino-3,4,6-tri-O-acetyl-2-deoxy-1-O-N-(ethan-1-il-1-iliden)-α-D-glucopyranoside
A solution of compound 1.14 (14.0 g, 36.0 mmol, 1.0 equiv) in 168.0 mL of anhydrous DCE was stirred at 50 °C under inert atmosphere in a flame-dried shlenk. After 10 minutes acid catalyst TMSOTf (1.21 mL, 41.0 mmol, 1.14 equiv) was added and the resulting pink solution was stirred 20 hours at 50 °C. The reaction mixture was neutralized by Et3N
(2.0 mL), transferred in a round-bottom flask and the evaporation of the organic solution with addition of about 0.5 mL of Et3N afforded a crude product (26.5 g), which was
subjected to flash chromatography. Elution with hexane/AcOEt 1:9 + 0.1% of Et3N mixture
yielded compound 1.15 (13.4 g, yield 95%), pure as a yellow syrup with physical and chemical properties consistent with the properties described in the literature:[20] [α]20
D = +50 (c 1.4; CHCl3); Rf = 0.43 (CHCl3/MeOH 95:5); 1H NMR (CDCl3): δ 5.93 (d, 1H, J = 7.4 Hz), 5.22 (t, 1H, J = 2.4 Hz), 4.90 (bd, 1H, J = 9.41 Hz), 4.14-4,00 (m, 3H), 3.59-3.52 (m, 1H), 2.07 (s, 3H), 2.05 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H). 13C NMR (CDCl 3): δ 169.7, 168.7, 168.4 (3C), 165.8, 98.6, 69.5, 67.7, 66.7, 64.2, 62.6, 20.1, 19.9, 13.1.
NHI 2-acetamido-2-deoxy-3,4,6-tri-O-acetyl-β-D-glucopyranoside
Procedure a: activated molecular sieves 4 Å (0.215 g),
compound 1.15 (0.230 g, 0.69 mmol, 1.0 equiv) and NHI-2
1.9 (0.250 g, 0.75 mmol, 1.1 equiv) were dissolved in 3.5
mL of anhydrous DCE and the obtained mixture was stirred at 80 °C until the complete dissolution of NHI-2 1.9. Then Lewis acid TMSOTf (0.06 mL, 0.34 mmol, 0.5 equiv) was added at room temperature. The reaction mixture was heated at 80 °C and stirred overnight at the same temperature. Then the mixture was diluted with CH2Cl2 and filtered; the
filtered solution was neutralized by saturated aqueous NaHCO3 and washed with saturated
aqueous NaCl. Evaporation of the washed organic solution afforded a crude product (0.300 g) consisting of compound 1.16 and unreacted NHI-2 1.9, which was subjected to flash chromatography. Elution with a 9:1 CHCl3/Acetone mixture yielded compound 1.16 (0,140
1.15 O AcO AcO AcO N O O AcO AcO AcO NHAc N COOMe CF3 Ph O 1.16
freshly distilled Et2O; the resulting solution was cooled at 0 °C, then a solution of NHI-2
1.9 (0.090 g, 0.27 mmol, 3.0 equiv) in 0.3 mL of Et2O was added to the resulting reaction
mixture. Subsequently Lewis acid BF3·Et2O (8.0 μL, 0.063 mmol, 0.7 equiv) was added
and the reaction mixture was stirred for 48 h at room temperature. Evaporation of the washed (NaHCO3, saturated aqueous NaCl) organic solution afforded a crude product
(0.085 g). Purification by preparative TLC (CHCl3/Acetone 9:1) yielded compound 1.16
(0.020 g, 10% yield), pure as a pale yellow solid: mp 222-225 °C; [α]20
D = +26.9 (c 0.79; CHCl3); Rf = 0.3 (9:1 CHCl3/Acetone); 1H NMR (CDCl3): δ 8.10 (s, 1H), 7.74 (s, 2H), 7.63 (d, 2H, J = 7.7 Hz), 7.49-7.36 (m, 3H), 7.33 (s, 1H), 6.70 (d, 1H, J = 8.58 Hz), 5.52 (d, 1H, J = 9.05, H-1), 5.28-5.16 (m, 2H), 4.55 (q, 1H, J = 9 Hz), 4.26 (dd, 1H, J = 12.4, 4.68 Hz), 4.05 (s, 1H), 4.04 (d, 1H, J = 1.98), 4.00 (s, 1H), 3.95 (s, 3H), 3.75-3.69 (m, 1H), 2.07 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.77 (s, 3H). 13C NMR (CDCl 3): δ 171.0, 171.0, 170.6, 169.4, 161.0, 140.2, 140.0, 139.6, 129.2, 128.3, 127.4, 124.3 (q, J = 272.4 Hz), 123.77 (q, J = 33.0 Hz), 120.0 (q, J = 4.8 Hz), 119.03 (q, J = 4.7 Hz), 117.4 (q, J = 2.1 Hz), 114.3, 108.4, 105.8, 73.6, 72.5, 68.0, 61.8, 52.5, 52.4, 23.3, 20.8, 20.7, 20.4.
NHI 2-acetamido-2-deoxy-β-D-glucopyranoside
Compound 1.16 (56.5 mg, 0.087 mmol, 1.0 equiv) was dissolved in 4 mL of a mixture 2:3 of CH2Cl2 (1.6 mL) and
MeOH (2.4 mL) and cooled at 0 °C. 0.05 mL of a freshly prepared 0.33 M solution of MeONa/MeOH were added to the resulting solution and the reaction mixture was stirred 4 h at room temperature. Then the mixture was neutralized with acidic resin Amberlite and filtered. Evaporation of the filtered solution afforded 0.031 g of a crude product. Crystallization from MeOH of the crude yielded compound 1.12 (0.020 g, yield 50%), pure as a pale yellow solid: mp: 190-194 °C; [α]20
D = -36.9 (c 0,17; MeOH); Rf = 0.15 (9:1 AcOEt/MeOH); 1H NMR (DMSO-d 6): δ 8.15 (s, 1H), 8.06 (d, J = 9.0 Hz, 1H, NH), 7.91-7.82 (m, 3H), 7.55-7.43 (m, 3H), 7.11 (s, 1H), 5.21-5.18 (m, 2H, 2OH), 5.11 (d, J = 8.7 Hz, 1H, H-1), 4.41-4.38 (m, 1H, OH), 3.92 (s, 3H), 3.80 (dd, J = 18.7, 9.0 Hz, 1H), 3.67-3.41 (m, 4H), 3.19-3.11 (m, 1H), 1.94 (s, 3H); 13C NMR (DMSO-d 6): δ 170.0, 159.9, 139.5, 138.2, 137.9, 130.4, 129.6, 128.6, 127.8, 124.7 (q, J = 271.0 Hz), 122.4 (q, J = 32.3 O HO HO HO NHAc N COOMe CF3 Ph O 1.12
Hz), 119.4 (q, J = 4.7 Hz), 117.2 (q, J = 2.0 Hz), 114.2, 106.9 (C-1), 104.6, 77.5, 74.1, 70.2, 61.2, 54.4, 52.8, 23.8.
LC-MS analytical conditions:
• Waters X-bridge 2.1 cm column;
• 10 µL of a 100 µM compound solution in methanol was injected; • Flow rate of 200 µL/min;
• Positive ionization mode; • UV detector set to 254 nm
• Solvent A: 95% H2O, 5% ACN, 0.1% formic acid
• Solvent B: 95% ACN, 5 % H2O, 0.1% formic acid
Time (min) % A % B
0 100 0
10 50 50
4. APPENDIX
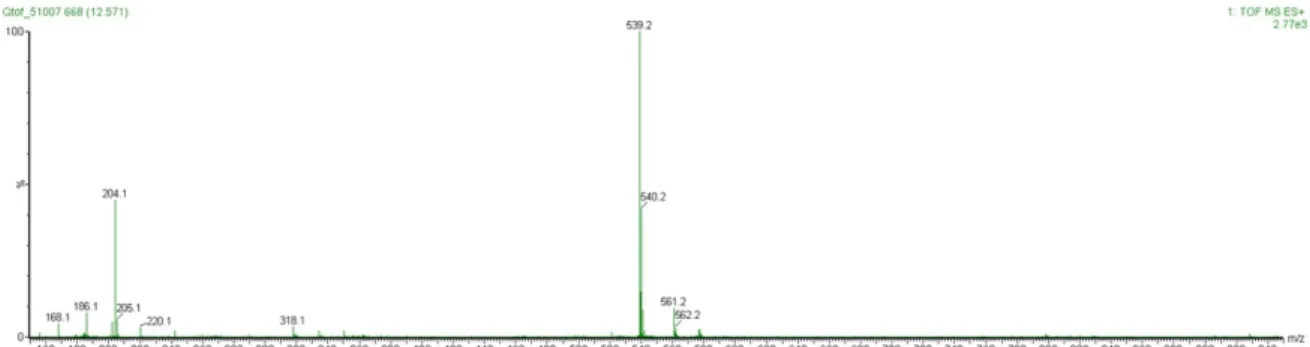
4.1 LC-MS Analysis of Glc-NAc conjugate 1.12
Figure I: UV Trace at 254 nm and TIC/ESI in positive mode of Glc-NAc conjugate 1.12.
MW = 538.47 O HO HO HO NHAc N COOMe CF3 Ph O 1.12
5. REFERENCES
1. Zhiyuan, M.; Vosseller, K.; Springer Verlag, 2013 and references therein. 2. Hanahan, D.; Weinberg, D. A.; Cell 2011, 144, 646-674.
3. Warburg, O.; Science 1956, 123, 309.
4. Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y.; J. Cell.
Biol. 2007, 178, 93-105.
5. Wells, L.; Vosseller, K.; Hart, G. W.; Cell. Mol. Life Sci. 2003, 60, 222-228. 6. Myatt, S. S.; Lam, E. W.; Nat. Rev. Cancer 2007, 7, 847-859.
7. Lynch, T. P.; Ferrer, C. M.; Jackson, S. R.; Shahriari, K. S.; Vosseller, K.; Reginato, M. J.; J. Biol. Chem. 2012, 287, 11070-11081.
8. Yu, Z.; Schmaltz, R. M.; Bozeman, T. C.; Paul, R.; Rishel, M. J.; Tsosie, K. S.; Hecht, S. M.; J. Am. Chem. Soc. 2013, 135, 2883-2886.
9. Chan, D. A.; Sutphin, P. D.; Nguyen, P.; Turcotte, S.; Lai, E. W.; Bahn, A.; Reynolds, G. E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D. E.; Bonnet, M.; Flanagan, J. U.; Bouley, D. M.; Graves, E. E.; Denny, W. A.; Hay, M. P.; Giaccia, A. J.; Sci.
Transl. Med. 2011, 3.
10. Altenberg, B.; Greulich, K. O.; Genomics 2004, 84, 1014-1020.
11. Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; Funel, N.; Leòn, L. G.; Giovannetti, E.; Peters, G. J.; Palchaudhuri, R.; Calvaresi, E. C.; Hergenrother, P. J.; Minutolo, F.; J. Med.Chem 2011, 54, 1599-1612.
12. Calvaresi, E. C.; Granchi, C.; Tuccinardi T.; Di Bussolo, V.; Huigens, R. W. III; Lee, H. Y.; Palchaudhuri, R.; Macchia, M.; Martinelli, A.; Minutolo, F.; Hergenrother, P. J; ChemBioChem 2013, 14, 1-5.
13. Esposto C.; Master Thesis “Glicosidi dell'unita ripetitiva del polisaccaride
capsulare dello Streptococcus Pneumoniae 14 (SP14)” A.A. 2009/2010.
14. Křenek, K.; Ŝimon, P.; Weignerová, L.; Fliedrová, B.; Kuzma, M.; Křen, V.;
Beilstein J. Org. Chem. 2012, 8, 428-432.
15. Weissmann, B.; J. Org. Chem. 1966, 31, 2505-2509.
39, No. 1.
17. Liu, J.; Gin, D. Y.; J. Am. Chem. Soc. 2002, 124, 9789-9797.
18. Frau, I. PhD Thesis, University of Pisa, School of graduate studies “Scienza del farmaco e delle sostanze bioattive”, 2013, p. 197.
19. Coxon, B.; Carbohydr. Res. 2005, 340, 1714.