4
1. DISTURBI DELL’UMORE
Il Disturbo Bipolare e la depressione unipolare rappresentano una delle più frequenti espressioni di malattia mentale. La prevalenza lifetime della depressione unipolare nella popolazione generale è stimata tra 4.6% e 17%; la prevalenza a 12 mesi risulta compresa tra 3% e 10.3%. La depressione è circa due volte più frequente nel sesso femminile. La prevalenza del Disturbo Bipolare I nell’arco della vita nei paesi industrializzati è di 0.5% e le stime salgono a 1.1-5.3% considerando anche il Disturbo Bipolare II (Cassano e coll., 2006). La depressione maggiore interessa dal 5% al 13% dei pazienti che si sottopongono a visite ambulatoriali di medicina generale (Coyne e coll., 1994), tuttavia, è spesso non diagnosticata e non trattata, oppure trattata inadeguatamente, quando pure correttamente diagnosticata (Goldman e coll., 1999).
Frequenti e gravi sono anche le complicanze, come l’abuso di alcool e sostanze stupefacenti, e il rischio di suicidio, stimato, almeno nelle forme bipolari, circa 30 volte superiore a quello della popolazione generale. Si tratta quindi di disturbi estremamente diffusi, caratterizzati non soltanto da isolati episodi di depressione e/o mania, ma anche da un’ aumentata morbilità e mortalità. Ciò si evince non solo da un tasso di suicidio che si attesta intorno al 15 % per entrambi i disturbi, ma anche dall’esistenza di una significativa comorbidità medica e di una notevole compromissione del funzionamento sociale e lavorativo (Evans e coll., 2003). E’ stato osservato che pazienti con diabete mellito, malattie respiratorie croniche, epilessia o cardiopatia ischemica affetti anche da depressione maggiore ottengono risultati terapeutici inferiori rispetto a quelli non depressi (Ciechanowski e coll., 2000; Jiang e coll., 2002). Anche la mortalità per malattie internistiche sembra essere maggiore in pazienti depressi (Angst e coll., 2002). E’
5
stato dimostrato che il trattamento antidepressivo può ridurre la mortalità o migliorare i risultati terapeutici dopo infarto acuto del miocardio (Taylor e coll., 2005) o ictus (Jorge e coll., 2003) e ridurre il rischio di suicidio (Gibbons e coll., 2005).
1.1 ASPETTI CLINICI
Lo stato attuale delle conoscenze consente di prospettare una visione unitaria in base alla quale i disturbi dell’umore costituiscono un continuum (spettro maniaco depressivo) che comprende a un estremo i temperamenti affettivi, come la ciclotimia e la distimia, e le patologie più lievi, come la depressione unipolare, all’altro quelle più gravi, come il Disturbo Bipolare di tipo I. Questo inquadramento, che tiene conto non solo della sintomatologia dei singoli episodi, ma anche del temperamento premorboso, delle modalità di esordio, delle caratteristiche di decorso e delle fasi intervallari, ha mostrato un elevato valore euristico sia sul piano clinico, per quanto riguarda la diagnosi ed il trattamento a breve e a lungo termine, che su quello della ricerca scientifica, per quanto riguarda gli studi sulla trasmissione genetica e sui markers biologici.
La depressione è una sindrome clinica caratterizzata da umore depresso, alterazioni cognitive e psicomotorie nonché una serie di sintomi neurovegetativi che riflettono una disregolazione limbico-diencefalica.
La diagnosi di Episodio Depressivo Maggiore si basa su criteri clinici tra cui quelli proposti dalla American Psychiatric Association e pubblicati nel DSM-IV-TR (2000). Questi criteri includono umore depresso o perdita di interessi o di piacere per almeno due settimane, più quattro tra altre sette caratteristiche: perdita o aumento di almeno il 5% del peso corporeo in un mese o persistente alterazione
6
dell’appetito, insonnia o ipersonnia, alterazioni psicomotorie, affaticabilità o mancanza di energia, sentimenti di svalutazione o di colpa eccessivi o immotivati, riduzione della concentrazione o indecisione, ideazione suicidaria o tentativi di suicidio. E’ necessario che i suddetti sintomi siano tali da causare disagio significativo o compromissione del funzionamento sociale e lavorativo. Devono essere escluse condizioni mediche generali quali neoplasie, ictus, malattie demielinizzanti, epilessia, anemia, etc, in grado di causare quadri depressivi secondari del tutto simili al disturbo primario.
Sintomi psicotici quali deliri di colpa, di rovina o somatici complicano più del 14% degli episodi depressivi maggiori (Johnson, 1991).
Si distinguono diversi sottotipi di depressione sulla base della rilevazione di determinati clusters sintomatologici: questi sottotipi (depressione melanconica, reattiva, psicotica, atipica, etc), comunque, sono basati solo su differenze sintomatologiche e non devono essere considerati entità biologiche distinte (Akiskal, 2000).
Nei sistemi internazionali di classificazione dei disturbi mentali i criteri per la diagnosi della depressione bipolare e unipolare sono gli stessi benché si sia ipotizzato che le due forme potrebbero rappresentare entità nosologiche distinte, sottese da un differente substrato biologico, e comunque richiedano un diverso approccio terapeutico. Diversi Autori hanno tentato di differenziare dal punto di vista fenomenologico le fasi depressive dei disturbi bipolari dalla depressione maggiore ricorrente. I risultati più replicati indicano la prevalenza di labilità emotiva, rallentamento motorio, irritabilità, sintomi atipici (iperfagia e ipersonnia), sintomi psicotici e comorbidità con disturbi d’ansia e abuso di sostanze nelle forme bipolari, di agitazione, ansia intraepisodica, somatizzazioni e inappetenza con calo ponderale in quelle unipolari (Goodwin e Jamison, 2007). L’utilità clinica di queste osservazioni è comunque limitata in quanto i dati sono
7
spesso contraddittori e la maggior parte deriva da studi di confronto tra pazienti con depressione bipolare I e depressione ricorrente essendo stati esclusi quelli con depressione bipolare II. A rigore, quindi, in assenza di una fase (ipo)maniacale spontanea in anamnesi la diagnosi dovrebbe essere di depressione unipolare, anche se ormai ricercatori e clinici concordano nel collocare nell’ambito dello spettro bipolare anche le depressioni seguite da fasi (ipo)maniacali scatenate da antidepressivi, come pure le depressioni a esordio nel post-partum o a elevata ricorrenza, le depressioni resistenti al trattamento e quelle che si verificano in soggetti con familiarità per disturbo bipolare o con temperamento premorboso di tipo ipertimico o ciclotimico (Cassano e Tundo, 2008).
Un discorso a sé richiede la depressione resistente ai trattamenti. I dati relativi alla prevalenza della depressione resistente al trattamento indicano che circa 1/3 dei pazienti trattati per depressione maggiore non risponde in modo soddisfacente a un primo trial con antidepressivi (Thase e Rush, 1997); dati ancora più pessimistici emergono dallo studio STAR-D (Trivedi e coll., 2006) che ipotizza un 50% di non risposta a un trial con antidepressivo e meno del 30% di remissione. Osservazioni di follow-up rivelano inoltre che circa il 20% rimane sintomatico 2 anni dopo l’esordio del disturbo (Paykel, 1994) e che, anche dopo molteplici interventi terapeutici, fino al 10% dei pazienti è ancora depresso (Souery e coll., 1999). La diagnosi corretta, comprensiva della valutazione delle eventuali comorbidità mediche e psichiatriche e della tipizzazione della depressione con particolare attenzione al riconoscimento dei markers di bipolarità, rimane l’elemento essenziale per evitare la somministrazione di trattamenti inadeguati e quindi inefficaci. Per declinare i gradi di resistenza è fondamentale porre l’attenzione sull’adeguatezza del trattamento non solo per quanto concerne la scelta della molecola, ma anche dei tempi e delle dosi di
8
assunzione dello stesso. In molti casi si assiste infatti a errori terapeutici che condizionano l’efficacia della terapia, quali per esempio la prescrizione di dosaggi inadeguati di farmaco, o i tempi delle cure insufficienti, inferiori a 4-6 settimane. Occorre infine menzionare l’esistenza di fattori legati al paziente come determinanti della scarsa risposta terapeutica: la bassa compliance in primis, l’eventuale presenza di comorbidità medica non nota o non dichiarata, le variazioni individuali della farmacocinetica, l’errata assunzione della terapia prescritta. Tra i più importanti sistemi proposti per la stadiazione della depressione resistente al trattamento si fa spesso riferimento a quello proposto da Thase e Rush (1997) che individua sei diversi stadi:
• Stadio 0 (pseudoresistenza): mancata risposta a un primo trial con antidepressivo di provata efficacia inadeguato per dosi e tempi
• Stadio I: mancata risposta a un trial con antidepressivo a dosi e con
modalità adeguate
• Stadio II: mancata risposta a 2 trials con antidepressivi di classi differenti
• Stadio III: mancata risposta a 2 o più trials di cui almeno 1 con
triciclici
• Stadio IV: mancata risposta a 2 o più trials di cui almeno 1 con IMAO
• Stadio V: mancata risposta a 2 o più trials di cui almeno uno associato a un ciclo di TEC bilaterale (considerata il gold standard per la depressione farmaco-resistente).
9 1.2 ASPETTI PATOGENETICI
La storia dei disturbi dell’umore ha origini remote e numerose sono state le descrizioni da parte di studiosi e letterati nel corso dei secoli.
Lontani riferimenti sono attribuiti alla “tristezza” di Bellerofonte nell’Iliade omerica e alle descrizioni di fluttuazioni dell’umore nelle culture antiche, da quella egiziana a quella babilonese, a quelle ebraica. Ippocrate, nel IV secolo a.C., considerava il cervello sede dell’affettività e di ogni manifestazione psichica: a lui si deve la prima descrizione clinica dei disturbi dell’umore, così come il termine melancòlia, affezione causata dall’azione patogena della bile nera sul cervello. Areteo di Cappadocia, nel I secolo d.C., colse per la prima volta il legame tra depressione e mania, considerandole fasi opposte di una stessa condizione morbosa e descrivendo l’andamento ricorrente degli episodi in uno stesso soggetto.
Con il tramontare della impostazione organicistica della medicina greco-romana, sarà necessario attendere il secolo illuminista perché il ruolo principale, nell’interpretazione dei disturbi mentali, ritorni al cervello. In realtà già nel 1600 Robert Burton, nei suoi scritti, formulò le prime ipotesi relative a fattori costituzionali implicati nella patogenesi della melanconia, insieme all’alcool, alla dieta e ai ritmi biologici. Emil Kraepelin (1856-1926), responsabile, con la distinzione tra daementia precox e psicosi maniaco-depressiva, di una svolta nella classificazione dei disturbi mentali, teorizzò la presenza di anomalie cerebrali nei pazienti maniaco-depressivi, sebbene gli studi autoptici non li avessero evidenziati. Sempre nella prima metà del Novecento, lo psichiatria americano Adolf Meyer (1866-1950), cui si fa risalire l’origine del termine “depressione”, ipotizzò che la malattia fosse il risultato dell’interazione tra fattori biologico-genetici e fattori ambientali stressanti. Le acquisizioni attuali confermano le teorie di Meyer: la depressione ha dimensioni psico-socio-biologiche. Gli stressors,
10
cioè gli eventi traumatizzanti di ordine sociale o psicologico, possono scatenare, alimentare e mantenere il disturbo in soggetti costituzionalmente predisposti. E’ stato osservato che esiste un rapporto inverso tra predisposizione biologica e ruolo degli eventi scatenanti: nei soggetti con un carico biologico importante, in cui il disturbo ha spesso un esordio precoce, sono sufficienti stimoli ambientali di lieve entità per indurre un episodio depressivo; al contrario, quando il peso dei fattori biologici è minore, occorre il sommarsi di numerosi eventi stressanti per stimolare la comparsa di un disturbo dell’umore. E’ proprio alla luce di questa predisposizione biologica, più o meno marcata ma comunque necessaria, che negli ultimi decenni si è assistito al superamento della classica distinzione tra depressione “endogena” e “reattiva”.
1.2.1 Ipotesi monoaminica
Il primo vero progresso nella comprensione della neurobiologia dei disturbi dell’umore, e in particolare della depressione, si deve alla scoperta per serendipity, circa 50 anni fa, delle prime molecole ad azione antidepressiva. L’iproniazide e l’imipramina, due farmaci registrati pressoché nello stesso periodo ma in maniera indipendente e per differenti impieghi, essendo il primo un antitubercolare, l’altro un antistaminico sperimentale con una struttura triciclica, si rivelarono entrambi in grado di elevare l’umore nei pazienti che li assumevano. I ricercatori fecero quindi chiarezza sul meccanismo d’azione dei due farmaci nel cervello: si scoprì che entrambi determinano un incremento della concentrazione extracellulare di due importanti neurotrasmettitori, serotonina e noradrenalina, rispettivamente l’imipramina attraverso il blocco del loro re-uptake a livello delle terminazioni pre-sinaptiche e l’iproniazide attraverso l’inibizione del principale enzima metabolizzante, la monoamino-ossidasi (MAO). Fu proposto, pertanto, che la depressione potesse dipendere da un deficit di serotonina e noradrenalina
11
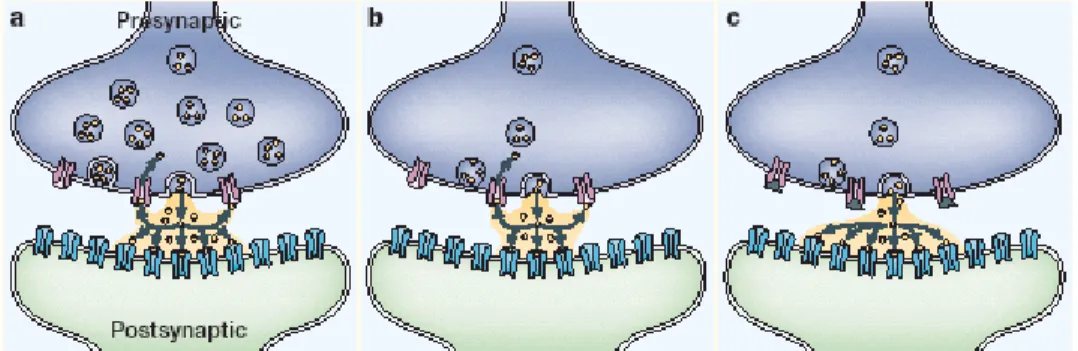
a livello di importanti siti recettoriali nel cervello, una teoria che è nota come ipotesi monoaminergica della depressione (Burney e coll., 1965; Wong e coll., 2004) [Fig. 1.1].
Figura 1.1- Ipotesi monoaminergica della depressione. a: neurotrasmissione in condizioni di normalità. b: riduzione della neurotrasmissione nella depressione. c: aumento della concentrazione extracellulare di neurotrasmettitori dopo blocco dei siti di re-uptake. (modificato da Castren E, 2005)
L’ipotesi monoaminica indirizzò l’interesse dell’industria farmaceutica sul metabolismo delle monoamine per decine di anni: l’imipramina è stata progressivamente affiancata da varie molecole ad azione più selettiva. In breve tempo, però, sulla base di molte osservazioni, si riconobbe l’inconsistenza della semplice correlazione tra concentrazione di monoamine e depressione. Per esempio, sebbene gli effetti degli antidepressivi sul metabolismo delle monoamine si osservino subito dopo la loro somministrazione, è ormai noto che servono diverse settimane di trattamento continuato perché compaia la risposta clinica (Nestler, 1998). La scoperta che il trattamento a lungo termine con farmaci antidepressivi produce cambiamenti adattativi nei recettori delle monoamine e nei meccanismi di trasduzione del segnale a valle (Sulser e coll., 1978), ha spostato il focus della ricerca sugli effetti cronici che gli antidepressivi esercitano sulla concentrazione di neuropeptidi, di fattori di crescita e rispettivi
12
recettori, e di molecole coinvolte nei processi di trasduzione (Duman e coll., 1997; Manji e coll., 2001; Coyle e coll., 2003) . L’ipotesi monoaminica è evoluta in quella che oggi è nota come ipotesi molecolare della depressione.
1.2.2 Depressione e neuroplasticità
Soprattutto nel corso dell’ultimo decennio, quindi, il progresso neuroscientifico, favorito dall’integrazione di competenze mediche, biologiche, chimiche e fisiche, nonché dallo straordinario sviluppo delle tecniche di neuroimaging, ha consentito di definire significativamente i correlati neurobiologici dei disturbi psichiatrici. Fondamentali sono state le acquisizioni in tema di neuroplasticità, sia a livello cellulare che a livello di reti neuronali: in senso stretto, qualsiasi modificazione di tipo trascrizionale o trasduzionale può considerarsi una forma di modulazione della plasticità neuronale, che assume aspetti eclatanti nel caso di modificazioni strutturali, quali ad esempio neoformazione di spine dendritiche o di varicosità assonali in rapporto a stimolazioni di tipo comportamentale, elettrofisiologico o farmacologico. Inoltre, il “dogma centrale” che i neuroni del SNC adulto non possano andare incontro a duplicazione cellulare, è stato superato dalla scoperta da parte di Gould e collaboratori (1997) dell’esistenza di neurogenesi in alcune aree del SNC di primati adulti, in particolare a livello del giro dentato dell’ippocampo. I fenomeni neurogenetici sarebbero favoriti da trattamenti psico e farmacoterapeutici, e avverrebbero persino spontaneamente come reazione a insulti traumatici ed ischemici. Le reti neuronali non sono rigide e fisse ma estremamente plastiche e soggette a profonde modificazioni che, nel corso della vita, consentono l’adattamento a nuove situazioni.
L’osservazione in una percentuale significativa di depressi di alterazioni di tipo atrofico, a livello ippocampale e paraippocampale, frontale e prefrontale, ha suggerito l’ipotesi che lo stress e/o la depressione, siano in grado di determinare
13
tali alterazioni attraverso un’ attivazione protratta dell’ asse ipotalamo-ipofisi-surrene con conseguente morte cellulare nell’ippocampo e soppressione della neurogenesi (Sapolsky e coll., 1990, 1994, 1996; Duman e coll., 1997, 2004). Studi post-mortem, infatti, hanno dimostrato una riduzione del numero e del volume di neuroni e cellule gliali in diverse aree corticali (Ongur e coll., 1998; Rajkowska e coll., 1999; Cotter e coll., 2001). La riduzione del numero delle cellule gliali consente di escludere che la perdita neuronale sia in parte dovuta a un processo infiammatorio, evenienza in cui si osserverebbe gliosi, mentre l’aspetto atrofico delle popolazioni cellulari suggerisce l’ipotesi della morte cellulare per apoptosi. La disregolazione del suddetto asse ipotalamo-ipofisi-surrene, era già stata documentata da tempo (Holsboer e coll., 1986). Nuove osservazioni hanno, però, ne hanno sottolineato la valenza patogenetica: gli steroidi surrenali determinano una deplezione cellulare di glucosio, rendendo i neuroni particolarmente sensibili all’incremento di neurotrasmettitori eccitatori come il glutammato.
Nel cervello, il rilascio di fattori neurotrofici, tra cui il Brain Derived Neurotrophic Factor (BDNF), avviene secondo un meccanismo attività-dipendente: il neurone che innerva sufficientemente un altro neurone target, induce la produzione ed il rilascio della proteina (Thoenen, 1995; Poo, 2001). E’ bene precisare che l’evento cruciale nella plasticità attività-dipendente non è la formazione di contatti sinaptici, ma è la selezione e la stabilizzazione di quelle sinapsi che mediano segnali utili, unitamente alla riduzione selettiva di quelle che producono rumore (Hua e coll., 2004). Non è esatto, quindi, considerare vantaggiosa la neurogenesi e dannose l’apoptosi e l’eliminazione selettiva di sinapsi: è la combinazione dei due fenomeni, guidata dai livelli di attività neuronale e quindi dal maggiore o minore rilascio di neurotrofine, a favorire il processo adattativo di neuroplasticità.
14
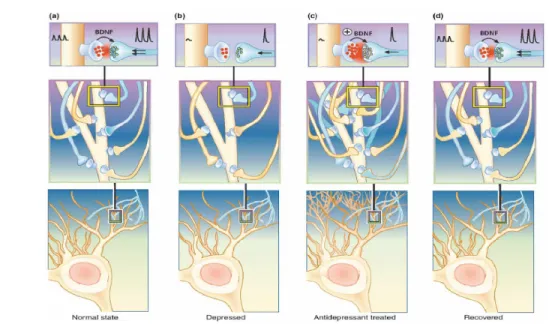
In questa ottica, gli antidepressivi, stimolando la trasmissione sinaptica e quindi il rislascio di BDNF attività-dipendente, avvierebbero un processo di autoriparazione in cui la plasticità neuronale e le variazioni della neurotrasmissione andrebbero di pari passo verso il ripristino della normotimia. L’eziopatogenesi della depressione risiederebbe nell’alterazione di quei meccanismi intracellulari di trasduzione del segnale dal neurotrasmettitore al neurone post-sinaptico, che si concluderebbero con la produzione di BDNF e, quindi, la regolazione dei processi di neuroplasticità e di sopravvivenza cellulare (Manji coll., 2001; Popoli e coll., 2002). In condizione di stress il gene del BDNF verrebbe represso, determinando apoptosi dei neuroni ippocampali e conseguente atrofia del distretto, portando infine allo sviluppo di depressione [Fig. 1.2].
Figura 1.2- Un modello dell’effetto della depressione e del trattamento antidepressivo sulla connettività attività-dipendente. a: Nello stato di normalità l’attività presinaptica determina il rilascio attività dipendente di BDNF a livello post-sinaptico e la stabilizzazione delle connessioni sinaptiche. b: Nella depressione la connettività sinaptica è gradualmente ridotta a causa dell’insufficiente rilascio attività-dipendente di neurotrofina. c: Il trattamento cronico antidepressivo supporta la sinaptogenesi attraverso il rilascio di BDNF ed eleva l’umore. d: La potatura uso-dipendente dei contatti sinaptici inattivi gradualmente, nell’arco di mesi, porta alla stabilizzazione del network neuronale funzionale e al recupero completo della normotimia.
15 1.2.3 Ipotesi neurotrofica: studi preclinici e sull’uomo
L’ipotesi neurotrofica dei disturbi affettivi appare supportata dai numerosi dati di neuropsicofarmacologia sperimentale. Studi su modelli comportamentali animali di depressione hanno dimostrato che l’infusione cerebrale di BDNF ha un effetto sovrapponibile a quello delle molecole antidepressive (Siuciack e coll., 1997; Shirayama e coll., 2002; Hoshaw e coll, 2005). D’altra parte, la somministrazione di antidepressivi o l’elettroconvulsione causano un aumento dell’espressione del BDNF nella corteccia dell’animale da esperimento (Tardito e coll., 2006; Nibuya e coll., 1995). La somministrazione cronica di antidepressivi induce, infatti, una modificazione nei meccanismi di trasduzione, potenziando la via del cAMP con conseguente aumento dei livelli di espressione del fattore di trascrizione CREB (cAMP Response Element Binding protein) e quindi del BDNF nei neuroni ippocampali e corticali (Popoli e coll., 2002). Il ruolo fisiologico di CREB è quello di modulare l’espressione di molteplici geni, in particolare di quello per il BDNF, e il suo livello è ridotto in individui depressi [Fig. 1.3]. Il trattamento cronico con litio e valproato induce l’espressione di proteine antiapoptotiche (come la Bcl-2) e determina un aumento dei livelli di espressione di BDNF nell’ippocampo e nella corteccia cerebrale (Fukumoto e coll., 2001). Anche l'anticonvulsivante lamotrigina, ampiamente indicato nel trattamento della depressione bipolare, ha dimostrato, se somministrato cronicamente nel ratto, la capacità di aumentare l'espressione del BDNF in ippocampo e corteccia frontale così come di contrastare la riduzione dell'espressione della neurotrofina stress-indotta (Li e coll., 2010). Un’azione modulante positiva, inoltre, è stata osservata con gli antagonisti del recettore NMDA (memantina), anch’essi con una possibile efficacia antidepressiva (Martanova e coll., 2001).
16
Analisi post-mortem sull’ ippocampo dimostrano che l’espressione di BDNF è ridotta nei pazienti depressi suicidi (Chen e coll., 2001; Dwivedi e coll., 2003) mentre risulta aumentata nei pazienti in trattamento antidepressivo al momento del decesso (Karege e coll., 2005). Analogamente, l'espressione del BDNF a livello prefrontale in adolescenti suicidi è risultata significativamente inferiore rispetto a controlli sani (Pandey e coll., 2008). Infine, un recente studio di Keller e coll., (2010) ha rilevato in campioni post-mortem di cervello di suicidi un incremento significativo della metilazione a carico di una regione del promotore del gene del BDNF suggerendo un legame tra comportamento suicidario e alterazioni epigenetiche dell'espressione del BDNF.
Per quanto riguarda le analisi condotte a livello periferico, diversi studi dimostrano che i livelli sierici di BDNF risultano significativamente ridotti in pazienti depressi e inversamente correlati con la gravità della sintomatologia depressiva (Karege e coll., 2002, 2005; Shimizu e coll., 2003; Piccinni e coll., 2008), durante le fasi depressive e maniacali di pazienti bipolari (Cunha e coll., 2006), in pazienti bipolari sia depressi che in fase maniacale e sia trattati farmacologicamente che drug-free (de Oliveira e coll., 2009), in pazienti unipolari in fase depressiva o eutimica e in pazienti bipolari I o II eutimici (Monteleone e coll., 2008), nonché in volontari sani con tratti depressivi di personalità (Lang e coll., 2004). Altri studi dimostrano che il trattamento antidepressivo è in grado di aumentare i livelli sierici di BDNF in pazienti depressi (Aydemir e coll., 2004, 2006; Gonul e coll., 2005; Yoshimura e coll., 2007; Huang e coll., 2007). Anche una recente metanalisi ha confermato che i livelli sierici di BDNF sono inferiori nei pazienti depressi rispetto ai controlli sani e che risentono positivamente del trattamento antidepressivo (Sen e coll., 2008). Al contrario, un solo studio ha descritto un sottogruppo di pazienti in cui i livelli sierici della neurotrofina diminuiscono dopo il trattamento (Gervasoni e coll., 2005). Anche la TEC su
17
pazienti depressi è stata associata all'incremento dei valori sierici di BDNF (Bocchio-Chiavetto e coll., 2006; Okamoto e coll., 2008; Hu e coll., 2010).
Pochi Autori hanno indagato i valori di BDNF nel plasma in soggetti sani (Fujimura e coll., 2002; Radka e coll., 1996; Rosenfeld e coll., 1995). Livelli plasmatici ridotti di BDNF sono stati riscontrati in pazienti depressi (Karege e coll., 2005; Piccinni e coll., 2008), in pazienti bipolari in fase maniacale non trattati farmacologicamente (Machado-Vieira e coll., 2007), in pazienti bipolari al primo episodio di malattia (Palomino e coll., 2006), in pazienti con depressione geriatrica a esordio tardivo (Shy e coll., 2010). I livelli plasmatici di BDNF nei pazienti depressi risultano correlati negativamente con la gravità della sintomatologia, con il numero di episodi nonché con i sintomi più rappresentativi della disfunzione a carico dell’asse HPA (ipotalamo-ipofisi-surrene) come le alterazioni del pattern ipnico ed il rallentamento psicomotorio (Dell’Osso e coll., 2010). I livelli plasmatici nei pazienti depressi, così come si è visto per quelli sierici, risentono positivamente della TEC (Marano e coll., 2007; Piccinni e coll., 2009): in particolare, lo studio di Piccinni e coll. (2009) ha dimostrato un incremento dei livelli di BDNF solo nei pazienti che hanno risposto clinicamente alla TEC e che erano i pazienti con i livelli basali meno bassi. In pratica questi risultati suggeriscono una possibile utilità dei livelli plasmatici di BDNF al baseline come un predittore di risposta alla TEC in pazienti depressi drug-resistant.
Una metanalisi recente ha confermato che i pazienti bipolari hanno livelli di BDNF in fase depressiva e maniacale ma non in fase eutimica inferiori rispetto a controlli sani: ciò lascia speculare circa un possibile ruolo del BDNF periferico come biomarker stato-dipendente nel disturbo bipolare (Lin, 2009).
Il principale limite degli studi sul siero o sul plasma risiede nella difficile interpretazione del significato dei livelli periferici di BDNF. Tuttavia, in
18
Letteratura c’è l’evidenza che altri fattori di crescita periferici, tra cui il VEGF e l’IGF-1, hanno accesso al cervello (Pan e coll, 1998; Trejo e coll., 2001; Fabel e coll., 2003) e che i livelli cerebrali e sierici di BDNF subiscono variazioni parallele durante i processi di crescita e sviluppo dei ratti (Karege e coll.,2002), pertanto, anche il BDNF periferico potrebbe influenzare la neurogenesi e le funzioni del SNC.
L’ipotesi neurotrofica nell’eziopatogenesi dei disturbi dell’umore richiede, comunque, ulteriori prove sperimentali, al fine di chiarire alcune osservazioni che allo stato attuale delle conoscenze potrebbero apparire come contraddittorie. La più importante tra queste osservazioni riguarda le azioni opposte che il BDNF dimostra in diverse regioni del cervello. Sebbene la somministrazione della neurotrofina nel mesencefalo, nell’ippocampo e nei ventricoli laterali produca un effetto antidepressivo in modelli animali di depressione, l’iperespressione del BDNF in altre regioni può avere l’effetto contrario. Nel sistema dopaminergico mesolimbico, infatti, l’iperespressione del BDNF a livello dell’area ventrale tegmentale produce un incremento dei comportamenti simil-depressivi, mentre l’iperespressione di una forma non funzionante del recettore TrkB a livello del nucleo accumbens determina una risposta antidepressiva (Eisch e coll., 2003). Si tratta comunque di un complesso di dati che rende evidente che i fenomeni di neuroplasticità cerebrale possono svolgere un ruolo centrale sia nell’origine dei disturbi affettivi che nei meccanismi di azione dei farmaci efficaci nella loro terapia. Appare altrettanto chiaro come lo sviluppo delle conoscenze in questo contesto possa condurre a una visione dell’azione farmacologica fondata su modelli non più legati alla semplice azione a livello recettoriale, ma estesi all’azione sugli eventi intracellulari della risposta.
19
Figura 1.3- Modello esemplificativo dell’azione molecolare della 5-HT. L’aumento cronico della 5-HT intrasinaptica, come a seguito di trattamento cronico antidepressivo, porta alla
down-regulation di alcuni recettori serotoninergici (5-HT1A, 5-HT4, 5-HT6, 5-HT7, 5-HT2A). D’altra parte, è stata osservata una up-regulation dei componenti post-recettoriali della cascata dello cAMP, inclusi i livelli di attività dell’enzima adenilato ciclasi, i livelli di PKA, e l’espressione di CREB. Ciò è possibile perché i recettori in questione, pure espressi in numero minore sulla membrana cellulare, conservano comunque un livello di attività minimo a fronte di livelli decisamente e cronicamente più alti di 5-HT: il bilancio totale depone a favore di un’attivazione della via dello cAMP. CREB è un target comune delle vie di trasduzione a valle dei diversi recettori della 5-HT, poiché non solo i recettori 5-HT4, 5-HT6 e 5-HT7 sono accoppiati alla via dello cAMP sopra descritta, ma anche i recettori 5-HT2A e α1 della noradrenalina inducono
l’espressione di CREB tramite l’attivazione di kinasi calcio-calmodulina-dipendenti. D’altra parte, il sistema dello cAMP, e quindi l’espressione di CREB, sono regolati negativamente dai recettori 5-HT1A (accoppiati ad una subunità α della proteina G di tipo inibitorio).
L’incremento dell’attività e dell’espressione di CREB risulta nella regolazione di geni bersaglio tra cui quelli del BDNF e del suo recettore TrkB. (modificato da Duman, 1998)