TYROSINE KINASE RECEPTORS (RTKs)
Protein tyrosine kinases represent a wide family of homologous enzymes, both transmembranous and cytoplasmic, that catalyze the transfer of the γ-phosphate group from ATP to a hydroxy group of selected tyrosine residues in target protein substrates. Under physiological conditions, tyrosine phosphorylation represent a fundamental signal transduction mechanism that proceeds hierarchically in a ordered sequence of protein interactions, ensuring cross-talk between cells and regulating key aspects of cell life such as proliferation, differentiation, metabolism, and apoptosis.1
Approximately 90 TK receptors have been identified, 58 of which are transmembrane receptor type (eg: EGFR, Epidermal Growth Factor Receptor) and 32 the cytoplasmic “non receptor” type.
Transmembrane RTKs are floating protein in the double layer phospholipid membrane, all of which share a similar structure characterized by three regions: a cytosolic, an extracellular, an intramembrane portion. For their different structural features of domains approximately 20 RTK classes have been identified (Figure 1). The best characterized are:2
RTK class I (Epidermal Growth Factor (EGF) receptor family) RTK class II (Insulin receptor family)
RTK class III (Platelet-derived Growth factor (PDGF) receptor family) RTK class IV (Fibroblast Growth Factor (FGF) receptor family)
RTK class V (Vascular Endothelial Growth Factor (VEGF) receptors family) RTK class VI (Hepatocyte Growth Factor (HGF) receptor family)
RTK class VII (Tropomyosin-receptor-kinase (Trk) receptor family) RTK class XI (Angiopoeitin (TIE) receptor family)
Figure 1: Human Receptors Tyrosine Kinase2
The extracellular region exposes N-terminal domain to the interstitial fluid and presides over the recognition and interaction with the ligand.
Hydrophilic intracellular portion, C-terminal, is equipped with sites that govern signal transduction after ligand-receptor interaction. This portion is characterized by tyrosine residues in specific positions for any receptor and after interaction with the ligand undergo autophosphorylation. It’s the specific arrangement that influence the interaction with the cytoplasmic proteins and its phosphorylation, because those exposed on their surface domains enable to recognize specific phosphorylated aminoacid sequences of the catalytic domain of intracellular portion of the receptor.3
On the contrary, cytoplasmic receptors are devoid of the extracellular portion and generally allow the signal transduction within the cell only after the activation of transmembrane receptors.
Normally, TK receptors are encoded by protoncogenes, and when they undergo mutations or are overexpressed, become oncogenes and activate enzymes prematurely, leading to the appearance of tumors.
The basis of many cancers are anomalies involving two important classes of genes:
• tumour suppressor genes, that normally inhibit cell growth, may undergo mutations that are usually recessive with loss of function;
• protoncogenes, that stimulate the cell to advance its cell cycle, are affected by dominant mutations with gain of function, which leads to increased or uncontrolled activity of the product, or oncogenes.
So inactivated tumour suppressor genes lead to a lack of controller proteins preventing the excessive cell proliferation, while protoncogenes mutation leads to production of excessive amounts or of an excessively active form of the protein growth promoters.
In both cases the result is the same: an uncontrolled cell growth that degenerates into a tumour. The difference between oncogenic and normal genes may also be related to the replacement of a single amino acid, which often results in a radical change in protein function.
While normal cytoplasmic receptors encoded by protoncogenes are activated by transmembrane receptors, in turn activated by their ligand, those resulting from the respective oncogenes are constitutively endowed with enzymatic activity.
Under physiological conditions the kinase activity is very low, while it’s increased in tumor cells: in fact, the increase in intracellular phosphotyrosine, associated with increased cell proliferation, represents a real tumor marker status.
In the last years some RTK have become the targets of anti-tumor drugs, which, inhibiting the TK activity, could block the proliferation of cancer resulting from signal transduction.3
THE EPIDERMAL GROWTH FACTOR RECEPTOR EGFR
The Epidermal Growth Factor Receptor (EGFR) is one of the most well-studied tyrosine kinases receptor: it’s coded by c-erbB-1 protoncogenes, located on chromosome 7, and is constitutively expressed in some tissues of epithelial, mesenchymal and neural origin. The receptor acts as a result of binding with the specific growth factor EGF (Epidermal Growth Factor) or less specific ligands.1
There are four known EGFR family members called ErbB or HER (Human
ErbB-Receptors Synonyms
HER1 erbB-1, EGFR
HER2 erbB-2
HER3 erbB-3
HER4 erbB-4
Table 1. Synonyms of HER family members
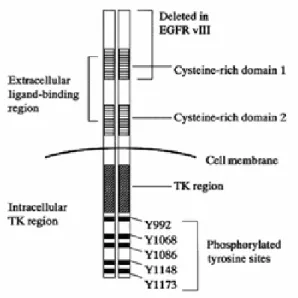
The EGFR is a transmembrane glycoprotein of 170 kDa formed by a single polypeptide chain containing 1186 amminoacids, and is characterized by three regions (Figure 2):4
Figure 2. EGFR structure
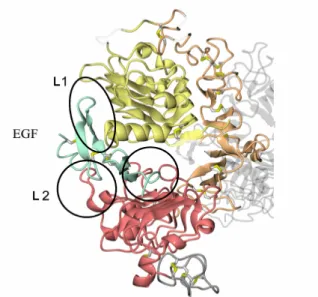
• A part of amino-terminal extracellular domain (or ectodomain) which consists of 621 amino acids and comprises four sub-domains. Early studies have shown that the major binding site of the receptor for growth factors is located in a fragment of domain III (or L2), between amino acids 295 and 543. Subsequent studies, instead, have revealed that a portion of domain I (or L1) plays a role,
albeit minor, in the binding of growth factors. Domains L1 and L2 are rich in cysteine (Figure 3). Domains II (or CR1) and IV (or CR2) are characterized by several modules linked together through one or two disulfide bridges. Behind the CR1 domain a loop protrudes that binds to another receptor of CR1 during dimerization. The extracellular portion is very variable within the HER family, being characterized by a homology of 30-50% (Figure 4).5,6
Figure 3. Extracellular domain to bind EGF
• The transmembrane region consists of 23 amino acids, from 622 to 644, with hydrophobic characteristics, folded into a single α-helix. This latter extends up to amino acid 647, thus suggesting its continuation even in the internal domain close to the membrane.
• The cytoplasmic domain consists in a kinase activity portion extending from amino acid 644 to 955 and is characterized by high homology between members of the HER family (59-82%) and in a carboxyl terminus (between amino acids 956 and 1186), more variable (homology around 30%), containing sites for tyrosine phosphorylation (indicated by Y or Tyr): Y 992, Y 1068, Y 1148 and Y 1173.4-6
The catalytic domain consists of about 250 amino acids folded to form two distinct lobes. The minor lobe has the function to link ATP, allowing γ-phosphate transfer, the larger lobe recognizes the substrate and properly orients the γ-phosphate. The minor lobe is characterized by β-sheets, while the major is dominated by α-helices with a small β-sheet, near the cavity between the two lobes.
The more conserved structure among HER family members is the ATP binding site, composed of a lysine (Lys 721), a Gly-X-Gly-X-X-Gly sequence and a tyrosine (Tyr 845). The sequence rich in glycine is deputed to the formation of hydrogen bonds with the γ-phosphate of ATP, the nucleotide is further stabilized by the presence of Lys 721 and Glu 738. Adenine is inserted into a hydrophobic pocket and makes hydrogen bonds at N6 and N7.7
GROWTH FACTORS AND RECEPTOR ACTIVATION
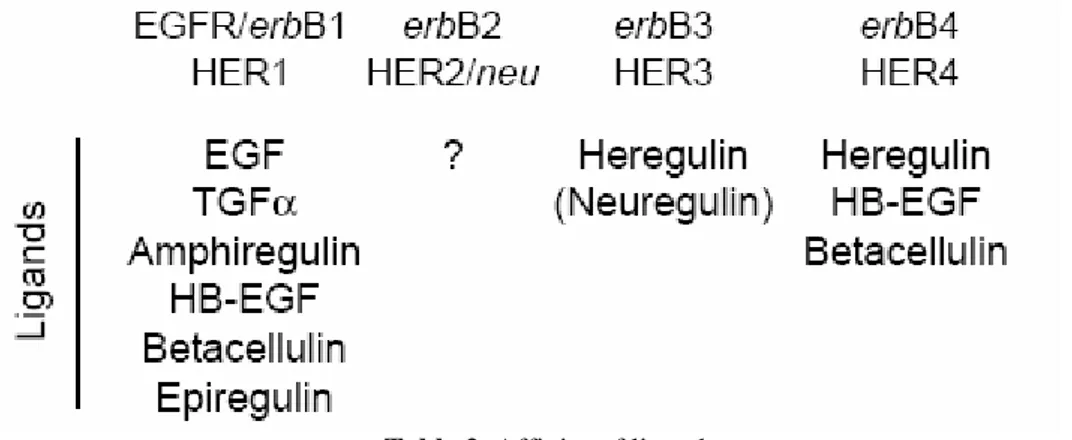
Growth factors bind to HER family members with different affinities, only HER2 has no specific ligands. Usually they are divided into three groups: the first includes EGF (Epidermal Growth Factor), TGF-α (Trasforming Growth Factor-α) and AR (anfiregulin) that bind only to EGFR, the second includes factors that make a specific link both with EGFR and with ErbB4, as BTC (betacellulin), HB-EGF (Heparin-binding HB-EGF), EPR (epiregulin) and the third group includes NRG
(neuroregulin), which can be divided into two subgroups according to their ability to bind ErbB3 and ErbB4 (NRG-1, NRG-2) or only ErbB4 (NRG-3 and NRG-4) (Table).5,6
Table 2. Affinity of ligands
The presence of HER receptors in the basolateral membrane of epithelial cells, and the discovery of many HER ligands such as EGF, TGFα, AR, BTC and NRGs in the extracellular matrix suggest that HER play an important role in mediating the signal between the epithelium and stroma.
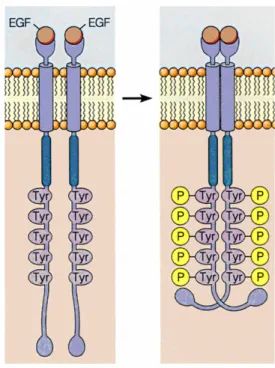
Interaction with the ligand in both domain I and III induces a change in the receptor conformation, allowing to expose an interface that was previously hidden and activating the process of dimerization. (Figure 5).8
In the inactive state the receptor exists as a monomer on the surface of the cell membrane. Binding of growth factor stabilizes the dimeric form (active) and causes its autophosphorylation: the catalytic site of each chain phosphorylates tyrosine residues located in the carboxy-terminal portion of the other chain. (Figure 6).
Figure 6. Dimerization and autophosphorylation of receptor
The interaction with the ligand may promote homo-dimerization (between monomers of the same receptor, eg HER1/HER1) or hetero-dimerization between the receptor and other members of the HER family (eg HER1/HER2), increasing its tyrosine kinase activity. Quantitative studies at the cell surface have shown that one of the most frequent association is the complex HER1/HER2.8
In both types of dimerizations the autophosphorylated receptors activate (through phosphorylation), other substrates that recruit the cytoplasmic adapter proteins and trigger a cascade of signals that drive important cellular functions like cell growth, proliferation, cytoskeleton rearrangement, apoptosis, motility, changes of gene expression and angiogenesis.
EGFR SIGNALLING PATHWAYS 6,9
The most studied signal transduction mechanism include the Ras/Raf/MEK/ERK and PI3K/PDK1/Akt cascade, but also the phospholipase PLC-γ and JAK /STAT can be engaged by activation of EGFR through overlapping or independent mechanism. It is still not clear which are the principal pathways in cancer initiation and progression.
In the Ras dependent EGFR activation the key role is played by GRB-2 (Growth Factor Receptor Bound protein 2) adapter protein, through its ability to bind directly to the receptor in its portions Y1068 and Y1086 or indirectly through phosphorylated Shc protein .
GRB-2 and Shc mediate the activation of transduction of different signals, depending on the type of ligand, the level of expression of the receptor and on the type of EGFR receptor dimerizes with. The study of this cellular process suggested that the association of Shc with EGFR through its domains, which involves tyrosine phosphorylation and recruitment of GRB-2, may be a critical step in the mechanism of activation of EGFR by the employee Ras / MAPK .
The molecules at the top of the cascade of intracellular signal transduction are characterized by the presence of regions of homology with the src oncogene, the so-called SH2 domains (src homology 2) and SH3 (src homology 3). These domains are essential for the interaction between molecules in the chain of transmission.
SH2 domains consist of amino acid sequences that bind phosphorylated tyrosine residues, the SH3 domains are composed of amino acid sequences that recognize sites rich in proline.
The SH3 domains of GRB-2 recognize proline-rich sequences present in the guanidine nucleotide exchange factor Sos (Son of Sevenless), leading to the formation of GRB-2/Sos complex being moved inside the membrane. Sos can activate the Ras protein, a small G protein, inducing the exchange of GDP for GTP. This causes, consecutively, activation of the Raf family of kinases (a-Raf, B-Raf and Raf-1), MEK (Mitogen activated extracellular signal regulated
kinases) and subsequently of Erk1 and Erk2 (Extra-cellular signal-regulated
kinases).
Finally, Erk1/2 kinases, members of the family of MAPK (Mitogen-Activated
Protein Kinases) positively regulate cell proliferation by activating major transcription factors associated with cell proliferation such as c-Myc and isoforms of the RSK (Ribosomal Subunit Kinase) family. In modulating the balance between cell proliferation, apoptosis and senescence, the EGFR/PI3K/Akt pathway is crucial in promoting an intense proliferation through EGFR/Ras/Raf/MEK/ERK cascade (Figure 7).
Figure 7. EGFR signal transduction pathways9
The C-terminal domain of EGFR contains the Y 920 residue, which directly provides the site for the binding of p85 subunit of PI3K (phosphoinositol-3 kinase) either directly or indirectly through binding to GRB-2.
PI3K generates PIP3 (phosphatidyl inositol-3,4,5-triphosphate), which recruits and activates the protein kinase Akt, also known as protein kinase B (PKB), and PDK-1 (phospho inositol-dependent kinase) by binding their PH domain to PIP3.
Phosphorylated Akt protein is able to control the programmed cell death through phosphorylation and consequent inhibition of Bad (a member of the family of pro apoptotic Bcl-2) and Caspase 9 (an enzyme dependent on Fas receptor membrane belonging to the extrinsic activation of apoptosis). The final result is the inhibition of their apoptotic activity and thus promotion of cell survival.
Akt also activates several relevant transcription factors as: HIF-1α, NFkB and CREB that cause an increasing of anti-apoptotic genes. In addition to survival mechanism regulated by Akt, EGFR may activate STAT (signal and activator of transcription) pathways through a JAK (Janus Kinase) -dependent or a JAK-independent mechanism.
Stimulation of EGFR induces phosphorylation of STAT1 and the beginning of the formation of the system STAT1 and STAT2 with JAK1 and JAK2. This allows STAT to be moved into the nucleus within only 15 minutes and this may contribute to cancer cell survival through effects on gene expression. A JAK independent mechanism of activation has been proposed for STAT5b which has a direct docking site at EGFR (Tyr 845).
EGFR-mediated signal transduction also contributes to the regulation of both angiogenesis and metastasis. Angiogenesis, responsible for tumour progression, is mediated by EGFR through up-regulation of VEGF (Vascular Endothelial
Growth Factor) and MMPs (metal protease). Furthermore, the phosphorylation of cytoplasmic domain of EGFR Tyr 992, activating PLCγ (phospholipase Cγ) can directly influence the development of metastases. Tyr 992 is indeed the binding site of PLCγ.
EGFR AND CANCER
The increased activity of EGFR is associated with many human cancers like Non Small Cell Lung Cancer (NSCLC), colorectal adenocarcinoma, glioblastoma, Head and Neck Squamous Cell Carcinoma (HNSCC) and tumours of the pancreas, breast, ovaries, prostate and stomach, with a percentage that varies depending on the type of cancer (Table 3).1,10
Kind Of Tumor
% Of EGFR Expression
NSCLC 40-80
Head and Neck 80-100
Colorectal 25-100 Stomac 33-81 Pancreas 30-50 Ovaries 35-70 Breast 15-37 Prostate 40-90 Glioma 40-92
Table 3. EGFR expression in human tumor
The oncogenic activation of EGFR can occur by various mechanisms: over-expression of ligand or receptor, activating mutations, failure of the mechanisms of inactivation or transactivation by dimerization of the receptor.
Mutated EGFR were identified: among them EGFR vIII (variant III) has been found in most human cancers and is characterized by the absence of the cysteine rich domain.4
EGFR vIII presents a modified binding region and therefore is not able to interact with the ligand and to dimerize, so its kinase activity is constitutive.
Another EGF receptor devoid of the extracellular domain and of 32 amino acids in the carboxy-terminal portion is the product of the viral oncogene v-erb B, that is one of two oncogenes of avian erythroblastosis; also in this case the receptor is constitutively active.11
GATING EGFR 12
The function of all proteins is regulated by a mechanisms of attenuation or gating. Upon activating of many tyrosine kinase, including EGFR, a rapid decrease both in the receptor number at the cell surface and in the cellular content of activated receptors occurs: this process is known as “down-regulation”.
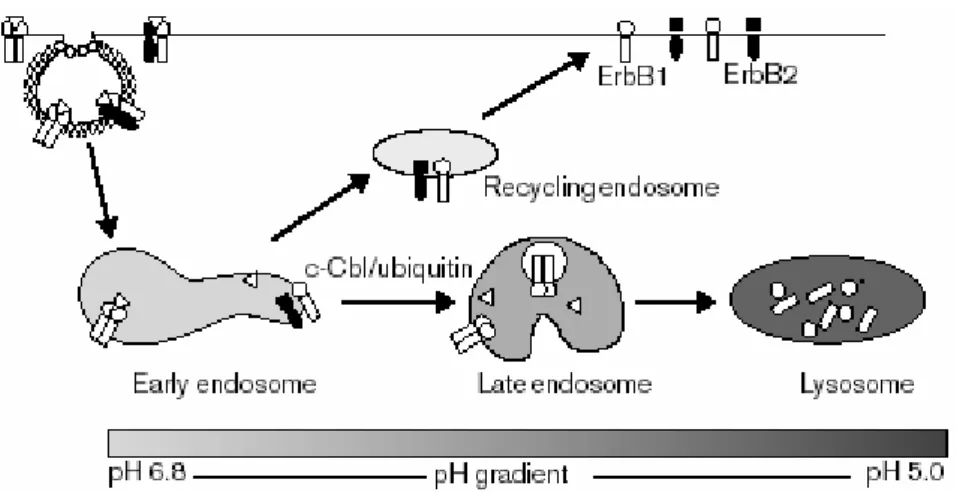
The formation of the ligand-activated receptor complex, in fact, is followed by a membrane invagination that allows the incorporation of the complex itself into the cell (endocytosis).
During this process Cbl plays a key role, by direct interaction with EGFR or indirectly through the aforesaid Src. Their function is to encourage the formation of vesicles incorporating the complex. The vesicles are coated with clathrin, a protein that after ligand-receptor interaction undergoes proliferation forming clathrin-coated pits that allow the entry of vesicles inside the cell; here they merge to form a primary endosome. Primary endosome maturation to late endosome allows the dissociation of the receptors from their ligands: some receptors are recycled and brought back to the plasma membrane, other, are delivered toward lysosomal degradation pathway (Figure 8). The above mentioned mechanism is altered in mutated receptors, they are in fact resistant to lysosomal degradation.
Figure 8. HER receptor endocytosis
About this mechanism, it’s important to observe that exists an activated three-dimensional structure of HER2 that is ligand-independent: this receptor dimerizes preferentially with EGFR, determinig a reduction in EGFR endocytosis and, consequently, in its degradation .
Besides being involved in the mechanism of ligand-mediated endocytosis, EGFR undergoes a spontaneous metabolic turnover, with a half-life of approximately 10-14 hours both in fibroblasts and in epithelial cells and of 20-48 hours in transformed cells. An understanding of the mechanisms governing negatively growth factors and endocytosis signals could lead to identification of new therapeutic targets for the treatment of cancer.
THE EPIDERMAL GROWTH FACTOR EGF
EGF is a 6kDa peptide, consisting of 53 amino acids, discovered in 1962 by Levi Montalcini and Cohen in studies on the factor responsible for the growth of nerve fibres (NGF, Nerve Growth Factor) (Figure 9).13
Figure 9. EGF structure
It was noticed that the injection of extracts of submaxillary gland in newborn mice induced the premature opening of eyelids and accelerating incisor eruption, effects associated to stimulation of epithelial growth and differentiation.
The epidermal growth factor plays a crucial role in promoting the transition of cells from G0 to G1 phase, in which the synthesis of the enzymatic and structural proteins kit necessary for subsequent DNA replication occurs (S phase) (Figure 10).
EGF not only induces increase in the number of sensitive cells but also modulates cellular differentiation, with effects varying depending on the tissue. Structurally, the fundamental characteristic of EGF is a pattern of 6 cysteine residues, highlighted in yellow in Figure 6, located at definite intervals in a 40 amino acids framework sequence that determine the formation of three intramolecular disulfide bridges, thus helping to define the three-dimensional peptide structure.11 (Figure 11)
Figure 11. Amino acid sequence of EGF
The disulfide bonds allow a protein folding that ensures interaction with the specific receptor. EGF is expressed by glandular and lining epithelia and is present in the oral cavity and gastrointestinal tract, where limits the gastric secretion, regulates tropism and mucosal integrity and promotes epithelial regeneration processes. It is also widely expressed in the mammary gland and its presence in the milk suggests an important role in the gastro-intestinal epithelium maturation of the newborn. EGF is present in neglectable concentrations in plasma and in significant amounts in platelets, where it is released after stimuli inducing agglutination and degranulation. Therefore it may be assumed that
locally EGF could contribute to repair processes of wounds and injuries. So, in physiological conditions, EGF plays an important functional role in regulating growth and cellular differentiation.11
DESIGN OF TYROSINE KINASE INHIBITORS
The EGFR activity begins with the interaction of the growth factor on the receptor extracellular region, that activates tyrosine phosphorylation in the receptor intracellular domain by binding with ATP, triggering the cascade of signal transduction.
Tyrosine kinase inhibitors that act at the intracellular domain are small molecules able to across cell membranes, that block or compete with the binding of ATP on the catalytic site, preventing the phosphorylation of tyrosine residues. The catalytic site consists of two kinase domains separated by a deep cleft, where the molecule of ATP is located. The smaller N-terminal domain contains the binding site of ATP and is mainly composed of an antiparallel β-strand and an α-helix. The largest C-terminal domain has a predominantly helical structure and functionally presides to catalysis.14
Conceptually, the binding site of ATP can be divided into five regions (Figure 12):
Adenine region: An hydrophobic region that accomodates the purine ring of ATP, which joins the lobes through the formation of two hydrogen bonds with the polypeptide chain, in which are involved the nitrogen atom N1 and the amino group in the 6 position, which behave as donor and acceptor of hydrogen. This region is also called the "hinge" region. In addition to these polar interactions, the purine ring also forms non-polar interactions with hydrophobic residues located at the N- and C-terminal lobes. The region of adenine’s binding is not characterized by large amino acid variability and therefore the interaction with this site doesn’t provide many useful elements for selectivity towards specific protein kinases.
Sugar Region: Where the riboside portion of ATP is located, whose 2'-OH group forms a hydrogen bond with a polar residue located at the beginning of the C-terminal lobe.
Phosphate region: This region accepts the triphosphate group and is mainly composed of a glycine-rich flexible loop, and an alpha helix structure that properly orients the phosphate group of ATP for catalysis. In most crystal structures of ATP-kinase, a hydrogen bond between the α and β phosphate groups of ATP and a residue of lysine has been found. The γ-phosphate group instead interacts with an arginine residue.
Buried region and solvent accessible region: ATP does not occupy these two regions. They constitute the main source of structural and sequence diversity among the members of the kinase superfamily. The buried region consists in a lipophilic pocket variable in shape and dimension, opposite to the sugar region.
The shape of solvent accessible region depends, however, on the presence or absence of a glycine residue, which can cause a conformational change of the protein between the hinge region and the initial portion of the C-terminal tail.15,16
Inside the EGFR ATP binding site, typical amino acid residues stand out as important structural elements, some of which are common features of the entire RTK family.17 Hydrogen bonds between the 6-NH2 and the backbone carbonyl of Gln 767 and between N1 and the backbone amide of Met 769 fix the purine onto the extended coil stretch, that connects the N-terminal lobe with the C-terminal lobe of the enzyme. A third hydrogen bond between N7 of adenine and the hydroxyl group of Thr 830 located on strand 8 preceding the activation loop may be less important because it is not conserved in all protein kinases.
Non-polar interactions occur between the purine and amino acids Val 702, Ala 719 and Leu 768, which form part of the conserved glycine-rich flap. The ribose and the triphosphate moieties extend towards the opening of the cleft where phosphorylation occurs. The ribose 2'-OH may form a hydrogen bond with Cys 773, either directly or indirectly through water. Despite the high degree of homology, there are some significant amino acids differences in the ATP binding site of EGFR, compared to other tyrosine kinases. The entrance of the binding pocket, which includes the Cys 773 residue, is more hydrophobic than in other kinases. The ATP binding site of EGFR includes three sulfur-containing amino acid residues (Cys 751, Met 769, Met 742) and, in particular, the residue Cys 751 is present only on EGFR and is part of the hydrophobic pocket adjacent to the ATP site. This pocket is present also in other tyrosine kinases as well, but is more shallow due to the exchange of Cys 751 for Val. In the EGFR binding pocket we find also a Thr 766 residue, which seems to be an important centre of interaction of enzyme inhibitors.17
Since the early studies, the design of new ATP-competitive inhibitors selective towards one or a group of protein kinases turned out as a difficult goal, because of the similarities of the binding site inside the kinases family. Despite this, several potent inhibitors with an acceptable degree of selectivity have been reported in the literature.
Chemically heterogeneous compounds, represented by heterocycles both of natural and synthetic origin, have proved to be able to inhibit the tyrosine kinase activity of EGFR.
EGFR INHIBITORS
Over twenty years ago, EGFR has been proposed as a target for cancer treatment for a number of reasons: first the fact that the receptor was over-expressed in various types of cancer cells; in addition, the growth of such cells was inhibited by a series of EGFR monoclonal antibodies both in vitro and in vivo. Preclinical and clinical studies confirmed the possibility of an EGFR-targeted therapy and the clinical activity of EGFR inhibitors was established and approved. There are three major classes of EGFR inhibitors: products of natural origin, such as quercetin and genistein, monoclonal antibodies, directed against the extracellular domain of the receptor, such as cetuximab and "small molecules", competitive inhibitors of the ATP binding site on the tyrosine kinase domain of EGFR, such as gefitinib and erlotinib. Therapeutic options should also be remembered as the use of antisense oligonucleotides or ribozymes that decrease the expression of EGFR.9,18
During the first research done in this area, the products of natural origin quercetin and genistein have proved to effectively inhibit EGFR.19,20 They show inhibitory properties in micromolecular concentration against a high range of kinase proteins and therefore have a low specificity.
The biflavonoid quercetin is both free and in conjugated form in many fruits and vegetables, in vitro it is able to inhibit the growth and proliferation of malignant cells, aerobic glycolysis of Ehrlich ascites cells and phosphorylation of oncogene src of Rous sarcoma virus,19 src is an example of a viral oncogene that
can induce tumour development in the host cell through the codification of the known phosphoprotein pp60v-src (PM60000).
The isoflavon genistein, which occurs mainly in soy, is a potent tyrosine kinase inhibitor: it blocks the autophosphorylation of EGFR (IC50 2.6 µM), src (IC50 26 µM) and other tyrosine kinase.20
The second approach to the inhibition of EGFR is represented by monoclonal antibodies; they are able to interact with the extracellular domain of the receptor by competing with its ligand and thus blocking activation. This process results in inhibiting cell growth.
Cetuximab (IMC-225), for example, is a G1 immunoglobulin (IgG1), that interacts with the domain III of EGFR, partially occluding the ligand binding region on this domain. Although ligand-binding to domain I is unaffected, growth factors must engage sites on both domains I and III for high-affinity binding, so blockade of either one is sufficient to prevent signalling. Cetuximab is also able to promote EGFR internalization. Once internalized, the receptor is then degraded without phosphorylation and/or activation, resulting in a down regulation from cell surface and consequent reduction of EGFR-dependent signalling pathways.9
Another action of cetuximab is blocking the conformational change of the receptor, which is essential for dimerization and proliferation of pro-angiogenic factors. Cetuximab has been shown to inhibit various human tumour cell lines, action due to cell cycle arrest in G1 phase and/or apoptosis. This antibody has been evaluated in clinical trials both as single agent and in combination with conventional chemotherapy and radiation therapy. Because inhibition of EGFR and conventional therapies act potentially through different cytotoxic mechanisms, theoretically a combination of therapies offers the potential advantage of synergism of activity, without overlapping of side effects.6,18
Moreover, there is another class of anti-EGFR monoclonal antibodies that prevent receptor dimerization, such as pertuzumab (2C4). This antibody binds the HER2 receptor and prevents its hetero-dimerization with other HER family members.
of the receptor, thus blocking its catalytic activity. Some of them induce the formation of inactive EGFR homodimers or EGFR/HER2 heterodimers. Such molecules, in addition to the ability in inactivating of both receptors belonging to the HER family, also inhibit the mutated receptors avoid of the extracellular domain.
Monoclonal antibodies and small molecules have different mechanisms of action, although they lead to the same effects on signal transduction, effectively blocking EGFR signal transduction, including MAPK and PI3K/Akt and the Jak/Stat cascade. A significant difference between monoclonal antibodies and small molecules is that the former are selective inhibitors against EGFR, while the latter are less selective inside HER family members; in fact, if some cancer cells previously inhibited by small molecules are treated with monoclonal antibodies, the anti-tumour activity increases. This suggests a possible combined therapy of the two different types of receptor inhibitors.18
The first important class of synthetic heterocyclic inhibitors was that of anilinoquinazolines, with PD153035 (discovered by Parke-Davis Pharmaceutical
Research, in 1994) being the most active compound. In fact this agent selectively inhibits EGFR with an IC50 of 5pM and blocks other tyrosine kinase only at higher concentrations, around 50µM. PD 153035 prevents the receptor autophosphorylation in fibroblasts and human epidermal carcinoma cells, and blocks cellular processes such as mitogenesis, gene over-expression and oncogenic transformation.21 N N NH Br O O PD153035
Based on the structure of this active molecule, another series of active compounds has been developed. The structural requirements identified inside this class are the presence of electron donating substituents in 6 and 7 positions of quinazolines, a small lipophilic substituent (a halogen atom) in the meta position of aniline, a NH group in the 4 position and free CH groups in the 2, 5, 8 positions.
Among the new synthetic molecules, an anilinoquinazoline derivative, ZD1839 (AstraZeneca),22 has emerged and was the first to be approved by the FDA in 2003, with the name of gefitinib, for the treatment of non-small cell lung cancer (NSCLC). Gefitinib is active when administered orally and is a reversible competitive type, potent and selective EGFR inhibitor.6,23
N N NH F Cl O N O O Gefitinib
The activity of ZD1839 on EGFR in vitro was 23nM, thus lower if compared to that shown by PD153035, but the good bioavailability properties and the ability to induce marked regression in some tumours have led to the development of this molecule as a drug.
On the contrary, in vivo, because of high intracellular concentration of ATP, higher doses are needed to lead the inhibition of other tyrosine kinase as HER2. In fact, gefitinib inhibits HER2 autophosphorylation with an IC50 of 1µM in breast cancer cells in which this receptor dimerizes with EGFR.6
In order to increase activity in vivo the methoxyl groups of PD153035 were modified, by the introduction of, an alkoxyamine side chain, that improved the physical properties of basicity, lipophilicity and solubility. Moreover, the introduction of a fluorine atom in the para position gives greater metabolic
In vitro effects of gefitinib as a single agent were mainly cytostatic, although cytotoxic effects have been observed in a few cases. It has been suggested that gefitinib could favour several mechanisms involving pro-apoptotic members of Bcl2 protein family such as Bad.25
Gefitinib did not confer an overall survival advantage; however, gefitinib treated patients survived longer than those placebo-treated in two specific patient subsets: individuals of Asian origin (median, 9.5 versus 5.5 months, respectively) and never-smokers (median, 8.9 versus 6.1 months, respectively).26
Gefitinib appears to indirectly inhibit angiogenesis, the growth of cancer cells in human colon, breast, ovaries and stomach, in vivo accompanied by reduced production of VEGF and TGF-α. Treatment with gefitinib is associated with a rapid regression of the mentioned tumours, but if the administration is discontinuous the tumour grows, so a long-term drug administration is required to maintain a response in patients.6
Gefitinib has been combined with chemotherapy with a variety of cytotoxic agents, resulting in enhanced anti-tumour effects in cultured cells and in vivo
models, except in combination with gemcitabine. In addition, gefitinib treatment resulted in synergistic effects in combination with radiation. Importantly, sequence-dependent effects were reported in cells treated with combinations of gefitinib with radiation or chemotherapy (cisplatinand/or 5-fluorouracil), with the best results observed whengefitinib was administered before radiation and before or during chemotherapeutic treatments; whereas some antagonistic effects were observedwhen gefitinib was given after cytotoxic treatments.25 Side effects of this molecule are: diarrhoea, nausea, vomiting and rash.
Erlotinib (OSI774,OSI/Genentech, Tarceva®), lapatinib (GW572016, Glaxo
Smith-Kline)27 and canertinib (IC 1033, Pfizer)28 are other 4-anilinoquinazoline derivatives with potent inhibitory activity against this tyrosine kinase, with IC50 values in the nanomolar order.
N N NH O O O O HC Erlotinib N N NH Cl O CH2NHCH2CH2SO2CH3 O F Lapatinib N N NH F Cl NHCOCH=CH2 O N O Canertinib
While erlotinib is rather selective for EGFR (IC50 = 2 nM) and requires one
order of magnitude higher concentrations for the inhibition of HER-2, lapatinib is active on both receptors with IC50 values comparable. EGFR and HER2 have
homologous domains and their simultaneous inhibition represents a viable therapeutic opportunity for patients with cancer. An over-expression of HER2 was observed in breast cancer and a co-over-expression of both receptors in patients with ovarian cancer.29
Although the mechanism of action is equal to that of gefitinib, erlotinib appears to be more active.26 Studies have demonstrated its inhibitory activity on many cancers such as renal carcinoma, colorectal carcinoma, NSCLC, pancreatic cancer. Prolonged treatment of patients with this drug leads to a reduction of
metastases by 30%. Side effects are similar to those caused by gefitinib, suggesting that these events are related to HER1 inhibition.4
Canertinib28 is an irreversible inhibitor of EGFR (IC50 1.5 nM) and HER2 and
HER3 kinase activity;28 from a structural point of view, is characterized by the presence of an acrylamide chain in the 6-position. It showed an inhibitory activity of autophosphorylation of EGFR and HER2 at concentration 7.4 nM and 9nM, respectively. Its ATP-competitive action is irreversible due to its ability to bind covalently to the 773 cysteine residue at the entrance of ATP binding pocket. To ensure the same bioavailability characteristics of gefitinib, two essential features, the morpholine group and the para-fluorine atom of aniline, are maintained.28
Erlotinib was approved as a drug for NSCLC and pancreatic cancer treatment; lapatinib and canertinib are currently undergoing phase I/II clinical studies.
NEWS ON THE CLINICAL USE OF EGFR INHIBITORS
EGFR inhibitors are a dynamic field of research and development, as they have characteristics that make them particularly versatile in combating tumour progression. So we can list the clinical benefits that can be derived by this new class of anti-cancer drugs:
• Effectiveness in many types of solid tumours. • Utility for long-term treatment.
• Possibility to use either alone or in combination with other drugs. • Good tolerability.
• Absence of haematotoxicity.
• Capability to reduce resistance to radiotherapy or hormone therapy.
• Improvement of the quality of life with respect to other types of anticancer drugs. • Pre-medication or monitored dosage are not requested.
Table 4 shows the description of some drugs used in the testing.29
Drug Action Target Tumour type Clinical stage
Gefitinib IRESSA ZD1839 Reversible inhibitor, ATP-competitive EGFR NSCLC Approved (2003) Erlotinib TARCEVA OSI-774 Reversible inhibitor, ATP-competitive EGFR NSCLC, pancreatic cancer Approved (2004) Lapatinib GW572016 TYKERB EGFR, ErbB2 Advanced ErbB2 positive breast cancer,
NSCLC, HNSCC
Approved (2007) Phase II Canertinib
CI-1033 Irreversible inhibitor
EGFR,ErbB2, ErbB3 NSCLC and breast cancer Phase I / II Cetuximab Erbitux IMC-225 Chimeric
human-murine mAb EGFR
CRC and pancreatic cancer Approved (2004) Phase II Pertuzumab rhumAb2C4 Omnitarg
Humanized mAb ErbB2 dimerization
Breast, prostate, ovarian
cancers Phase II / III
Table 4. EGFR inhibitors in clinical development
RESISTANCE TO "EGFR-TARGETING" THERAPIES
Differences have been observed in the responses of individual patients treated with EGFR inhibitors. The first difference emerged was a close dependence of therapeutic efficacy on the "contents" of EGFR, that is its expression rate in cells. Other differences in the response to therapy are concerned with the presence of mutations: these, depending on their location, may influence the effect of some EGFR inhibiting agents:
- The presence of mutations at the extra-cellular level affect therapy with monoclonal antibodies;
- Mutations affecting amino acids in the ATP-pocket on the intracellular tyrosine kinase domain affect response to drugs such as gefitinib: patients affected
by these mutations respond much better to therapy, at least in the initial phase of treatment.
Indeed it was noted that in some patients, who initially responded to administration of gefitinib, after a prolonged treatment EGFR acquires a second mutation that leads to a high resistance to these substances;18,30
- Mutations at intermediate factors, such as k-Ras, Raf, PI3K are elements that can influence the activity of EGFR-inhibitors.
This different sensitivity to various treatments depending on the factors listed above requires a careful selection of patients to proceed with an appropriate therapeutic protocol.
NEW TREATMENT GUIDELINES29
In the attempt to improve the effectiveness of cancer therapy more and more combination therapies are emerging, which may be essentially of two types:
- Combination therapies aimed at the same target, either using irreversible or reversible inhibitors, characterized by different spectrum of resistance induced by mutation. A clinical trial with lapatinib and trastuzumab has led to encouraging results,
- Therapy directed towards more different receptors; there are many tyrosine inhibitors very poorly selective against an unique receptor. This perspective can be exploited by the selection of agents targeting more kinases involved in tumour progression.
Recently new multi-target agents have been developed, including inhibitors of EGFR and other HER family members (as canertinib, inhibitor of EGFR, HER2 and HER3), or multi-target inhibitors of EGFR tyrosine kinases belonging to different groups.
INDIRECT AND DIRECT TARGETING OF ANGIOGENESIS PATHWAYS
The development of a vascular supply is essential not only for organ development and differentiation during embryogenesis, but also for wound healing and reproductive functions in the adult. Angiogenesis is also implicated in the pathogenesis of a variety of disorders: proliferative retinopathies, age-related macular degeneration, rheumatoid arthritis, and psoriasis and demonstrated to be a fundamental event in the process of tumour growth and metastatic dissemination. Hence, the molecular basis of tumour angiogenesis has been of keen interest in the field of cancer research. The vascular endothelial growth factor (VEGF) pathway is well established as one of the key regulators of this process.31 The EGFR and VEGFR pathways seem to be closely linked in solid tumours, particularly with respect to angiogenesis. Simultaneous inhibition of EGFR and VEGFR pathways might provide improved efficacy or anti-angiogenesis actions over blocking either pathway alone as a result of indirect and direct down-regulation of angiogenic pathways and direct tumour targeting with EGFR antagonists.5
VASCULAR ENDOTHELIAL GROWTH FACTORS AND RECEPTORS VEGF/VEGFR
The VEGF family and its receptors are key regulators of angiogenesis and are highly specific for endothelial cells.32
VEGF was isolated and cloned in 1989 by Ferrara, Plouet and co-workers33; the predominant member of this family is VEGF-A, a 45 kDa glycoprotein composed of 165 amino acids34 that interacts with two transmembrane tyrosine kinase receptors: VEGFR-1 and VEGFR-2. VEGF-A also exists in other isoforms characterized by a different number of amino acids.
Figure 13. VEFG structure
The VEGF-B protein trough its binding to VEGFR-1 exert a mitogenic effect on endothelial cells and moreover may link VEGF-A, increasing its action. VEGF-C and VEGF-D are structurally similar and may be considered as members of a subfamily (Figure 13). These proteins, interacting with VEGFR-2, induce a slight mitogenic effect on endothelial cells, while binding VEGFR-3 regulate lymphangiogenesis. At last, VEGF-E, the most recently identified factor, is specific only for endothelial cells of endocrine glands.
Human VEGFR-1 and VEGFR-2 consist of 1338 and 1356 amino acids, respectively. They are structurally similar and formed by four region: the extracellular ligand domain, a transmembrane domain, an intracellular kinase domain that is comprised of a regulary domain and a catalytic domain tethered togheter by a hinge region, and downstream carboxy-terminal region. The kinase domains of VEGFR-1 and VEGFR-2 are the most conserved regions, with about 70% homology, while the carboxy-terminus rapresents the most differentiated region.35,36 Both VEGFR-1 and VEGFR-2 are located on surface of vascular endothelial cells, and they bind VEGF with high affinity. More recently, a third receptor has been discovered, VEGFR-3, which is closely related to VEGFR-1
and -2 and activated by VEGF, but limited in action to lymphangiogenesis; its expression is associated with the dissemination of tumor cells to regional limph nodes.37
Figure 14. Representation of vascular growth factors and their receptor interaction38
VEGFR SIGNALLING39
The mechanism by which VEGFR-1 and VEGFR-2 receptors trasduce signal have been exstensively studied at the molecular level. The binding of VEGF to two proximal VEGFR-2 receptors causes their dimerization, following activation and autophosphorylation of the intracellular kinase domain, which catalyzed the phosphorylation of cytosolic substrate proteins and downstream cellular events (Figure 14). Among several tyrosine residues that have been shown to be phosphorylated, two major VEGF-dependant autophosphorylation sites have been
identified in VEGFR-2; however, only the autophosphorylation of Tyr 1175 has been identified as crucial for VEGF-dependant endothelial cell proliferation.40
Figure 15. VEGFR pathway
Activation of the MAPK pathway in response to VEGF has been observed in many types of endothelial cells. The PLC-γ-PKT pathway has also been implicated in the mitogen action of VEGF. VEGFR-1 interacts with the PLC-γ SH2 domain, inducing the phosphorylation and activation of PLC-γ leading to the hydrolysis of phosphatidylinositol-4,5-bisphosphate to diacylglycerol and inositol-1,4,5-trisphosphate. Inositol-1,4,5-trisphosphate is likely responsible for the increase in intracellular Ca2+ after VEGF stimulation, whereas diacyl-glycerol, in turn, activates PKC isoforms expressed in the target cells.
VEGF also activate PI3-K that activates Akt, a serine kinase involved in antiapoptotic signaling. Akt has also been reported to directly activate endothelian nitric oxide synthase, suggesting that Akt may regulate the increased production of nitric oxide in response to VEGF stimulation. STATs are latent cytoplasmic transcription factors. STAT activation by the VEGFRs has been studied in transient transfection assay. All three receptors were shown to be strong activators of STAT3 and STAT5, whereas STAT1 was not activated by the VEGFRs. However, the role of this pathway in endothelian cell biology is unknown(Figure 15).
Although very little is known about the specific signal transduction of VEGFR-3 in the lymphatic endothelium, mutations in VEGFR-VEGFR-3 have been linked with hereditary lymphedema, an autosomal dominant disorder of the lymphatic system.
Endothelial cell proliferation and survival in response to VEGF may require the association of VEGFR-2 with cell surface adhesive proteins. Activated VEGFR-2 was found in a complex with integrin αvβ3, an adhesion molecule specifically expressed on angiogenic endothelium; αvβ3 has been shown to be involved in the regulation of the cell cycle and the survival of endothelial cells. VE-cadherin, an endothelium-specific cell-adhesion protein, has also been implicated in molecular interactions with the VEGF.
VEGF/VEGFR IN ANGIOGENESIS41
Angiogenesis is the process that leads to the formation of new capillaries sprouting or splitting from pre-existing vessels, in fact, in order to grow beyond a size of 1-2 mm, tumors need new blood capillaries to create their own nutrient supply, to remove metabolic waste and to encourage metastasis formation.42 Hypoxic conditions in the centre of tumor mass stimulate the release of proangiogenic factors from tumor cells that are localized in the proximity of pre-existing blood vessels, and lead to the formation of new capillaries around the tumor mass.15
VEGF and its receptors have a central role in promoting cancer and a strategic position in the regulation of angiogenesis; although the precise role of VEGFR-1 function is still emerging, in recent years several studies have suggested that this receptor may play both negative and positive role in angiogenesis. In fact, VEGFR-1 has negative function, probably trapping VEGF-A in the embryo, while showing a positive role in adulthood in a tyrosine kinase-dependent manner. VEGFR-1 acts primarily as a decoy receptor, modulating the availability of VEGF for VEGFR-2, which is the principal receptor for VEGF signalling.43
Elevated VEGF expression, due to abnormal and inefficient tumor vasculature, with many blind ends, and it is difficult for convetional drug molecules to gain access to the tumor tissue; moreover, drug penetration is diminished by the high interstitial pressure of many tumor.44 Anti-VEGF therapy may produce vascular normalization, resulting in a decrease in vascular volume and interstitial pressure, leading to enhanced delivery of cytotoxic therapeutic agents.
So blockade of angiogenesis is one of the more attractive approaches for the treatment of both solid tumors and haematological malignancies.45
In human disease, VEGFR-2 was shown to have a major role in tumor angiogenesis and diabetic retinopathy, while VEGFR-1-dependent signalling was shown to play a role in angiogenesis of certain tumors, the progression of rheumatoid arthritis, and aterosclerosis.46,47
ANTI-VEGFR AGENTS
Therapeutic strategies targeted at blocking VEGF or its receptor signalling systems are an attractive approach for the treatment of different diseases, primarily tumors. Therapies molecularly targeted at angiogenesis are supposed to be less toxic for chronic administration, in comparison with conventional treatments such as chemotherapy, but it is thought that anti-VEGF agents alone will be not sufficient for cancer monotherapy; in fact, such molecules are often evaluated in clinical trials in combination with chemotherapy.48
Agents currently in development against VEGF or its receptors include neutralizing monoclonal antibodies (such as bevacizumab), and small molecules TK-inhibitors.
MONOCLONAL ANTIBODY
The most advanced approach to VEGF-dependent angiogenesis inhibition is the humanized monoclonal antibody directed against VEGF-A, bevacizumab, by Genentech.34,49 Bevacizumab prevents the binding of all VEGF isoforms to all VEGFRs; in preclinical animal model, it blocks the growth of a variety of human tumor xenografts. The results obtained in clinical trials on different malignancies such as: combination with 5-fluorouracil- and irinotecan-based chemotherapy in metastatic colorectal cancer, and association with paclitaxel in metastatic breast cancer, led to its approval by the FDA as a first-line treatment in combination with chemotherapy for metastatic colorectal cancer.
This antibody is generally well tolerated, even though toxicities such as gastrointestinal perforation and tromboembolic complications have been observed in a small percentage of cases.50
A different anti-VEGF-A monoclonal antibody, ranibizumab, has been developed by Genentech for the treatment of the age-related macular degeneration (AMD), charaterized by neovascularisation; this new agent maintains or improves vision and was approved by the FDA in June 2006.51
Aptamers are nucleic acids that recognize their target with high affinity and specificity.
Macugen® is a modified RNA developed by Eyetech Pharmaceuticals and Pfizer that inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF-A with extremely high affinity (Kd = 50 pM), and was approved
by USA (United States) FDA in December 2004 for the treatment of AMD.52 AngiozymeTM, by Chiron Corporation/Rybozyme Pharmaceuticals, is a stabilized rybozime against the pre-mRNA of the VEGF-1 gene that reduces the
and the truncated soluble forms in plasma.53 Recent phase I clinical trials showed that Angiozyme is well tolerated, with minimal toxicities and good bioavailability. Phase II trials for specific tumor types are ongoing to further asses the biological and clinical activity of Angiozyme.54
A different anti-angiogenic approach is based on the use of a soluble recombinant decoy receptor, called VEGF Trap, a protein constructed from the VEGF receptor-binding domain linked to an immunoglobulin. It posses a high affinity for VEGF and demonstrated marked efficacy in inhibiting angiogenesis and the growth of tumors in preclinical animal models. Currently, it is being studied in phase I clinical trials in humans with advanced solid malignancies55 and for treatment of patients with intraocular neovascularisation due to AMD.56
The negative aspects of both anti-VEGF and anti-VEGFR based therapies are the high cost of manufacturing and the necessity for parental application.48
SMALL MOLECULES TK INHIBITORS
Another important class of angiogenesis inhibitors currently in development is that of the small-molecule tyrosine kinase inhibitors. Generally these compounds have dual activity inhibiting both VEGFR-1 and VEGFR-2 in vascular endothelial cells.
Among ATP site-directed inhibitors of VEGFR, indolin-2-ones, identified in 1993, represent an important class of anti-angiogenesis agents.57,58 The oxindole derivative sunitinib (SU11248) is an inhibitor of VEGFR-1, VEGFR-2, VEGFR-3 (IC50 values 15, 38, 30 nM respectively), but also inhibits PDGFRs
(Platelet-Derived Growth Factor Receptors), Kit, Flt3, RET (REarranged during Transfection), and CSF-1R (Colony Stimulating Factor 1 Receptor) with similar efficacy, and is approved multinationally for the treatment of advanced renal cell carcinoma (RCC) and imatinib-resistant or imatinib-intolerant gastrointestinal stromal tumour. In January 2006, it was approved by FDA for the treatment of gastrointestinal and kidney cancer.
F N H O N H O NH NEt 2 Sunitinib
Sunitinib is characterized by a 5-fluoro-substituted indolinone structure, bearing a diethylaminoethyl group that confer good solubility. The co-crystal X-ray structure of the catalytic domain of the FGF (Fibroblast Growth Factor) receptor with several oxindoles suggests that the compound bind in the ATP pocket with the indolin-2-one core participating in key H-bond donor/acceptor capacities with the carbonyl of Glu 915 and the NH Cys 917, typical residues of the hinge region of VEGFR-2. Other key SAR features include the preference of a Z methylidene geometry that can be enforced by a heteroaromatic ring capable of participating in intramolecular H-bond interaction with the indolin-2-one core.57,58 Another important class of angiogenic inhibitors is that of anilinophthalazines disclosed by Novartis. These compounds were selective for human VEGFR.
The anilinophtalazine derivative valatanib52 (PTK787/ZK222584) is one of the most potent and selective first-generation VEGFR kinase inhibitors, with IC50
values of 110, 43, 195 nM against VEGFR-1, VEGFR-2, VEGFR-3, respectively. It also inhibits other kinases including PDGFR-β and c-Kit, but at higher concentrations. This compound is currently in phase III clinical trials for metastatic colorectal cancer.
Valatanib was docked in a model of ATP binding site of VEGFR-2 constructed using the available X-ray structures of the domain of FGF receptor 1.
According to Authors’ hypothesis, valatanib does not form direct hydrogen bonds with the peptide backbone of the hinge region as does ATP and many reported kinase inhibitors, but rather occupies the hydrophobic regions of the binding site.
N N HN Cl N COOH COOH Valatanib
The aniline moiety is located in a hydrophobic pocket, while the phthalazine bicycle also makes hydrophobic contacts with other amino acids. Although none direct hydrogen bond with the hinge region is established, the aniline NH group forms water-mediated hydrogen bonds with Glu 915 and Cys 917 of the hinge region of VEGFR, and the inhibitor pyridil nitrogen is assumed to form a hydrogen bond with Lys 1060, a residue of the kinase activation loop.52
In the context of the same structural class of gefitinib (anilinoquinazolines) appropriate modifications of the substituents have led to the identification of vandetanib, (AstraZeneca) a novel drug, that joins two actions with a dual pharmacological profile, is an orally active tyrosine kinase inhibitor that exhibits potent agonist activity toward VEGFR-2 and, to a lesser extent, to EGFR.
N N NH Br F O O N CH3 Vandetanib
The clinical development of vandetanib is being progressed, as in vivo tests have demonstrated the ability to inhibit, in a dose-dependent way, the growth of a
wide range of cancers. Its administration in mice leads to inhibition of VEGF signal, of angiogenesis, of neuro-vascularization induced by the tumour, and the tumour growth. Vandetanib is undergoing phase I clinical testing in patients who have advanced solid tumours.39,40