53
CAPITOLO 3: RISULTATI E DISCUSSIONE
3.1 NOMENCLATURA
La nomenclatura dei poliuretani utilizzati si basa sulla natura dei segmenti costituenti . La prima lettera indica l’estensore di catena (C per CDM e L per L-lisina etil estere), la seconda lettera indica il diisocianato (L per LDI), le lettere e cifre seguenti si riferiscono al poliolo. Le sigle C-1250 o C-2000 indicano il PCL diolo di peso molecolare medio numerale rispettivamente di 1250 o 2000, la sigla CE-6XX si riferisce al copolimero a tre blocchi PCL-PEG-PCL in cui il segmento di PEG ha peso molecolare 600 e contenente una percentuale molare di unità ossietileniche pari a XX. Ad esempio la sigla CLCE-650 indica un poliuretano i cui segmenti costituenti sono: CDM, LDI e il copolimero a tre blocchi con il 50% di unità ossietileniche e contenente PEG 600 come blocco centrale.
Estensori di catena:
CDM
L-lisina etil estere
Diisocianato: LDI HO CH2 CH2 OH NH2 O OEt NH2 O C N CH2 CH N C O COOEt 4
54 Polioli:
PCL diolo
PCL-PEG-PCL
Per il copolimero a blocchi è stata mantenuta la sigla C27 utilizzata in letteratura
55
3.2 PREPARAZIONE E CARATTERIZZAZIONE DELLE
MICROSFERE
Le microsfere sono state preparate con la tecnica di evaporazione-estrazione del solvente ad emulsione singola olio in acqua come riportato nel paragrafo 2.4. I parametri sperimentali quali rapporto fase acquosa/fase organica, velocità e tempo di agitazione meccanica e modalità di isolamento delle particelle dall’ambiente di reazione, sono stati mantenuti costanti al fine di attribuire ogni differenza nella morfologia e nelle proprietà delle microparticelle alle diversità strutturali delle matrici polimeriche, alla quantità di farmaco impiegato e ai diversi tipi emulsionanti utilizzati. La temperatura di lavoro è stato l’unico parametro non monitorato: è stato infatti scelto di lavorare a temperatura ambiente per cercare di ottenere microsfere di dimensioni non troppo elevate [53]. Il rischio connesso con tale modo di operare è il favorire eventuali fenomeni di coalescenza delle particelle che, come è noto, vengono inibiti alle basse temperature.
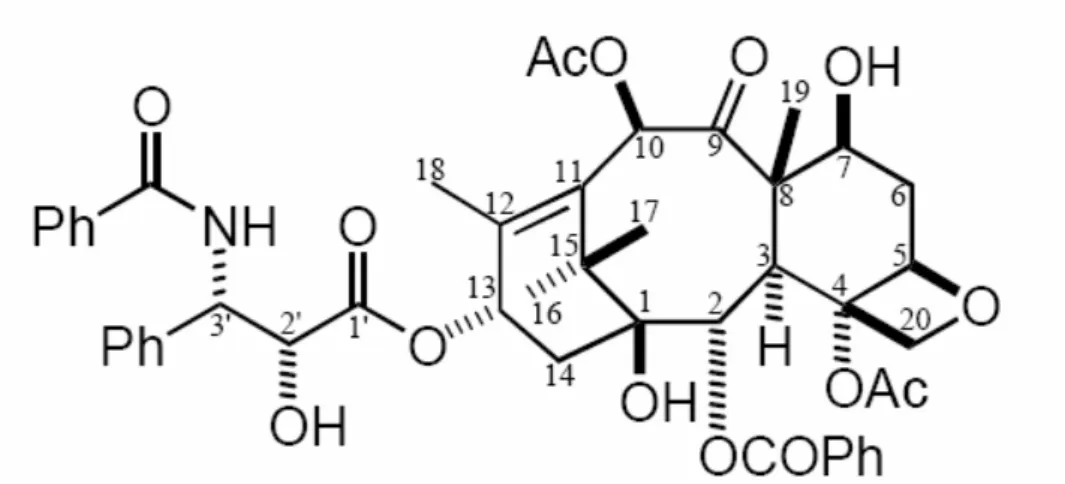
Come agente farmacologico è stato utilizzato il Paclitaxel (PXT), un composto appartenente alla famiglia dei Tassani ricavato dalla Taxus brevifolia. Il PXT è un farmaco antiproliferativo ampiamente usato nei trattamenti antitumorali e recentemente impiegato come agente antirestenoico. Nella figura 3.0 viene riportata la struttura del PXT.
56 Ogni tipo di polimero utilizzato, è stato sciolto, insieme al farmaco fortemente idrofobo, in 5 ml di diclorometano e tale fase organica è stata dispersa in 100 ml di acqua distillata contenente un emulsionante, ottenendo così un rapporto fase organica/fase acquosa 1:20. Per ottenere una microemulsione, le due fasi sono state sottoposte ad agitazione meccanica a 800 rpm per due ore. La velocità di agitazione utilizzata è idonea per ottenere particelle di dimensioni micrometriche. È stato inoltre mantenuto un tempo di agitazione discretamente lungo, per cercare di ottenere delle particelle con una dispersione monomodale anche se tale proposito è difficile da raggiungere con la tecnica utilizzata.
A causa dell’elevata volatilità del solvente organico, durante la fase di agitazione l’ambiente di reazione è stato isolato dall’ambiente esterno per minimizzare perdite premature di DCM in seguito all’agitazione stessa. Il requisito fondamentale della tecnica di evaporazione-estrazione del solvente è infatti la lenta velocità con cui il solvente stesso deve essere allontanato dall’ambiente di reazione. Ciò è necessario per garantire, tra l’altro, una buona morfologia particellare e un adeguato controllo sulla porosità superficiale, parametri che influenzano sensibilmente il profilo cinetico di rilascio [60].
Dopo la fase di agitazione a 800 rpm, la velocità è stata diminuita a 250 rpm e il recipiente di reazione è stato aperto per permettere l’evaporazione del solvente. La microemulsione è stata quindi lasciata sotto agitazione a tale velocità per tutta la notte. Dopo tale periodo la dispersione finale presentava un aspetto lattiginoso tipico di uno stato colloidale.
Le particelle sono state infine isolate dall’ambiente di reazione mediante centrifugazione selettiva. Una primo ciclo di centrifugazione è stato effettuato a 5000 rpm. Il surnatante è stato successivamente ricentrifugato a 14000 rpm. Tale doppia centrifugazione è stata effettuata per separare quanto più possibile, le microsfere di dimensioni maggiori da quelle di dimensioni minori. La maggior parte delle particelle è stata recuperata dalla prima centrifugazione, mentre il surnatante derivante dalla seconda centrifugazione è stato scartato benché non si presentasse completamente limpido. Successivamente le particelle sono state accuratamente lavate diverse volte con acqua distillata per eliminare l’eccesso di emulsionante. Dopo l’ultimo lavaggio, le particelle sono state riprese e ridisperse con la minor quantità di acqua possibile, e sono state poste in liofilizzatore per poterle recuperare in forma di polvere. Infine, sono state
57 conservate in essiccatore per evitare l’adsorbimento e l’assorbimento di umidità, fenomeni particolarmente pronunciati per i sistemi particellari [52].
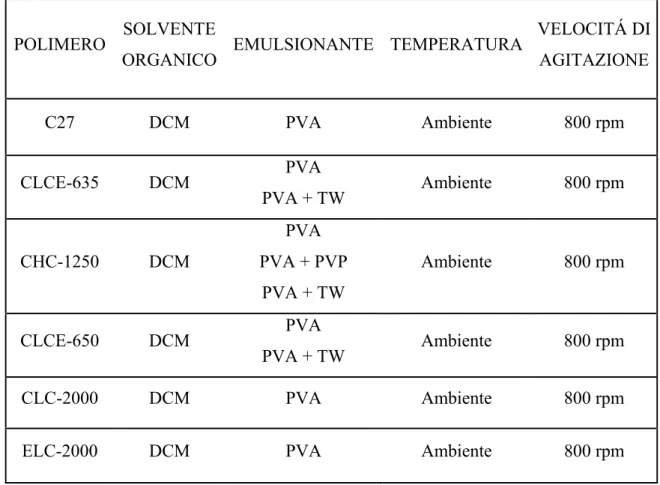
In tabella 3.1 vengono riassunti i parametri operativi per la preparazione delle microsfere insieme ai diversi polimeri ed emulsionanti utilizzati.
TABELLA 3.1 Parametri e materiali utilizzati per la preparazione delle microsfere
POLIMERO SOLVENTE
ORGANICO EMULSIONANTE TEMPERATURA
VELOCITÁ DI AGITAZIONE C27 DCM PVA Ambiente 800 rpm CLCE-635 DCM PVA PVA + TW Ambiente 800 rpm CHC-1250 DCM PVA PVA + PVP PVA + TW Ambiente 800 rpm CLCE-650 DCM PVA PVA + TW Ambiente 800 rpm CLC-2000 DCM PVA Ambiente 800 rpm
ELC-2000 DCM PVA Ambiente 800 rpm
3.2.1 ANALISI MORFOLOGICA E DIMENSIONI DELLE
MICROSFERE
La morfologia delle microsfere, è stata studiata mediante analisi al microscopio a scansione elettronica (SEM) e al microscopio a forza atomica (AFM).
Le immagini, relative alle microsfere recuperate dalla prima centrifugazione a 5000 rpm (Figure 3.1-3.11), mostrano particelle con una buona sfericità e una superficie non macroporosa ad eccezione delle microsfere di ELC-2000 che mostrano un certo grado di porosità. La figura 3.12 rappresenta invece l’immagine relativa alle particelle di C27 ottenute dalla centrifugazione a 14000 rpm.
58 FIGURA 3.1 Microsfere di C27 (Paclitaxel 20% w/w)
59 FIGURA 3.3 Microsfere di CLCE-635 (PVA) (Paclitaxel 5% w/w)
60 FIGURA 3.5 Microsfere di CHC-1250 (PVA) (Paclitaxel 5% w/w)
61 FIGURA 3.7 Microsfere di CHC-1250 (PVA+Tween80) (Paclitaxel 5% w/w)
62 FIGURA 3.9 Microsfere di CLCE-650 (PVA+Tween80) (Paclitaxel 5% w/w)
63 FIGURA 3.11 Microsfere di ELC-2000 (PVA) (Paclitaxel 5% w/w)
64 Come si può vedere confrontando le figure 3.1 e 3.2, la quantità di farmaco utilizzato durante la preparazione, non influisce sulla dimensione delle particelle. Risultati analoghi sono stati trovati per le altre tipologie di microsfere. Le particelle preparate a partire dai polimeri contenenti dei blocchi di PEG in catena e utilizzando PVA come emulsionante (figure 3.1, 3.2, 3.3, 3.8), mostrano una morfologia migliore rispetto a quelle preparate da poliuretani aventi poli(caprolattone) come segmento soft. A titolo di esempio in figura 3.13 viene riportata l’immagine di una singola microsfera di CLCE-650 dalla quale si può apprezzare con maggior chiarezza l’assenza di porosità e l’aspetto rugoso della superficie.
FIGURA 3.13 Ingrandimento di una microsfera di CLCE-650 (PVA)
Con l’analisi AFM, è in generale difficile ottenere immagini ottimali per campioni di micro e nanoparticelle. Come esempio vengono riportate nelle figure 3.14-15 le immagini ottenute all’AFM per le microsfere di C27 e CLCE-635 (PVA) nelle quali si può notare ancora una volta, la superficie non porosa delle particelle.
65 FIGURA 3.14 Immagine AFM delle microsfere di C27 (Paclitaxel 5% w/w)
66 La presenza o meno di PEG nelle macromolecole influenza sensibilmente le dimensioni delle microsfere: quelle preparate a partire da C27, CLCE-635 e CLCE-650 presentano mediamente dimensioni inferiori, a parità di emulsionante, rispetto alle particelle di CHC-1250, CLC-2000 e ELC-2000. Ciò può essere attribuito alla maggiore o minore stabilità dei sistemi colloidali ottenuti durante la formulazione delle microsfere. La stabilità delle microgocce di soluzione organica disperse in fase acquosa dipende dalla superficie totale esibita dalla fase organica e dalla tensione interfacciale che prende origine dal fatto che la superficie della goccia è sede di una discontinuità di campo elettrico, dovuta alla diversa entità di questo all’interno della goccia organica e nella fase acquosa [11]. Tale stabilità viene espressa quantitativamente dall’energia superficiale (Es) della massa di soluzione organica ed è data dal prodotto della tensione
interfacciale specifica (tensione interfacciale per cm2) per la superficie totale della soluzione organica:
Es = γσ dove:
γ = tensione interfacciale specifica (erg*cm-2) σ = superficie totale (cm2)
L’energia superficiale tende a raggiungere il minor valore possibile a causa della tendenza di ogni sistema a portarsi allo stato con minor contenuto di energia.
L’energia superficiale può diminuire sia per diminuzione di γ che di σ. Il valore di γ è strettamente correlato alla natura della fase organica e al tipo di emulsionante utilizzato. Considerando che l’agitazione meccanica imposta al sistema riduce l’iniziale massa di fase organica in microgocce, aumentando sensibilmente il valore di σ, segue che l’energia superficiale può assumere un valore accettabile, tale da conferire stabilità al sistema, solo per le fasi organiche che presentano intrinsecamente un basso valore di γ, a parità di emulsionante utilizzato. In caso contrario, l’energia superficiale deve essere minimizzata da σ o, in altri termini, ci deve essere la formazione di gocce di dimensioni maggiori le quali forniscono, rispetto alla formazione di gocce più piccole, una superficie totale di fase organica minore. In definitiva, le soluzioni organiche dei polimeri contenenti segmenti di PEG in catena, sono in grado di formare, durante la fase di agitazione, microgocce di dimensioni minori a causa delle interazioni favorevoli dei segmenti di PEG con la fase acquosa che si traducono in una diminuzione di γ, portando infine alla formazione di microsfere più piccole. Contrariamente, le soluzioni dei polimeri non contenenti segmenti di PEG in catena presentano delle interazioni
67 sfavorevoli con la fase acquosa che si traducono in un elevato valore di γ. Conseguentemente, le microgocce di piccole dimensioni tendono a coalescere portando alla formazione di gocce di maggiori dimensioni, allo scopo di ridurre l’energia superficiale riducendo il valore di σ. In seguito all’evaporazione del solvente, si ottengono quindi microsfere di dimensioni maggiori. L’effetto dell’emulsionante sul valore di γ viene dimostrato da un confronto tra le figure 3.5-3.7 dalle quali si evidenzia una significativa diminuzione delle dimensioni delle microsfere di CHC-1250, che non presenta segmenti di PEG in catena, in seguito all’utilizzo di Tween80 o di PVP.
3.2.2 INDICE DI RISOSPENDIBILITÁ
Una valutazione semi-quantitativa della disperdibilità delle microsfere, è stata effettuata con il metodo riportato nel paragrafo 2.4.4 esprimendo i risultati in termini di Indice di Risospendibilità (R.I.) [61]. Tale procedura consente, in prima approssimazione, di valutare la natura colloidale (intrinseca o estrinseca) del sistema in esame. Il metodo consiste nel valutare la quantità di particelle che una volta seccate, possono essere disperse quando vengono messe a contatto con la fase acquosa. A tale scopo una quantità nota di particelle è stata sospesa in acqua e sottoposta ad agitazione con il vortex per 5 minuti. Successivamente, la sospensione è stata lasciata a riposo per un breve periodo di tempo al fine di permettere la sedimentazione spontanea degli aggregati. Dopo tale periodo, il surnatante è stato cautamente rimosso, e su di esso, è stata ripetuta la procedura precedente altre due volte per permettere una completa sedimentazione degli eventuali aggregati rimasti. Gli aggregati sedimentati sono stati infine liofilizzati e pesati. Il surnatante ottenuto dopo il terzo periodo periodo di sedimentazione è stato centrifugato e il residuo ottenuto è stato liofilizzato e pesato. L’Indice di Risospendibilità è stato calcolato secondo la formula:
R.I. = Mr/(Mr + Ma) dove:
Mr = massa risospesa Ma = massa aggregata
L’intera procedura è stata condotta su tre replicati. In tabella 3.2 vengono riportati i valori medi di R.I. relativi alle diverse tipologie di microsfere.
68 TABELLA 3.2 Indici di Risospendibilità (R.I.) delle microsfere
POLIMERO Mn % PEG EMULSIONANTE R.I. %
C27 203700 34 (Mn 35000) PVA 100 CLCE-635 34500 35 PVA 88,9 CHC-1250 77300 0 PVA PVA + PVP 18,8 37,5 CLCE-650 21100 50 PVA PVA + TW 100 100 CLC-2000 178000 0 PVA 48,2 ELC-2000 92200 0 PVA 70,2
Le microsfere preparate da polimeri contenenti segmenti di PEG in catena, mostrano i più alti R.I. In particolare, un confronto tra il CLCE-635 e il CLCE-650, aventi pesi molecolari confrontabili, mostra che la risospendibilità delle particelle aumenta all’aumentare del contenuto di PEG in catena. Ciò è plausibile considerando l’elevata idrofilia del PEG che rende le microsfere più affini alla fase disperdente conferendo così ai colloidi una natura intrinseca. Per quanto riguarda le microsfere di C27, nonostante l’alto peso molecolare del polimero che spesso porta ad una disperdibilità meno soddisfacente [62], la natura anfifilica del polimero stesso fa si che le macromolecole presentino una distribuzione superficiale dei segmenti PEG e un ripiegamento dei segmenti di PCL verso l’interno della sfera. In questo modo, le particelle acquisiscono una notevole idrofilia e vengono facilmente idratate dalla fase acquosa.
Le restanti tre tipologie di microsfere, esibiscono valori di R.I. nettamente inferiori, comportamento che può essere primariamente attribuito alla mancanza di segmenti PEG nelle catene macromolecolari. La presenza di gruppi esterei laterali nelle macromolecole costituenti le microsfere influenza positivamente la loro disperdibilità compensando, almeno in parte, la mancanza di segmenti PEG in catena principale. Questo effetto è più marcato per le particelle di ELC-2000 che contengono gruppi esterei laterali sia nei segmenti dell’estensore di catena, sia in quelli del diisocianato.
69 Il più basso valore di R.I. si riscontra per le microsfere preparate da CHC-1250 utilizzando PVA come emulsionante. Questo basso valore è in accordo con quanto mostrato in figura 3.5 dove si può notare un parziale fenomeno di coalescenza delle particelle più piccole. L’utilizzo di una miscela di PVA e PVP come emulsionante, produce un discreto miglioramento della disperdibilità, aumentando R.I. da 18.8% a 37.5%. Tale miglioramento è già stato apprezzato nelle figure 3.6-3.7 dove si nota una morfologia sferica maggiormente definita.
A causa del basso valore di R.I. esibito dalle particelle di CHC-1250, le caratterizzazioni descritte in seguito sono state effettuate sulle rimanenti 5 tipologie di microsfere.
3.2.3 EFFICACIA DI INCAPSULAMENTO
La quantità di farmaco incapsulato dalle microsfere è stata espressa in termini di efficacia di incapsulamento (EE) [61]. Tale quantità è stata determinata solubilizzando le particelle in DCM e portando a secco la soluzione. Il residuo è stato ripreso con acetonitrile in cui risulta solubile il farmaco ma non il polimero. La sospensione risultante è stata analizzata, dopo filtrazione e diluizione, mediante HPLC. Per valutare l’efficienza di tale metodologia, miscele fisiche di microsfere placebo e farmaco con gli stessi rapporti in peso polimero/PXT delle microsfere cariche, sono state sottoposte alla medesima procedura al fine di determinare un eventuale fattore di correzione per le EE. La riprecipitazione dei polimeri in acetonitrile, potrebbe infatti causare l’occlusione di una certa quantità di farmaco, portando ad errori in difetto nella determinazione delle EE. Per ogni campione, sono analizzati tre replicati e l’efficacia di incapsulamento, è stata espressa come valor medio delle tre determinazioni.
In tabella 3.3 sono riportate le EE corrette, per le particelle preparate a partire dai polimeri C27, CLCE-635, CLCE-650, CLC-2000 e ELC-2000.
70 TABELLA 3.3 Efficacia di Incapsulamento (EE) delle microsfere
POLIMERO EMULSIONANTE PXT CARICATO
(W/W %) EE % C27 PVA 5 20 87,4 82,7 CLCE-635 PVA 5 20 84 79,1 CLCE-650 PVA PVA+TW 5 ; 20 5 ; 20 97,2 ; 95,7 26,9 ; 26,6 CLC-2000 PVA 5 20 56,3 52,8 ELC-2000 PVA 5 20 68,2 64,0
In generale i diversi tipi di microsfere preparate utilizzando PVA come emulsionante, esibiscono alti valori di EE. In accordo con quanto riportato da Ruan e Feng [61] un’alta EE di un farmaco idrofobo come il PXT, è ottenibile abbastanza facilmente preparando le microsfere con la tecnica di evaporazione-estrazione del solvente ad emulsione singola olio in acqua, a causa della perdita ridotta di PXT nella fase acquosa durante il processo di preparazione, rispetto a farmaci idrofili.
Sorprendentemente, i dati di EE mostrano un andamento opposto rispetto a quello atteso nei riguardi dell’idrofilia dei polimeri costituenti le particelle: il 97% di incapsulamento viene raggiunto dalle particelle preparate a partire da CLCE-650 (con PVA come emulsionante) che contiene una maggiore percentuale di PEG in catena rispetto al CLCE-635 il quale mantiene comunque un alto valore di EE. Tale comportamento può essere messo in relazione con la disposizione assunta dalle macromolecole durante il processo di formazione delle microsfere. Assumendo che i segmenti di PEG tendano a distribuirsi preferibilmente sullo strato superficiale delle sfere a causa della loro idrofilia, il farmaco idrofobo manifesterebbe la tendenza a rimanere incapsulato all’interno delle particelle piuttosto che adsorbito sulla superficie delle stesse a causa di interazioni del tipo molecola idrofobo/strato idrofilo poco conveniente. In questo modo
71 la quantità di farmaco adsorbito è trascurabile e non si hanno perdite nelle fasi di lavaggio.
Una drastica diminuzione del valore di EE per le microsfere di CLCE-650 si osserva in seguito all’utilizzo di Tween 80 nel processo di preparazione evidenziando come la scelta dell’emulsionante sia determinante per ottenere un soddisfacente incapsulamento del farmaco. Una tale diminuzione del valore di EE si trova in accordo con il ragionamento espresso in precedenza per il fatto che il Tween 80 può formare uno strato oleoso maggiormente idrofobo sulla superficie delle sfere favorendo l’adsorbimento del PXT che viene perso durante i ripetuti lavaggi. L’effetto di vari emulsionanti sull’efficacia di incapsulamento relativamente al tipo e alle quantità utilizzate, è stato riportato da Feng e Huang [58] che hanno attribuito minori valori di EE a possibili interazioni tra il farmaco e l’eccesso di emulsionante utilizzato.
Le microsfere preparate da CLC-2000 e ELC-2000 esibiscono un discreto valore di EE comunque inferiore a quello mostrato dalle altre tipologie di particelle a parità di emulsionante utilizzato. Due fattori possono essere presi in considerazione per spiegare tale comportamento: una minore idrofilia superficiale delle particelle di CLC-2000 e ELC-2000 tenderebbe a favorire un certo grado di adsorbimento del PXT limitandone l’incapsulamento; d’altra parte, la maggiore dimensione particellare soprattutto per le microsfere di CLC-2000, come mostrato in figura 3.10, tende a favorire l’incapsulamento del farmaco a causa della diminuzione di area superficiale per unità di volume. La sommatoria dei due effetti risulta comunque in una EE inferiore rispetto a quella esibita dalle altre microsfere indicando come la presenza di PEG in catena sia il fattore determinante per l’incapsulamento. Infine, è da sottolineare che dai dati di tabella 3.3, si osserva in tutti i casi un innalzamento dei valori di EE al diminuire della quantità di farmaco utilizzato. Ciò risulta in accordo con quanto riportato da Liggins e Burt relativamente a microsfere preparate da poli(L-acido latico) caricate con PXT [63].
72
3.2.4 STATO FISICO DELLA MATRICE POLIMERICA E DEL
FARMACO INCAPSULATO
Lo stato fisico della matrice polimerica e del farmaco incapsulato è stato analizzato mediante calorimetria a scansione differenziale (DSC). Mediante analisi di farmaco, materiale polimerico e microsfere prodotte, è possibile ottenere informazioni circa la natura del farmaco incapsulato nella matrice polimerica che può presentarsi in uno stato amorfo o cristallino [52]. Tale tecnica fornisce inoltre delle indicazioni su come il farmaco, una volta incapsulato, influenzi lo stato fisico del materiale polimerico convertito in forma microparticellare [53].
In tabella 3.4 vengono riassunte le proprietà termiche dei polimeri utilizzati
TABELLA 3.4 Proprietà termiche dei polimeri utilizzati per la preparazione delle microsfere POLIMERO Tg (°C) Tm (°C) ∆H (J/g) C27 CLCE-635 -41,5 52,8 91,9 CLCE-650 -41,5 53,0 90,2 CLC-2000 n.d. 41,8 62,3 ELC-2000 n.d. 45,5 54,3
Come riportato in seguito, l’analisi calorimetrica è stata condotta sulle microsfere preparate dai polimeri C27, CLCE-635, CLCE-650, CLC-2000 e ELC-2000 contenenti PXT al 20% w/w, e sulle relative microsfere placebo. Per poter effettuare dei confronti diretti tra le microsfere caricate con PXT e le relative microsfere placebo, è necessario che i due tipi di particelle presentino la stessa “storia termica”. A tale scopo, le microsfere placebo sono state preparate parallelamente a quelle caricate con PXT ed entrambe hanno subito, sempre in parallelo, gli stessi trattamenti per lo stesso intervallo di tempo. In figura 3.16viene riportato il termogramma del PXT puro.
73 FIGURA 3.16 Termogramma del Paclitaxel puro
Il termogramma evidenzia un’endoderma di fusione a 223° C in accordo con il risultato riportato da Liggins e collaboratori [55]. Nelle figure 3.17-3.21 vengono riportati i termogrammi relativi alle microsfere di C27, CLCE-635, CLCE-650, CLC-2000 e ELC-2000 caricate con PXT insieme ai termogrammi delle relative microsfere placebo. Per il CLCE-650 viene riportato, come confronto, solo il termogramma delle microsfere placebo preparate utilizzando PVA come emulsionante, dato che il termogramma delle particelle placebo preparate con PVA+Tween80, presentava lo stesso andamento con una leggera differenza per i valori di Tm e ∆H (tabella 3.5).
74 FIGURA 3.17 Termogramma in prima scansione delle microsfere placebo di C27 (1) e delle microsfere di C27 caricate con PXT al 20% w/w (2)
FIGURA 3.18 Termogramma in prima scansione delle microsfere placebo di CLCE-635 (1) e delle microsfere di CLCE-635 caricate con PXT al 20% w/w (2)
75 FIGURA 3.19 Termogramma in prima scansione delle microsfere placebo di CLCE-650 (1), delle microsfere di CLCE-650 caricate con PXT al 20% w/w con PVA come emulsionante (2) e delle microsfere caricate con PXT al 20% w/w con PVA+Tween80 come emulsionante (3)
FIGURA 3.20 Termogramma in prima scansione delle microsfere placebo di CLC-2000 (1) e delle microsfere di CLC-2000 caricate con PXT al 20% w/w (2)
76 FIGURA 3.21 Termogramma in prima scansione delle microsfere placebo di ELC-2000 (1) e delle microsfere di ELC-2000 caricate con PXT al 20% w/w (2)
In tabella 3.5 vengono riassunti i dati relativi alle proprietà termiche delle microsfere caricate con PXT e placebo, ricavati dai termogrammi sopra riportati.
TABELLA 3.5 Proprietà termiche delle microsfere caricate con PXT (20% w/w) e delle microsfere placebo
POLIMERO MICROSFERE
CARICATE CON PXT MICROSFERE PLACEBO ∆H (J/g) Tm (°C) ∆H (J/g) Tm (°C) C27 66,3 56,3 84,9 60,3 CLCE-635 40,7 51,3 43,3 52,7 CLCE-650 (PVA) 24,9 45,3 44,0 51,2 CLCE-650 (PVA+Tween80) 41,8 50,3 44,2 51,3 CLC-2000 49,8 47,3 52,0 50,2 ELC-2000 64,3 48,0 73,1 55,7
77 In generale, per tutte le tipologie di microsfere, si osserva una diminuzione del valore del ∆H, e un conseguente abbassamento del grado di cristallinità, per le microsfere caricate con PXT rispetto a quelle placebo, attribuibile oltre che al processo di preparazione, all’influenza esercitata dal farmaco incapsulato sulla possibilità di riarrangiamento delle macromolecole in strutture ordinate. La maggiore diminuzione si registra per le microsfere di CLCE-650 (PVA) e per le microsfere di C27. Le variazioni di ∆H relative alle altre tipologie di particelle sono meno accentuate. A parità di condizioni sperimentali, tali maggiori o minori variazioni di ∆H possono essere primariamente attribuite alla presenza del farmaco che interagisce maggiormente, una volta incapsulato, con il CLCE-650 e con il C27. Ciò si trova in accordo con i dati di EE riportati nella Tabella 3.3 relativi ai sudetti polimeri e con la teoria proposta per il meccanismo dell’incapsulamento. Un’ulteriore conferma viene fornita dai valori di ∆H relativi alle microsfere di CLCE-650 (PVA+Tween80) per le quali si registra una diminuzione di ∆H pari a 2,4 (J/g) nel passare dalle particelle caricate con PXT a quelle placebo. L’utilizzo di Tween80 abbassa drasticamente il valore di EE, come riportato in tabella 3.3, e di conseguenza le macromolecole vengono influenzate meno a causa della minor quantità di farmaco incapsulato, e possono riarrangiarsi in strutture ordinate tali che le particelle possano esibire un valore di ∆H e un conseguente grado di cristallinità, paragonabile alle rispettive microsfere placebo. Per quanto riguarda lo stato fisico del farmaco incapsulato, nessun picco viene osservato, nei termogrammi relativi alle microsfere caricate, nella regione di temperatura attorno a 223°C, temperatura a cui il PXT esibisce la sua endoderma di fusione (figura 3.16). Ciò indica che il farmaco, una volta incapsulato, si presenta in forma amorfa o comunque disperso a livello molecolare nella matrice polimerica, perdendo il suo grado di cristallinità [51]. Lo stesso comportamento del PXT è stato riscontrato da Mu e Feng [52] e da Ruan e Feng [53] rispetto ad altri materiali polimerici e a differenti metodi di preparazione delle microsfere. Lo stato amorfo del farmaco incapsulato è spesso desiderabile, a causa di un rilascio facilitato dalla matrice polimerica e di un profilo cinetico di rilascio più idoneo [53].
78
3.2.5 RILASCIO DI PACLITAXEL IN VITRO
Il rilascio di PXT dai sistemi microparticellari è stato valutato in soluzione tampone (pH =7,4) a 37°C, per le microsfere caricate con PXT al 20% w/w e al 5% w/w recuperate dalla centrifugazione a 5000 rpm, per una durata di 34 giorni. Le cinetiche di rilascio sono state seguite prelevando, ad intervalli di tempo diversi, la soluzione di rilascio e determinando la concentrazione di PXT mediante HPLC. A tale proposito, le particelle sono state centrifugate a 6000 rpm per ottenere una soluzione surnatante limpida. Aliquote misurate di tale soluzione sono state prelevate e diluite 1:1 con acetonitrile. La diluizione è stata effettuata allo scopo di prevenire un’eventuale precipitazione in colonna dei sali presenti in soluzione, essendo stata scelta come fase mobile una miscela acqua/acetonitrile 58/42. Le concentrazioni delle soluzioni di PXT sono state ricavate dalla retta di taratura riportata in appendice costruita con 10 soluzioni standard di PXT puro. Dopo ogni prelievo le particelle sono state ridisperse in soluzione tampone fresca. Le quantità di farmaco trovate per ogni volume prelevato da ogni singolo campione, sono state progressivamente sommate per valutare la percentuale di PXT rilasciato nel tempo. Per ogni campione di microsfere, l’analisi è stata condotta su tre replicati; i valori di rilascio riportati nelle tabelle 3.6-3.11 rappresentano la media delle tre determinazioni. I dati sono stati approssimati alla terza cifra significativa. Per ogni tabella vengono inoltre riportate le rappresentazioni grafiche dei profili di rilascio (figure 3.22-3.27) da cui si osserva una cinetica più lenta per le microsfere caricate con la maggior quantità di farmaco. Un andamento simile si trova in accordo con quanto riportato da Ruan e Feng [54]e Wang e collaboratori [56] che hanno attribuito le differenti cinetiche di rilascio al cambiamento della diffusione del farmaco causata dalla maggiore o minore quantità di incapsulamento. Un andamento opposto è stato invece riscontrato da Liggins e collaboratori [52].
79 TABELLA 3.6 Rilascio di Paclitaxel dalle microsfere di C27
MICROSFERE DI C27
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 - 0 - 1 1,42 0,67 3 2,07 0,87 6 2,93 1,38 8 3,37 1,64 10 3,88 1,97 13 4,35 2,17 15 4,85 2,48 17 5,36 2,77 20 5,83 3,12 22 6,30 3,27 24 6,79 3,46 27 7,21 3,70 29 7,64 3,89 32 7,91 4,02 34 8,15 4,10 0 1 2 3 4 5 6 7 8 9 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.22 Cinetica di rilascio delle microsfere di C27 Tempo/giorni
80 TABELLA 3.7 Rilascio di Paclitaxel dalle microsfere di CLCE-635
MICROSFERE DI CLCE-635
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 0 1 3,58 1,12 3 5,47 1,69 6 7,66 2,63 8 8,65 2,86 10 10,6 3,61 13 12,4 3,88 15 14,5 4,68 17 15,8 5,17 20 17,6 5,82 22 19,5 6,63 24 21,0 7,09 27 21,9 7,53 29 23,3 7,98 32 25,0 8,38 34 26,0 8,70 0 5 10 15 20 25 30 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.23 Cinetica di rilascio delle microsfere di CLCE-635 Tempo/giorni
81 TABELLA 3.8 Rilascio di Paclitaxel dalle microsfere di CLCE-650 (PVA)
MICROSFERE DI CLCE-650 (PVA)
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 0 1 3,45 2,08 3 6,07 3,57 6 8,44 4,95 8 9,72 5,72 10 10,7 6,32 13 12,1 7,10 15 13,1 7,71 17 14,2 8,37 20 15,0 8,85 22 15,9 9,33 24 16,6 9,76 27 17,4 10,2 29 18,5 10,9 32 19,3 11,4 34 19,9 11,7 0 5 10 15 20 25 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.24 Cinetica di rilascio delle microsfere di CLCE-650 (PVA) Tempo/giorni
82 TABELLA 3.9 Rilascio di Paclitaxel dalle microsfere di CLCE-650 (Tween80)
MICROSFERE DI CLCE-650 (PVA+Tween80)
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 0 1 4,76 2,07 3 9,61 4,17 6 14,4 6,28 8 18,7 8,14 10 22,8 9,93 13 26,6 11,5 15 30,3 13,2 17 33,5 14,6 20 36,5 15,9 22 39,3 17,1 24 41,7 18,1 27 43,9 19,1 29 46,5 20,0 32 47,7 20,7 34 49,1 21,4 0 10 20 30 40 50 60 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.25 Cinetica di rilascio delle microsfere di CLCE-650 (PVA+Tween80) Tempo/giorni
83 TABELLA 3.10 Rilascio di Paclitaxel dalle microsfere di CLC-2000
MICROSFERE DI CLC-2000
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 0 1 0,46 0,16 3 0,84 0,35 6 1,06 0,49 8 1,21 0,56 10 1,26 0,62 13 1,40 0,71 15 1,51 0,78 17 1,58 0,86 20 1,69 0,94 22 1,76 0,99 24 1,80 1,01 27 1,88 1,05 29 1,92 1,07 32 2,00 1,10 34 2,05 1,12 0 0,5 1 1,5 2 2,5 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.26 Cinetica di rilascio delle microsfere di CLC-2000 Tempo/giorni
84 TABELLA 3.11 Rilascio di Paclitaxel dalle microsfere di ELC-2000
MICROSFERE DI ELC-2000
PACLITAXEL 5% w/w PACLITAXEL 20% w/w Giorni Rilascio cumulativo% S.D. Rilascio cumulativo% S.D.
0 0 0 1 3,17 1,36 3 5,50 2,39 6 6,75 2,90 8 7,51 3,25 10 8,27 3,57 13 9,21 3,98 15 10,2 4,42 17 10,8 4,68 20 11,7 5,00 22 12,4 5,35 24 12,8 5,50 27 13,3 5,73 29 13,6 5,89 32 14,1 6,08 34 14,4 6,21 0 2 4 6 8 10 12 14 16 0 5 10 15 20 25 30 35 40 Giorni % P X T r ila sc ia to PXT 5% w/w PXT 20% w/w
FIGURA 3.27 Cinetica di rilascio delle microsfere di ELC-2000 Tempo/giorni
85 Per tutte le tipologie di microsfere, il rilascio di PXT presenta, dopo i primi 5 giorni un andamento quasi lineare, indicante una cinetica di rilascio praticamente di ordine zero. Un dato molto significativo per le particelle caricate al 5% w/w, è rappresentato dal basso “effetto burst” iniziale, quantificato in un rilascio di circa 2,9% per le microsfere di C27, 7,7% per le microsfere di CLCE-635, 8,4% per le microsfere di CLCE-650 (PVA), 1% per le microsfere di CLC-2000 e 6,7% per le microsfere di ELC-2000 nei primi 6 giorni. Valori nettamente inferiori vengono riscontrati per le microsfere caricate al 20% w/w. Un tale andamento è desiderabile per farmaci come il PXT che possono dare luogo a fenomeni di tossicità locale dovuti ad un accumulo eccessivo.
In figura 3.28 viene effettuato un confronto diretto tra i profili di rilascio delle microsfere caricate con PXT al 5% w/w.
0 5 10 15 20 25 30 0 5 10 15 20 25 30 35 40 Tempo/giorni % P X T r il asci at o CLCE-635 PXT 5% w/w CLCE-650 (PVA) PXT 5% w/w ELC-2000 PXT 5% w/w C27 PXT 5% w/w CLC-2000 PXT 5% w/w
FIGURA 3.28 Cinetiche di rilascio per le microsfere caricate con PXT al 5% w/w
Le particelle di CLC-2000 esibiscono il rilascio più lento che raggiunge un valore pari circa al 2% dopo 34 giorni. Tale cinetica di rilascio è strettamente correlabile con l’elevato peso molecolare, con le dimensioni delle particelle e con l’assenza di segmenti di PEG nelle catene macromolecolari. Microsfere preparate da polimeri ad alto peso molecolare mostrano, nei primi periodi, un rilascio di farmaco piuttosto lento imputabile ad un meccanismo di diffusione [59]. La diffusione del farmaco attraverso gli interstizi macromolecolari, viene inoltre rallentata dall’assenza di segmenti di PEG che faciliterebbero l’idratazione delle particelle dando luogo ad un maggiore rigonfiamento delle sfere, permettendo così una diffusione più rapida del farmaco attraverso gli
86 interstizi delle catene macromolecolari. Inoltre l’assenza di PEG, influenza significativamente la degradabilità delle microsfere a causa della difficoltosa idratazione delle stesse e della conseguente difficoltà di idrolisi dei gruppi esterei presenti in catena. Infine, l’elevata dimensione delle microsfere di CLC-2000 favorisce un rilascio più lento in accordo con quanto riportato da Narayani e Rao [57].
Le microsfere di C27 rilasciano poco più dell’8% di PXT nel periodo monitorato. I fattori limitanti per il rilascio da tali microsfere sono l’elevato peso molecolare del polimero e un grado di cristallinità maggiore rispetto alle altre tipologie di particelle come si può vedere confrontando i dati di ∆H riportati in tabella 3.5. In particolare, il maggiore grado di cristallinità, tende a rallentare i fenomeni di degradazione che sono più rapidi per le regioni amorfe [56]. Tali fattori vengono tuttavia in parte compensati dalla presenza di un’alta percentuale di PEG in catena e dalle dimensioni ridotte delle particelle. La sommatoria di questi fattori, rende possibile un rilascio di PXT più rapido rispetto alle microsfere di CLC-2000.
Un rilascio ancora più rapido viene evidenziato per le microsfere di ELC-2000 che raggiunge un valore pari a circa 14,4% il giorno 34. Una differenza tra le cinetiche di rilascio delle particelle di ELC-2000 e CLC-2000 così marcata può essere primariamente attribuita, oltre che alla grande differenza di peso molecolare e alle dimensioni inferiori delle microsfere di ELC-2000, al grado di porosità esibito da quest’ultime che facilita enormemente la diffusione del farmaco. Inoltre, la presenza di gruppi esterei laterali, in ELC-2000, facilita l’idratazione e il successivo rigonfiamento delle particelle, favorendo ancora una volta la diffusione del farmaco.
Un confronto tra le microsfere di CLCE-635 e CLCE-650 mostra un profilo cinetico di rilascio molto simile fino al dodicesimo giorno con una percentuale di PXT rilasciato, leggermente superiore per le particelle di CLCE-650. Successivamente, le cinetiche di rilascio tendono a differenziarsi con un andamento che evidenzia un rilascio più rapido per le microsfere di CLCE-635 che raggiunge il 26% il giorno 34 contro il 20% raggiunto dalle microsfere di CLCE-650 lo stesso giorno. Un andamento di questo tipo può apparire in prima analisi sorprendente dato che il CLCE-635 e il CLCE-650, aventi tra l’altro un peso molecolare confrontabile, differiscono per la diversa percentuale di PEG in catena che è maggiore per il CLCE-650. La più alta percentuale di PEG facilita l’idratazione e il successivo rigonfiamento delle particelle, fenomeni che permettono una maggiore diffusione del farmaco. Anche rispetto a meccanismi degradativi sarebbe da attendersi un rilascio più rapido per le microsfere di CLCE-650 a causa della
87 maggiore quantità di PEG che facilita l’idrolisi dei gruppi esterei presenti in catena. Tuttavia i profili cinetici di rilascio per tali microsfere, risultano in accordo con i dati relativi al DSC: dai termogrammi riportati nelle figure 3.18 e 3.19, si evidenzia che l’endoterma di fusione del CLCE-635 non viene influenzata dalla presenza del PXT, mentre una notevole influenza viene esercitata dal farmaco nei confronti del CLCE-650. Le maggiori interazioni del farmaco in fase amorfa con le più estese regioni amorfe delle particelle di CLCE-650, rendono più difficoltoso il rilascio rispetto alle microsfere di CLCE-635.
Un’indagine qualitativa della degradazione delle microsfere è stata condotta mediante analisi al SEM osservando la morfologia delle particelle alla fine del periodo di rilascio monitorato. Nelle figure 3.29 e 3.30 vengono mostrate le micrografie delle microsfere di CLCE-650 (PVA) e C27 caricate con PXT al 5% w/w alla fine del periodo di rilascio.
88 FIGURA 3.30 Microsfere di C27 dopo 34 giorni in soluzione tampone
Da un confronto tra la figura 3.29 con la figura 3.8 si nota chiaramente una netta diminuzione delle dimensioni particellari. Dopo il periodo di rilascio le microsfere di CLCE-650 (PVA) mantengono comunque una forma sferica lasciando presupporre una degradazione di tipo superficiale (surface erosion) caratteristica dei polimeri più idrofili. Inoltre, l’ottima morfologia mostrata in origine dalle microsfere, favorisce una degradazione omogenea della superficie, tale da rendere possibile il mantenimento della forma sferica. Rapportando il profilo cinetico di rilascio di PXT esibito dalle microsfere di CLCE-650 (PVA) con la loro variazione dimensionale dopo 34 giorni, lascia inoltre presupporre una massiccia concentrazione del farmaco nel nucleo delle particelle. Non è quindi da escludere, per periodi successivi a quello monitorato, un significativo cambiamento della cinetica di rilascio.
Un confronto tra la figura 3.30 con la figura 3.2 mostra una diminuzione delle dimensioni particellari meno evidente e una parziale perdita di sfericità. Ciò si trova in accordo con la lenta cinetica di rilascio mostrata dalle microsfere di C27 per le quali i fenomeni degradativi vengono rallentati dall’elevato grado di cristallinità esibito da tali particelle. Da ciò si deduce che per le microsfere di C27, nei primi periodi, il rilascio di farmaco sia controllato principalmente dalla diffusione.
89 È tuttavia da sottolineare che tali deduzioni, come affermato in precedenza, sono essenzialmente di tipo qualitativo perché basate sul giudizio visivo di immagini casuali fornite dal SEM, anche se in qualche modo supportate dai dati forniti dai profili di rilascio.
L’utilizzo di Tween80 nella fase di preparazione delle microsfere di CLCE-650, altera drasticamente la cinetica di rilascio. Come riportato in figura 3.31la quantità di farmaco rilasciato dalle particelle preparate dalla miscela di emulsionanti, raggiunge circa il 49% il giorno 34 contro il 20% raggiunto dalle particelle preparate con il solo PVA. Ciò potrebbe essere una conseguenza del vertiginoso calo del valore di EE registrato per le microsfere preparate con PVA+Tween80, essendo i due fattori, EE e cinetica di rilascio, strettamente collegati [55]. 0 10 20 30 40 50 60 0 5 10 15 20 25 30 35 40 Tempo/giorni % P X T r ila s c ia to CLCE-650 (PVA+Tween80) CLCE-650 (PVA)
FIGURA 3.31 Cinetiche di rilascio per le microsfere di CLCE-650 caricate con PXT al 5% w/w utilizzando PVA e PVA+Tween80 come emulsionanti
L’effetto di diversi emulsionanti sulla cinetica di rilascio del PXT da micro e nanosfere biodegradabili è stato riportato da Feng e Huang [55] e Mu e Feng [56] i quali hanno evidenziato significativi cambiamenti nei profili di rilascio al variare di quantità, emulsionante o miscela di emulsionanti utilizzati.
90
3.3 PREPARAZIONE E CARATTERIZZAZIONE DELLE
NANOSFERE
Le nanosfere sono state preparate con il metodo illustrato nel paragrafo 2.6. È stata utilizzata la stessa tecnica di preparazione impiegata per le microsfere ossia la tecnica di evaporazione-estrazione del solvente ad emulsione singola olio in acqua, variando alcuni parametri sperimentali. In particolare, sono stati variati:
• Concentrazione del polimero nella fase organica • Rapporto volumetrico fase organica/fase acquosa • Quantità di emulsionante utilizzato
• Velocità di agitazione
Le nanoparticelle sono state preparate utilizzando il C27, il CHC-1250, e il CLCE-650. La concentrazione polimerica nella fase organica è stata diminuita da 60 mg/ml, utilizzata per le microsfere, a 30 mg/ml. Una fase organica più diluita dovrebbe permettere di ottenere aggregati macromolecolari di dimensioni inferiori. Come emulsionante è stato utilizzato PVA la cui concentrazione è stata aumentata, rispetto alla formulazione delle microsfere, dal 2,5% fino ad un valore massimo del 4%. Vari autori riportano una diminuzione delle dimensioni particellari all’aumentare della quantità di emulsionante utilizzato [60,61]; inoltre considerevoli quantità di emulsionante sono necessarie per fornire un’adeguata stabilità colloidale soprattutto nella preparazione di sistemi nanoparticellari che, a parità di rapporto fase acquosa/fase organica, esibiscono un’area superficiale per unità di volume maggiore rispetto a quella dei sistemi microparticellari.
L’agitazione meccanica è stata imposta mediante omogenizzatore ad una velocità di 21500 rpm, e l’ambiente di reazione (un beaker da 250 ml) è stato mantenuto in un bagno acqua/ghiaccio per minimizzare la naturale tendenza delle particelle ad aggregarsi. La bassa temperatura rappresenta un fattore avverso per la preparazione delle nanosfere [63], tuttavia, è stato scelto di operare in tali condizioni, oltre che per il motivo sopra citato, anche per minimizzare la prematura perdita del solvente organico in seguito alla vigorosa agitazione. L’evaporazione del solvente è stata successivamente perseguita mediante l’utilizzo di un agitatore magnetico ad una velocità di circa 250 rpm. Le particelle sono state infine isolate dall’ambiente di reazione, per
91 centrifugazione a 14000 rpm a cui sono seguiti gli stessi stadi descritti precedentemente per le microsfere nel paragrafo 3.1.
L’analisi morfologica delle nanosfere di C27, CHC-1250 e CLCE-650 è stata effettuata mediante microscopio a scansione elettronica. Le rispettive micrografie sono riportate nelle figure 3.32-3.34.
92 FIGURA 3.33 Nanosfere di CHC-1250 (Paclitaxel 5% w/w)
93 Le micrografie mostrano dei risultati non del tutto soddisfacenti. Non essendo disponibili dati di light scattering, le dimensioni delle particelle possono essere valutate solo approssimativamente. Le figure mostrano una dimensione particellare attorno al micrometro, tuttavia risulta difficile valutare se tale dimensione è effettivamente inferiore ad un micrometro così da poter definire le particelle come “nanosfere”.
Le particelle di C27 e CLCE-650, mostrano una forma sferica mal definita ed evidenti fenomeni di coalescenza. Inoltre si può notare la presenza di un eccesso di PVA difficile da eliminare nonostante i ripetuti lavaggi. I maggiori fenomeni di coalescenza si notano per le nanosfere di CLCE-650, probabilmente favoriti rispetto alle particelle di C27, dalla natura elastomerica del materiale macromolecolare.
Le particelle di CHC-1250, sebbene mostrino una migliore forma sferica, successivamente alle fasi di lavaggio, si presentano sottoforma di un unico blocco di materiale gommoso non disgregabile in acqua anche se sottoposto a sonicazione.
In tabella 3.12 viene riportato l’indice di risospendibilità delle nanosfere determinato con la stessa procedura descritta nel paragrafo3.2.2.
TABELLA 3.12 Indici di Risospendibilità (R.I.) delle nanosfere
POLIMERO EMULSIONANTE R.I. %
C27 PVA 3,5% 48
CHC-1250 PVA 4% -
CLCE-650 PVA 3,8% 52
Il valore di R.I. per le nanosfere di CHC-1250 non è risultato determinabile per i motivi esposti in precedenza.
A causa delle insoddisfacenti morfologie e dei valori piuttosto bassi di R.I., non sono state effettuate ulteriori caratterizzazioni.
94
3.4 SINTESI E CARATTERIZZAZIONE DEL POLIURETANO
FUNZIONALIZZATO CON GRGDG
Come primo approccio per la preparazione di microsfere funzionalizzate con sequenze peptidiche è stato realizzato come sistema modello un poliuretano contenente la sequenza Gly-Arg-Gly-Asp-Gly. La sintesi del polimero è stata realizzata secondo una procedura in due stadi (figura 3.35) come riportato nel paragrafo 2.7.
FIGURA 3.35 Schema di sintesi dell’unità ripetitiva contenente il peptide GRGDG nel poliuretano funzionalizzato
OCN R NH C O O C NH R NCO O O C HNRNH O CO O OC O HNRNH C O NH G R G D G O NH NH H2C O n 4 NH2 G R (Pbf) G D (OtBu) G (OMe) O NH2 NH H2C O 4 1) 2) TFA, DCM
95 Il primo stadio ha portato alla formazione di un prepolimero per reazione di PCL diolo (Mn=2000) con LDI in rapporto molare 1:2. Il prepolimero ottenuto è stato
successivamente fatto reagire con L-lisina etil estere e un estensore di catena opportunamente sintetizzato contenente la sequenza peptidica GRGDG (H-Lys-Amc-Gly-Arg(Pbf)-Gly-Asp(OtBu)-Gly-OCH3). L’estensore è stato preparato secondo i
protocolli di sintesi peptidica in fase solida utilizzando in testa un residuo di L-lisina contenente i gruppi amminici liberi necessari per la reazione con i gruppi isocianato del prepolimero. E’ stato inoltre previsto l’inserimento di uno spaziatore (acido esamminopropanoico) tra il residuo di lisina e la sequenza peptidica, in modo da consentire alla stessa un’adeguata libertà conformazionale per l’interazione con il recettore cellulare. Dopo la polimerizzazione le protezioni del peptide sono state rimosse per trattamento con TFA in DCM.
L’avvenuta funzionalizzazione del poliuretano con la sequenza GRGDG protetta è stata evidenziata dall’analisi 1H-NMR confrontando gli spettri dell’estensore contenente il
peptide protetto (figura 3.36), del polimero non funzionalizzato (figura 3.37), e del polimero funzionalizzato con il peptide protetto stesso (figura 3.38)
FIGURA 3.36 Spettro 1H-NMR dell’estensore H-Lys-Amc-Gly-Arg(Pbf)-Gly-Asp(OtBu)-Gly-OCH3 a b c d e
96 FIGURA 3.37 Spettro 1H-NMR del polimero ELC-2000
FIGURA 3.38 Spettro 1H-NMR del polimero PU-GRGDG
a b c d e O S O O CH3 H3C H3C CH3 CH3 c c d e e
97 Lo spettro del polimero funzionalizzato è complesso, tuttavia si possono distinguere i segnali relativi ai gruppi protettivi in catena laterale, da confermare mediante analisi bidimensionale. In particolare i protoni del gruppo terbutossilico forniscono un segnale a 1.2 ppm, il gruppo metossilico sul C-terminale del peptide fornisce un segnale a 2.1 ppm e gli assorbimenti relativi al gruppo Pbf sono evidenziati in figura.
L’analisi FTIR (figura 3.39) ha evidenziato gli assorbimenti caratteristici della struttura ipotizzata: 3400 cm-1 (
ν
N-H) ; 2944 cm-1 (ν
a CH2) ; 2860 cm-1 (ν
s CH2) ; 1722 cm-1(
ν
C=O) ; 1528 cm-1 (δ
N-H) + (ν
C-N) ; 1166 cm-1(ν
C-O-C) .FIGURA 3.39 Spettri FTIR relativi a ELC-2000 (a) e PU-GRGDG (b)
Il confronto tra lo spettro del polimero contenente la sequenza peptidica e un polimero sintetizzato a partire dagli stessi monomeri eccetto l’estensore contenente il peptide non ha evidenziato differenze rilevanti (figura 3.39); tuttavia la presenza di un spalla adiacente al picco a 1722 cm-1 è stata attribuita allo stretching del legame N-H
ammidico del peptide, il cui spettro presenta un picco intenso a 1550 cm-1.
L’analisi SEC (cromatogramma in figura 3.40 ) ha mostrato che il polimero ottenuto ha un peso molecolare elevato e un indice di polidispersità accettabile per reazioni di polimerizzazione a stadi. 4000.0 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 650.0 53.4 56 58 60 62 64 66 68 70 72 74 76 78 80 82 84 86 88 90 92 94 96 98 99.8 cm-1 %T
a
b
δ
N-H98 FIGURA 3.40 Curva SEC del polimero PU-GRGDG
L’analisi UV-VIS, eseguita su soluzioni cloroformiche dei campioni di poliuretano contenenti il peptide prima (PU-GRGDG*, figura 3.41a) e dopo (PU-GRGDG, figura 3.41b) la deprotezione e sul polimero avente L-lisina etil estere come unico estensore (ELC-2000, figura 3.41c), ha consentito di ottenere informazioni qualitative sulla riuscita delle reazioni di polimerizzazione e sulla successiva reazione di deprotezione dei residui amminoacidici in catena laterale. Infatti solo nel caso del polimero PU-GRGDG*, si registra un intenso assorbimento a 256 nm dovuto al gruppo protettivo Pbf, secondo quanto riportato da C.G. Fields e G.B. Fields [74].
FIGURA 3.41 Spettro UV relativo a PU-GRGDG* (a), PU-GRGDG (b) e ELC-2000(c). 0 1000 2000 3000 4000 5000 6000 7000 0 500 1000 1500 2000 2500 3000 3500 Mn = 87300 Mn/Mw = 2.3 t/min
99
3.5 PREPARAZIONE E CARATTERIZZAZIONE DELLE
MICROSFERE DI PU-GRGDG
La preparazione delle microsfere a partire dal PU-GRGDG è stata effettuata con lo stesso metodo descritto nel paragrafo 3.1. Sono stati mantenuti gli stessi parametri operativi utilizzati per la preparazione delle microsfere di ELC-2000 per poter evidenziare l’influenza del peptide sulle caratteristiche delle particelle.
La morfologia delle particelle ottenute è stata studiata mediante analisi al SEM (figura 3.42)
FIGURA 3.42 Microsfere di PU-GRGDG (PXT 5%)
Da un confronto con la figura 3.11 si nota una forma sferica maggiormente definita per le microsfere di PU-GRGDG rispetto a quella mostrata dalle microsfere di ELC-2000. Inoltre le particelle funzionalizzate non mostrano alcuna macroporosità superficiale e presentano una dimensione inferiore rispetto a quelle di ELC-2000. Risulta quindi evidente l’influenza del peptide che impartisce maggiore idrofilia alle microsfere
100 rendendo così possibile una diminuzione delle tensione interfacciale, durante la fase di preparazione, portando infine ad ottenere particelle più piccole.
L’Indice di Risospendibilità e l’Efficacia d’Incapsulamento sono stati determinati con gli stessi metodi discussi nei paragrafi 3.2.2 e 3.2.3. Le determinazioni, eseguite su tre replicati, hanno fornito i seguenti valori:
R.I.% = 84,3 ± 4,6 (PXT 5% w/w) EE% = 71,5 ± 2,3 (PXT 5% w/w)
Confrontando tali valori con quelli relativi alle microsfere di ELC-2000 (tabelle 3.2 e 3.3) si riscontra un significativo innalzamento del valore di R.I. mentre una variazione meno accentuata si registra per EE. Il significativo aumento del valore di R.I. può essere attribuito al maggior carattere idrofilo imposto alle particelle dal peptide che diventano così più affini alla fase acquosa rispetto alle particelle di ELC-2000. L’innalzamento di EE è plausibile considerando un minore adsorbimento superficiale del farmaco, tuttavia tale effetto viene controbilanciato dalle minori dimensioni particellari che in definitiva rendono il valore di EE confrontabile con quello esibito dalle microsfere di ELC-2000. Un’indagine semi-quantitativa per evidenziare la presenza del farmaco all’interno delle microsfere di PU-GRGDG, è stata condotta mediante analisi FTIR. Per rendere possibile l’analisi sono state appositamente preparate microsfere di grandi dimensioni (circa 100 µm) con la procedura descritta al paragrafo 2.8. L’utilizzo di una quantità inferiore di emulsionante e una minore velocità di agitazione meccanica, hanno portato al risultato voluto. È stata quindi eseguita la mappatura di un campione di particelle da cui è stata isolata una singola sfera (figura 3.43-3.44)
101 FIGURA 3.43 Singola microsfera isolata dalla mappatura del campione
102 Successivamente è stato acquisito lo spettro in una zona interna della sfera concentrando l’attenzione su una banda di assorbimento caratteristica del PXT. L’individuazione di una banda di assorbimento caratteristica del farmaco, è stata effettuata per confronto tra lo spettro del PXT puro con lo spettro del PU-GRGDG e la scelta è caduta sulla banda di assorbimento a 1626 cm-1(figura 3.45)
FIGURA 3.45 Spettri FTIR del PXT e del PU-GRGDG
Lo spettro acquisito dall’analisi della microsfera (figura 3.46) evidenzia la presenza della banda di assorbimento a 1626 cm-1 attribuita allo stretching del legame C-C d’anello del PXT. 4000.0 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 650.0 64.5 66 68 70 72 74 76 78 80 82 84 86 88 90 92 94 96 98 99.2 cm-1 %T
PXT
PU-GRGDG
Banda del PXT 1626 cm-1103 FIGURA 3.46 Spettro FTIR della particella esaminata
Per valutare lo stato fisico del PXT incapsulato e il suo effetto sulla matrice polimerica funzionalizzata, sono state preparate microsfere placebo funzionalizzate parallelamente a microsfere funzionalizzate caricate con PXT al 20% w/w, e sottoposte ad analisi termica. I termogrammi ottenuti hanno mostrato dei risultati in linea con quelli ottenuti per le microsfere di ELC-2000 (tabella 3.5) sia per quanto riguarda il grado di cristallinità sia per l’assenza dell’endoterma di fusione del PXT incapsulato.
Lo studio sulla cinetica di rilascio in vitro è stato eseguito sulle microsfere caricate con PXT al 5% w/w per un periodo di 30 giorni. I dati ottenuti e il profilo cinetico vengono rispettivamente mostrati in tabella 3.13 e in figura 3.47.
4000.0 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 650.0 60.8 62 64 66 68 70 72 74 76 78 80 82 84 86 88 90 92 94 96 98 98.9 cm-1 %T PXT 1626 cm-1
104 TABELLA 3.6 Rilascio di Paclitaxel dalle microsfere di PU-GRGDG
MICROSFERE DI PU-GRGDG (PXT 5% w/w)
Giorni Rilascio cumulativo% S.D.
0 0 - 1 2,40 3 3,57 6 4,68 8 5,39 10 6,26 13 7,28 15 7,72 17 8,20 20 8,98 22 9,35 24 9,76 27 10,1 30 10,3 0 2 4 6 8 10 12 0 5 10 15 20 25 30 35 Tempo/giorni % P X T r il asci at o PU-GRGDG PXT 5% w/w
FIGURA 3.47 Cinetica di rilascio per le microsfere di PU-GRGDG caricate con PXT al 5% w/w
105 La figura mostra una cinetica di rilascio praticamente di ordine zero con un basso effetto burst quantificabile in un rilascio pari a 4,68% di farmaco nei primi 6 giorni. Poco più del 10% di PXT viene rilasciato nel periodo monitorato. Da un confronto tra tale profilo cinetico con quello esibito dalle microsfere di ELC-2000, si evince un’influenza trascurabile del peptide sulla cinetica di rilascio. Tuttavia le microsfere funzionalizzate mostrano un rilascio più lento rispetto alle particelle di ELC-2000. Ciò può essere attribuito alla mancanza di porosità delle particelle di PU-GRGDG che limita il rilascio di farmaco per diffusione. D’altro canto, altri due fattori dovrebbero indurre un rilascio più rapido rispetto alle microsfere di ELC-2000: la maggiore idrofilia imposta dal peptide, che facilita l’idratazione e il successivo rigonfiamento delle particelle, e la dimensione inferiore delle particelle stesse. La sommatoria dei vari fattori si traduce comunque in un rilascio più lento per le microsfere funzionalizzate indicando come la porosità sia il fattore determinante.
106
CONCLUSIONI E SVILUPPI FUTURI
Nel presente lavoro di tesi sono state ottenute matrici polimeriche in forma micro e nanoparticellare per il rilascio controllato di Paclitaxel, un farmaco antiproliferativo che ha trovato recente impiego come agente antirestenoico. La tecnica utilizzata per la formulazione delle microsfere (evaporazione-estrazione del solvente ad emulsione singola olio in acqua) si è dimostrata adeguata, rispetto ai materiali utilizzati, per l’ottenimento di particelle con una morfologia ben definita che rappresenta uno dei requisiti fondamentali per ottenere un rilascio controllato di farmaco. La stessa tecnica si è dimostrata meno soddisfacente per la formulazione delle nanosfere che hanno mostrato dei chiari fenomeni di coalescenza.
L’elevato potere di incapsulamento del farmaco esibito dalle microsfere, rappresenta un grande vantaggio nei confronti del Paclitaxel considerando il suo costo elevato. I sistemi mostrano inoltre la possibilità di ottenere un aumento di incapsulamento al diminuire della quantità di farmaco utilizzato nella fase di preparazione delle microsfere, permettendo così l’utilizzo di quantità minime di agente farmacologico con conseguente minimizzazione di inutili sprechi.
Giudizi più cauti possono invece essere dati nei confronti della stabilità colloidale di tali sistemi, essendo stata essa valutata in modo semi-quantitativo. È comunque da sottolineare che la disperdibilità e la conseguente stabilità colloidale dei sistemi microparticellari, cambia drasticamente al variare dell’ambiente circostante; di conseguenza una disperdibilità insoddisfacente in vitro potrebbe tradursi in una buona disperdibilità in ambiente fisiologico e viceversa.
I sistemi microparticellari formulati, sono risultati idonei per ottenere un rilascio controllato di Palitaxel. Il basso effetto burst mostrato nei profili di rilascio, è un fattore fortemente desiderabile per un farmaco come il Paclitaxel che da luogo a fenomeni di tossicità locale in seguito ad accumuli eccessivi. I profili cinetici evidenziano inoltre la possibilità di modulare l’entità del rilascio variando la composizione dei poliuretani e in particolare variando la composizione del segmento soft in modo da ottenere microsfere più o meno idrofile. Una volta stabilita un’adeguata finestra terapeutica per il Paclitaxel relativamente al suo impiego come agente antirestenoico, risulterebbe quindi possibile la sintesi di opportuni poliuretani, e la loro successiva conversione in sistemi microparticellari, semplicemente variando la composizione dei segmenti e in particolare del segmento soft.
107 La procedura sintetica utilizzata per la preparazione dei poliuretani, consente di ottenere poliuretani funzionalizzati con ligandi peptidici mediante l’utilizzo di estensori di catena contenenti tali ligandi. La successiva conversione di tali polimeri in sistemi microparicellari, permette di ottenere microsfere funzionalizzate per rilascio controllato e mirato di farmaco.
Un accurato studio su sequenze peptidiche effettuato tramite la tecnologia della libreria peptidica combinatoriale, ha consentito di individuare due peptici, denominati 5G6 (CNIWGVVLSWIGVFPEC) e 5E5 (CESLWGGLMWTIGLSDC) come molecole segnale per il legame selettivo delle VSMC [73]. Tali peptidi mostrano non solo affinità specifica verso le VSMC rispetto ad altri tipi di cellule testate (cellule endoteliali, macrofagi, monociti e fibroblasti) ma mostrano maggiore affinità verso le VSMC in proliferazione piuttosto che con le VSMC non proliferanti o in altre parole, sono in grado di distinguere il loro fenotipo contrattile da quello proliferativo. Ulteriori prove in vitro hanno evidenziato l’effettiva internalizzazione delle due sequenze da parte delle cellule bersaglio.
Le specifiche caratteristiche di legame dei peptidi 5G6 e 5E5 indicano un loro possibile impiego come molecole segnale in regioni affette da fenomeni di restenosi, la cui causa principale sembra essere la proliferazione incontrollata delle VSMC. La sintesi di un opportuno estensore di catena (figura A) e il suo utilizzo nella sintesi dei poliuretani (figura B) consentirebbe di ottenere, dopo la conversione delle matrici polimeriche in forma particellare, sistemi a rilascio controllato e mirato di tipo attivo contro la restenosi.
FIGURA A (a1-a2) Rappresentazione della struttura degli estensori funzionalizzati con la molecola segnale X. (S = spaziatore; X = 5G6 o 5E5)
108
FIGURA B Schema di sintesi di poliuretani funzionalizzati con la molecola segnale (S = spaziatore; X = 5G6 o 5E5)
OCN R NH CO O OC NH O R NCO + H O OH 2 O C N R NCO HO CH OH X S X S H2N CH NH2 C HN O R NH CO O OC O NH R NH C NH O R' NH n X S C HN O R NH CO O OC O NH R NH C n O O R' O X S Poliuretano Poliuretano-urea Prepolimero
109 Negli schemi di sintesi è stato previsto l’inserimento di uno spaziatore per fornire un certo grado di libertà conformazionale al ligando. Le microsfere funzionalizzate potrebbero essere utilizzate come rivestimento per stent coronarici i quali vengono attualmente impiegati come semplici supporti metallici o rivestiti da coatings polimerici contenenti un agente farmacologico, la cui cinetica di rilascio risulta poco controllabile. Infine l’ottimizzazione della tecnica per la formulazione delle nanosfere, consentirebbe di ottenere nanosfere funzionalizzate per il rilascio controllato e mirato di Paclitaxel così da unire l’orientamento di tipo attivo fornito dal ligando peptidico, con l’orientamento di tipo passivo in seguito all’effetto EPR (Enhanced Permeability and Retention) riscontrato nelle zone affette da restenosi e legato alle ridotte dimensioni delle particelle.